
Targeting Proliferation Signals and the Cell Cycle Machinery in Acute Leukemias: Novel Molecules on the Horizon


 ,
,  and
and 
Abstract
1. Introduction
2. Altered Cell Proliferation in Acute Leukemias
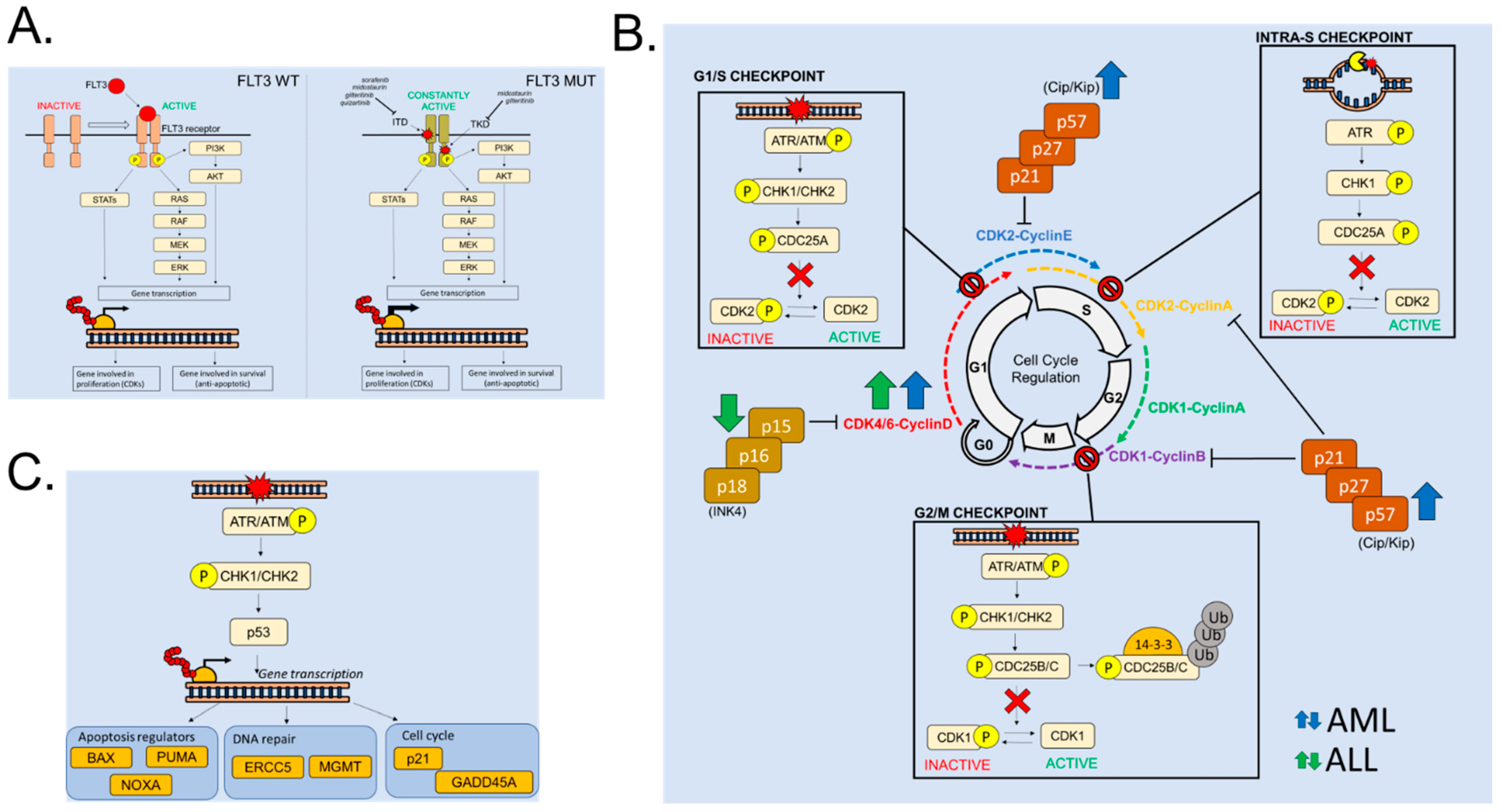
2.1. FLT3 Alterations in Acute Leukemia: A Driver of Cell Proliferation
2.2. Cell Cycle Regulation in Hematopoietic Cells and in Acute Leukemias
2.3. Cell Cycle Alterations in Acute Leukemia
3. Novel Molecules Targeting Proliferative Mechanisms in Acute Leukemias
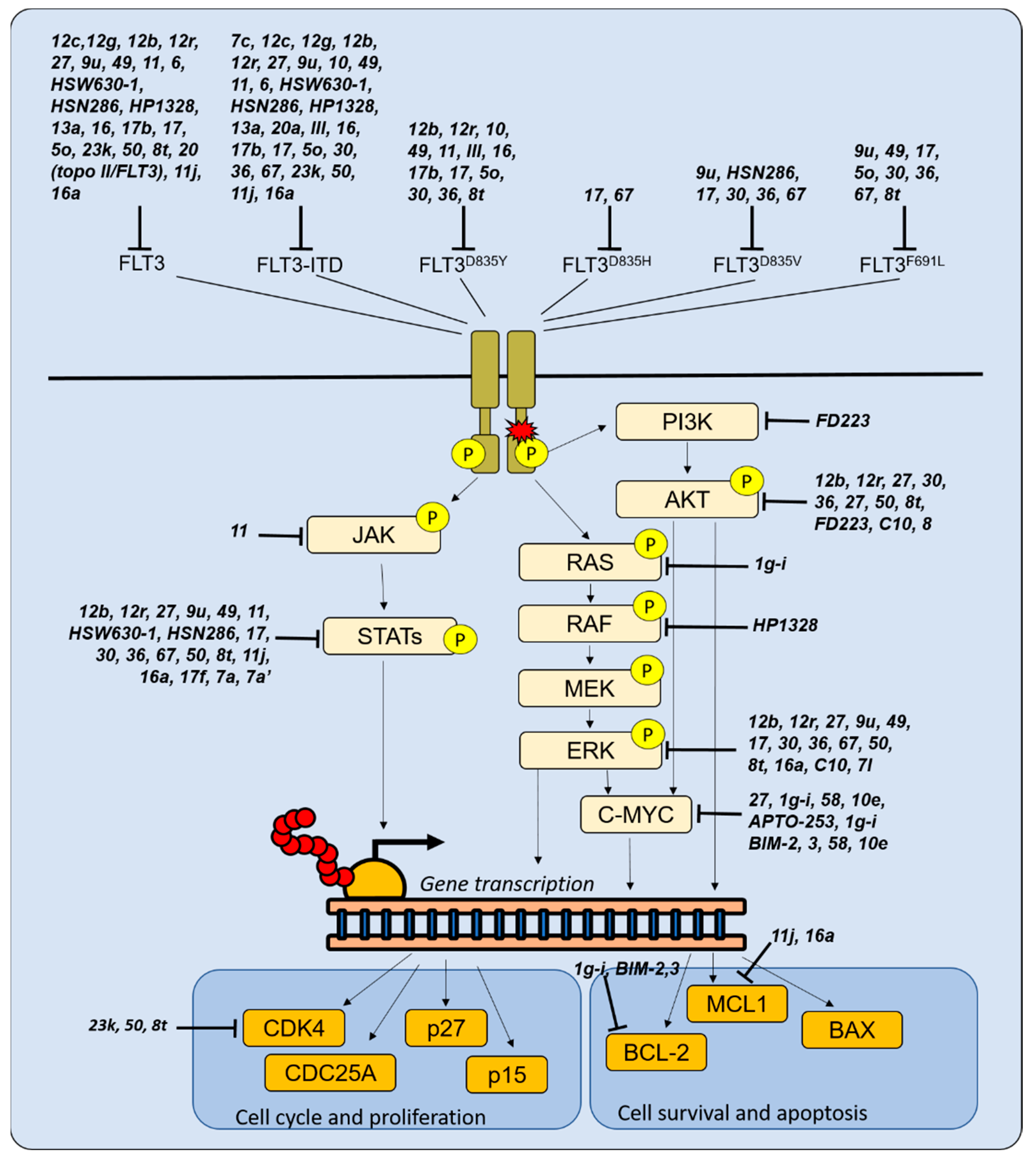
3.1. Novel Molecules Targeting FLT3
3.2. Targeting Other Proliferative Signals in AML
3.3. Novel Compounds with Unknown Targets That Inhibit Acute Leukemia Cell Proliferation
3.4. Novel Molecules Targeting the Cell Cycle Machinery
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.E.; Edwards, H.; Meshinchi, S.; Taub, J.W.; Ge, Y. “FLipping” the story: FLT3-mutated acute myeloid leukemia and the evolving role of FLT3 inhibitors. Cancers 2022, 14, 3398. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Rönnstrand, L. FMS-like tyrosine kinase 3/FLT3: From basic science to clinical implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, J.; Borge, O.J.; Bryder, D.; Theilgaard-Mönch, K.; Åstrand-Grundström, I.; Sitnicka, E.; Sasaki, Y.; Jacobsen, S.E.W. Upregulation of Flt3 expression within the bone marrow Lin-Sca1+c-Kit+ stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 2001, 15, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Buza-Vidas, N.; Woll, P.; Hultquist, A.; Duarte, S.; Lutteropp, M.; Bouriez-Jones, T.; Ferry, H.; Luc, S.; Jacobsen, S.E.W. FLT3 expression initiates in fully multipotent mouse hematopoietic progenitor cells. Blood 2011, 118, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Sitnicka, E.; Bryder, D.; Theilgaard-Mönch, K.; Buza-Vidas, N.; Adolfsson, J.; Jacobsen, S.E.W. Key role of Flt3 ligand in regulation of the common lymphoid progenitor but not in maintenance of the hematopoietic stem cell pool. Immunity 2002, 17, 463–472. [Google Scholar] [CrossRef]
- Karsunky, H.; Merad, M.; Cozzio, A.; Weissman, I.L.; Manz, M.G. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J. Exp. Med. 2003, 198, 305–313. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Wang, F.; Wang, M.; Liu, H.; Chen, X.; Cao, P.; Ma, X.; Teng, W.; Zhang, X.; et al. The mutational spectrum of FLT3 gene in acute lymphoblastic leukemia is different from acute myeloid leukemia. Cancer Gene Ther. 2020, 27, 81–88. [Google Scholar] [CrossRef]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 Recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters an analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Zhou, L.; Li, Y.; Yang, E.; Liu, Y.; Lv, N.; Fu, L.; Ding, Y.; Wang, N.; Fang, N.; et al. Profiling of somatic mutations and fusion genes in acute myeloid leukemia patients with FLT3-ITD or FLT3-TKD mutation at diagnosis reveals distinct evolutionary patterns. Exp. Hematol. Oncol. 2021, 10, 27–41. [Google Scholar] [CrossRef]
- Weisberg, E.; Roesel, J.; Furet, P.; Bold, G.; Imbach, P.; Flörsheimer, A.; Caravatti, G.; Jiang, J.; Manley, P.; Ray, A.; et al. Antileukemic effects of novel first- and second-generation FLT3 inhibitors: Structure-affinity comparison. Genes Cancer 2010, 1, 1021–1032. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3 -mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Cancer Discov. 2021, 2, 125–134. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Levis, M.J.; Perl, A.E.; Hill, J.E.; Rosales, M.; Bahceci, E. Molecular profile of FLT3-mutated Relapsed/refractory patients with AML in the phase 3 ADMIRAL study of Gilteritinib. Blood Adv. 2022, 6, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Frelin, C.; Xie, S.; Ferrari, R.; Dunant, C.F.; Zandi, S.; Neumann, A.; Plumb, I.; Doulatov, S.; Chen, J.; et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell 2015, 16, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Chen, C.; Cheng, T. Cell cycle regulation of hematopoietic stem or progenitor cells. Int. J. Hematol. 2016, 103, 487–497. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef]
- Lukas, J.; Lukas, C.; Bartek, J. Mammalian cell cycle checkpoints: Signalling pathways and their organization in space and time. DNA Repair 2004, 3, 997–1007. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 2003, 13, 261–291. [Google Scholar] [CrossRef]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Gudmundsson, K.O.; Du, Y. Quiescence regulation by normal haematopoietic stem cells and leukaemia stem cells. FEBS J. 2022; ahead of print. [Google Scholar] [CrossRef]
- Ren, S.; Rollins, B.J. Cyclin C/Cdk3 promotes Rb-dependent G0 exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef]
- Quereda, V.; Porlan, E.; Canãmero, M.; Dubus, P.; Malumbres, M. An essential role for Ink4 and Cip/Kip cell-cycle inhibitors in preventing replicative stress. Cell Death Differ. 2015, 23, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Blain, S.W.; Montalvo, E.; Massagué, J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor P27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J. Biol. Chem. 1997, 272, 25863–25872. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Aressy, B.; Ducommun, B. Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med. Chem. 2012, 8, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Boutros, R.; Froment, C.; Monsarrat, B.; Ducommun, B.; Dozier, C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J. Cell Sci. 2006, 119, 4269–4275. [Google Scholar] [CrossRef]
- Donehower, L.A. Phosphatases reverse P53-mediated cell cycle checkpoints. Proc. Natl. Acad. Sci. USA 2014, 111, 7172–7173. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: P53-P21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Della Ragione, F.; Borriello, A.; Mastropietro, S.; Della Pietra, V.; Monno, F.; Gabutti, V.; Locatelli, F.; Bonsi, L.; Bagnara, G.P.; Iolascon, A. Expression of G1-phase cell cycle genes during hematopoietic lineage. Biochem. Biophys. Res. Commun. 1997, 231, 73–76. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef]
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; Pathak, A.; Brown, N.; Deshpande, A.; et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-Cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Sexl, V. CDK6 and P16INK4A in lymphoid malignancies. Oncotarget 2013, 4, 1858–1859. [Google Scholar] [CrossRef]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Blaser, B.W.; Duchemin, A.-M.; Kusewitt, D.F.; Liu, T.; Caligiuri, M.A.; Briesewitz, R. Pharmacologic inhibition of CDK4/6: Mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood 2007, 110, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Bartram, C.R.; Seriu, T.; Miller, C.W.; Tobler, A.; Janssen, J.W.G.; Reiter, A.; Ludwig, W.D.; Zimmermann, M.; Schwaller, J.; et al. Analysis of a family of cyclin-dependent kinase inhibitors: P15/MTS2/INK4B, P16/MTS1/INK4A, and P18 genes in acute lymphoblastic leukemia of childhood. Blood 1995, 86, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Schirripa, A.; Sexl, V.; Kollmann, K. Cyclin-dependent kinase inhibitors in malignant hematopoiesis. Front. Oncol. 2022, 12, 4100. [Google Scholar] [CrossRef]
- Williams, R.T.; Roussel, M.F.; Sherr, C.J. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 6688–6693. [Google Scholar] [CrossRef]
- Berg, T.; Fliegauf, M.; Burger, J.; Staege, M.S.; Liu, S.; Martinez, N.; Heidenreich, O.; Burdach, S.; Haferlach, T.; Werner, M.H.; et al. Transcriptional upregulation of P21/WAF/Cip1 in myeloid leukemic blasts expressing AML1-ETO. Haematologica 2008, 93, 1728–1733. [Google Scholar] [CrossRef]
- Yokozawa, T.; Towatari, M.; Iida, H.; Takeyama, K.; Tanimoto, M.; Kiyoi, H.; Motoji, T.; Asou, N.; Saito, K.; Takeuchi, M.; et al. Prognostic significance of the cell cycle inhibitor P27Kip1 in acute myeloid leukemia. Leukemia 2000, 14, 28–33. [Google Scholar] [CrossRef]
- Simonetti, G.; Padella, A.; do Valle, I.F.; Fontana, M.C.; Fonzi, E.; Bruno, S.; Baldazzi, C.; Guadagnuolo, V.; Manfrini, M.; Ferrari, A.; et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer 2019, 125, 712–725. [Google Scholar] [CrossRef]
- Moison, C.; Lavallée, V.P.; Thiollier, C.; Lehnertz, B.; Boivin, I.; Mayotte, N.; Gareau, Y.; Fréchette, M.; Blouin-Chagnon, V.; Corneau, S.; et al. Complex karyotype AML displays G2/M signature and hypersensitivity to PLK1 inhibition. Blood Adv. 2019, 3, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2019, 144, 8–25. [Google Scholar] [CrossRef]
- Bruno, S.; Ghelli Luserna di Rorà, A.; Napolitano, R.; Soverini, S.; Martinelli, G.; Simonetti, G. CDC20 in and out of mitosis: A prognostic factor and therapeutic target in hematological malignancies. J. Exp. Clin. Cancer Res. 2022, 41, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ryu, H.; Song, J.Y.; Hwang, S.G.; Jalde, S.S.; Choi, H.K.; Ahn, J. Discovery of oxazol-2-amine derivatives as potent novel FLT3 inhibitors. Molecules 2020, 25, 5154. [Google Scholar] [CrossRef]
- Xu, Q.; Dai, B.; Li, Z.; Xu, L.; Yang, D.; Gong, P.; Hou, Y.; Liu, Y. Design, synthesis, and biological evaluation of 4-((6,7-dimethoxyquinoline-4-Yl)oxy)aniline derivatives as FLT3 inhibitors for the treatment of acute myeloid leukemia. Bioorg. Med. Chem. Lett. 2019, 29, 126630–126636. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Yu, M.; Ochnik, A.M.; Karanjia, J.D.; Basnet, S.K.; Kebede, A.A.; Kou, L.; Wang, S. Discovery of novel 4-azaaryl-N-phenylpyrimidin-2-amine derivatives as potent and selective FLT3 inhibitors for acute myeloid leukaemia with FLT3 mutations. Eur. J. Med. Chem. 2021, 213, 113215–113232. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, J.; Wang, C.; Carter-Cooper, B.; Yang, F.; Larocque, E.; Fine, J.; Tsuji, G.; Chopra, G.; Lapidus, R.G.; et al. Identification of new FLT3 inhibitors that potently inhibit AML cell lines via an azo click-it/staple-it approach. ACS Med. Chem. Lett. 2017, 8, 492–497. [Google Scholar] [CrossRef]
- Larocque, E.; Naganna, N.; Ma, X.; Opoku-Temeng, C.; Carter-Cooper, B.; Chopra, G.; Lapidus, R.G.; Sintim, H.O. Aminoisoquinoline benzamides, FLT3 and Src-family kinase inhibitors, potently inhibit proliferation of acute myeloid leukemia cell lines. Future Med. Chem. 2017, 9, 1213–1225. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, S.; Luo, B.; Yin, F.; Lu, W.; Li, Y.; Huang, K.; Liu, Q.; Huang, P.; Garcia-Manero, G.; et al. Identification of the benzoimidazole compound as a selective FLT3 inhibitor by cell-based high-throughput screening of a diversity library. J. Med. Chem. 2022, 65, 3597–3605. [Google Scholar] [CrossRef]
- Bharate, J.B.; McConnell, N.; Naresh, G.; Zhang, L.; Lakkaniga, N.R.; Ding, L.; Shah, N.P.; Frett, B.; Li, H.Y. Rational design, synthesis and biological evaluation of pyrimidine-4,6-diamine derivatives as type-II inhibitors of FLT3 selective against c-KIT. Sci. Rep. 2018, 8, 3722–3739. [Google Scholar] [CrossRef]
- Nemes, Z.; Takács-Novák, K.; Völgyi, G.; Valko, K.; Béni, S.; Horváth, Z.; Szokol, B.; Breza, N.; Dobos, J.; Szántai-Kis, C.; et al. Synthesis and characterization of amino acid substituted sunitinib analogues for the treatment of AML. Bioorg. Med. Chem. Lett. 2018, 28, 2391–2398. [Google Scholar] [CrossRef]
- Baska, F.; Sipos, A.; Őrfi, Z.; Nemes, Z.; Dobos, J.; Szántai-Kis, C.; Szabó, E.; Szénási, G.; Dézsi, L.; Hamar, P.; et al. Discovery and development of extreme selective inhibitors of the ITD and D835Y mutant FLT3 kinases. Eur. J. Med. Chem. 2019, 184, 111710–111722. [Google Scholar] [CrossRef] [PubMed]
- Sellmer, A.; Pilsl, B.; Beyer, M.; Pongratz, H.; Wirth, L.; Elz, S.; Dove, S.; Henninger, S.J.; Spiekermann, K.; Polzer, H.; et al. A series of novel aryl-methanone derivatives as inhibitors of FMS-like tyrosine kinase 3 (FLT3) in FLT3-ITD-positive acute myeloid leukemia. Eur. J. Med. Chem. 2020, 193, 112232–112251. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, X.; Song, Y.; Liu, J.; Ma, F.; Wang, P.; Liu, Y.; Zhao, L.; Kang, D.; Hu, L. Discovery of a potent and selective FLT3 inhibitor (Z)- N-(5-((5-fluoro-2-oxoindolin-3-ylidene)methyl)-4-methyl-1 H-pyrrol-3-Yl)-3-(pyrrolidin-1-Yl)propanamide with improved drug-like properties and superior efficacy in FLT3-ITD-positive acute myeloid leu. J. Med. Chem. 2021, 64, 4870–4890. [Google Scholar] [CrossRef]
- Zhang, L.; Lakkaniga, N.R.; Bharate, J.B.; Mcconnell, N.; Wang, X.; Kharbanda, A.; Leung, Y.K.; Frett, B.; Shah, N.P.; Li, H. Discovery of imidazo[1,2-a]pyridine-thiophene derivatives as FLT3 and FLT3 mutants inhibitors for acute myeloid leukemia through structure-based optimization of an NEK2 inhibitor. Eur. J. Med. Chem. 2021, 225, 113776–113787. [Google Scholar] [CrossRef]
- Tong, L.; Wang, P.; Li, X.; Dong, X.; Hu, X.; Wang, C.; Liu, T.; Li, J.; Zhou, Y. Identification of 2-aminopyrimidine derivatives as FLT3 kinase inhibitors with high selectivity over c-KIT. J. Med. Chem. 2022, 65, 3229–3248. [Google Scholar] [CrossRef]
- Zhi, Y.; Wang, Z.; Yao, C.; Li, B.; Heng, H.; Cai, J.; Xiang, L.; Wang, Y.; Lu, T.; Lu, S. Design and synthesis of 4-(heterocyclic substituted amino)-1h-pyrazole-3-carboxamide derivatives and their potent activity against acute myeloid leukemia (AML). Int. J. Mol. Sci. 2019, 20, 5739. [Google Scholar] [CrossRef]
- Liang, X.; Wang, B.; Chen, C.; Wang, A.; Hu, C.; Zou, F.; Yu, K.; Liu, Q.; Li, F.; Hu, Z.; et al. Discovery of N-(4-(6-acetamidopyrimidin-4-yloxy)phenyl)-2-(2-(trifluoromethyl)phenyl)acetamide (CHMFL-FLT3-335) as a potent FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutant selective inhibitor for acute myeloid leukemia. J. Med. Chem. 2019, 62, 875–892. [Google Scholar] [CrossRef]
- Yuan, X.; Chen, Y.; Zhang, W.; He, J.; Lei, L.; Tang, M.; Liu, J.; Li, M.; Dou, C.; Yang, T.; et al. Identification of pyrrolo[2,3-d]pyrimidine-based derivatives as potent and orally effective Fms-like tyrosine receptor kinase 3 (FLT3) inhibitors for treating acute myelogenous leukemia. J. Med. Chem. 2019, 62, 4158–4173. [Google Scholar] [CrossRef]
- Dayal, N.; Opoku-Temeng, C.; Hernandez, D.E.; Sooreshjani, M.A.; Carter-Cooper, B.A.; Lapidus, R.G.; Sintim, H.O. Dual FLT3/TOPK inhibitor with activity against FLT3-ITD secondary mutations potently inhibits acute myeloid leukemia cell lines. Future Med. Chem. 2018, 10, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Dayal, N.; Řezníčková, E.; Hernandez, D.E.; Peřina, M.; Torregrosa-Allen, S.; Elzey, B.D.; Škerlová, J.; Ajani, H.; Djukic, S.; Vojáčková, V.; et al. 3 H-pyrazolo[4,3- f]quinoline-based kinase inhibitors inhibit the proliferation of acute myeloid leukemia cells in vivo. J. Med. Chem. 2021, 64, 10981–10996. [Google Scholar] [CrossRef]
- Yen, S.-C.; Wu, I.-W.; Huang, C.-C.; Chao, M.-W.; Tu, H.-J.; Chen, L.-C.; Lin, T.E.; Sung, T.-Y.; Tseng, H.-J.; Chu, J.-C.; et al. O-methylated flavonol as a multi-kinase inhibitor of leukemogenic kinases exhibits a potential treatment for acute myeloid leukemia. Phytomedicine 2022, 100, 154061–154072. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.C.; Chen, L.C.; Huang, H.L.; Ngo, S.T.; Wu, Y.W.; Lin, T.E.; Sung, T.Y.; Lien, S.T.; Tseng, H.J.; Pan, S.L.; et al. Investigation of selected flavonoid derivatives as potent FLT3 inhibitors for the potential treatment of acute myeloid leukemia. J. Nat. Prod. 2021, 84, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, Y.; Zuo, M.; Liu, W.; Wang, L.; Zhu, W. Semisynthetic derivatives of fradcarbazole A and their cytotoxicity against acute myeloid leukemia cell lines. J. Nat. Prod. 2019, 82, 2279–2290. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, J.; Ren, J.; Chen, Y.; Wu, Y.; Cheng, J.; Jia, K.; Huang, F.; Cheng, Z.; Sheng, T.; et al. Discovery of a potent FLT3 inhibitor (LT-850-166) with the capacity of overcoming a variety of FLT3 mutations. J. Med. Chem. 2021, 64, 14664–14701. [Google Scholar] [CrossRef]
- Li, X.; Yang, T.; Hu, M.; Yang, Y.; Tang, M.; Deng, D.; Liu, K.; Fu, S.; Tan, Y.; Wang, H.; et al. Synthesis and biological evaluation of 6-(pyrimidin-4-Yl)-1H-pyrazolo[4,3-b]pyridine derivatives as novel dual FLT3/CDK4 inhibitors. Bioorg. Chem. 2022, 121, 105669–105689. [Google Scholar] [CrossRef]
- Wang, Y.; Zhi, Y.; Jin, Q.; Lu, S.; Lin, G.; Yuan, H.; Yang, T.; Wang, Z.; Yao, C.; Ling, J.; et al. Discovery of 4-((7H-pyrrolo[2,3-d]pyrimidin-4-Yl)amino)-N-(4-((4-methylpiperazin-1-Yl)methyl)phenyl)-1H-pyrazole-3-carboxamide (FN-1501), an FLT3- and CDK-kinase inhibitor with potentially high efficiency against acute myelocytic leukemia. J. Med. Chem. 2018, 61, 1499–1518. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Mohamed, F.E.A.; Lamie, P.F.; Bukhari, S.N.A.; Al-Sanea, M.M.; Musa, A.; Elmowafy, M.; Nayl, A.A.; Karam Farag, A.; Ali, S.M.; et al. Design, synthesis, and biological evaluation of novel pyrido-dipyrimidines as dual topoisomerase II/FLT3 inhibitors in leukemia cells. Bioorg. Chem. 2022, 122, 105752–105768. [Google Scholar] [CrossRef] [PubMed]
- Diab, S.; Abdelaziz, A.M.; Li, P.; Teo, T.; Basnet, S.K.C.; Noll, B.; Rahaman, M.H.; Lu, J.; Hou, J.; Yu, M.; et al. Dual inhibition of Mnk2 and FLT3 for potential treatment of acute myeloid leukaemia. Eur. J. Med. Chem. 2017, 139, 762–772. [Google Scholar] [CrossRef]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine kinase inhibitor—Induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10–30. [Google Scholar] [CrossRef]
- Alachkar, H.; Mutonga, M.; Malnassy, G.; Park, J.H.; Fulton, N.; Woods, A.; Meng, L.; Kline, J.; Raca, G.; Odenike, O.; et al. T-LAK cell-originated protein kinase presents a novel therapeutic target in FLT3-ITD mutated acute myeloid leukemia. Oncotarget 2015, 6, 33410–33425. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Huang, H.L.; Huangfu, W.C.; Yen, S.C.; Ngo, S.T.; Wu, Y.W.; Lin, T.E.; Sung, T.Y.; Lien, S.T.; Tseng, H.J.; et al. Biological evaluation of selected flavonoids as inhibitors of MNKs targeting acute myeloid leukemia. J. Nat. Prod. 2020, 83, 2967–2975. [Google Scholar] [CrossRef]
- Ozawa, Y.; Williams, A.H.; Estes, M.L.; Matsushita, N.; Boschelli, F.; Jove, R.; List, A.F. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk. Res. 2008, 32, 893–903. [Google Scholar] [CrossRef]
- Vin, H.; Ching, G.; Ojeda, S.S.; Adelmann, C.H.; Chitsazzadeh, V.; Dwyer, D.W.; Ma, H.; Ehrenreiter, K.; Baccarini, M.; Ruggieri, R.; et al. Sorafenib suppresses JNK-dependent apoptosis through inhibition of ZAK. Mol. Cancer Ther. 2013, 13, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J. A novel zinc finger protein, ZZaPK, interacts with ZAK and stimulates the ZAK-expressing cells re-entering the cell cycle. Biochem. Biophys. Res. Commun. 2003, 301, 71–77. [Google Scholar] [CrossRef]
- Reiter, K.; Polzer, H.; Krupka, C.; Maiser, A.; Vick, B.; Rothenberg-Thurley, M.; Metzeler, K.H.; Dörfel, D.; Salih, H.R.; Jung, G.; et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia 2018, 32, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kampa-Schittenhelm, K.M.; Heinrich, M.C.; Akmut, F.; Döhner, H.; Döhner, K.; Schittenhelm, M.M. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol. Cancer 2013, 12, 19–33. [Google Scholar] [CrossRef]
- Heng, H.; Wang, Z.; Li, H.; Huang, Y.; Lan, Q.; Guo, X.; Zhang, L.; Zhi, Y.; Cai, J.; Qin, T.; et al. Combining structure- and property-based optimization to identify selective FLT3-ITD inhibitors with good antitumor efficacy in AML cell inoculated mouse xenograft model. Eur. J. Med. Chem. 2019, 176, 248–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Riley-Gillis, B.; Han, L.; Jia, Y.; Lodi, A.; Zhang, H.; Ganesan, S.; Pan, R.; Konoplev, S.N.; Sweeney, S.R.; et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct. Target. Ther. 2022, 7, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Glaser, H.; Sassano, A.; Joshi, S.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Tallman, M.S.; Platanias, L.C. Negative regulatory effects of Mnk kinases in the generation of chemotherapy-induced antileukemic responses. Mol. Pharmacol. 2010, 78, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xu, C.; Li, Z.; Chen, Y.; Wu, T.; Hong, H.; Lu, M.; Jia, Y.; Yang, Y.; Liu, X.; et al. Bioisosteric replacements of the indole moiety for the development of a potent and selective PI3Kδ inhibitor: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2021, 223, 113661–113671. [Google Scholar] [CrossRef] [PubMed]
- Juen, L.; Brachet-Botineau, M.; Parmenon, C.; Bourgeais, J.; Hérault, O.; Gouilleux, F.; Viaud-Massuard, M.C.; Prié, G. New inhibitor targeting signal transducer and activator of transcription 5 (STAT5) signaling in myeloid leukemias. J. Med. Chem. 2017, 60, 6119–6136. [Google Scholar] [CrossRef] [PubMed]
- Polomski, M.; Brachet-Botineau, M.; Juen, L.; Viaud-Massuard, M.C.; Gouilleux, F.; Prié, G. Inhibitors targeting STAT5 signaling in myeloid leukemias: New tetrahydroquinoline derivatives with improved antileukemic potential. ChemMedChem 2021, 16, 1034–1046. [Google Scholar] [CrossRef]
- Guillon, J.; Denevault-Sabourin, C.; Chevret, E.; Brachet-Botineau, M.; Milano, V.; Guédin-Beaurepaire, A.; Moreau, S.; Ronga, L.; Savrimoutou, S.; Rubio, S.; et al. Design, synthesis, and antiproliferative effect of 2,9-bis[4-(pyridinylalkylaminomethyl)phenyl]-1,10-phenanthroline derivatives on human leukemic cells by targeting G-quadruplex. Arch. Pharm. 2021, 354, 2000450–2000463. [Google Scholar] [CrossRef]
- Hu, M.H.; Yu, B.Y.; Wang, X.; Jin, G. Drug-like Biimidazole Derivatives Dually Target c-MYC/BCL-2 G-Quadruplexes and Inhibit Acute Myeloid Leukemia. Bioorg. Chem. 2020, 104, 104264–104271. [Google Scholar] [CrossRef]
- Hu, X.; Cao, Y.; Yin, X.; Zhu, L.; Chen, Y.; Wang, W.; Hu, J. Design and synthesis of various quinizarin derivatives as potential anticancer agents in acute T lymphoblastic leukemia. Bioorg. Med. Chem. 2019, 27, 1362–1369. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, A.; Shi, J.; Zhou, D.; Shi, W.; Qiu, Q.; Liu, X.; Huang, W.; Li, J.; Qian, H.; et al. Design, synthesis, and biological activity evaluation of a series of novel sulfonamide derivatives as BRD4 inhibitors against acute myeloid leukemia. Bioorg. Chem. 2021, 111, 104849–104857. [Google Scholar] [CrossRef]
- Chen, P.; Yang, Y.; Yang, L.; Tian, J.; Zhang, F.; Zhou, J.; Zhang, H. 3-hydroxyisoindolin-1-one derivates: Synthesis by palladium-catalyzed C–H activation as BRD4 inhibitors against human acute myeloid leukemia (AML) cells. Bioorg. Chem. 2019, 86, 119–125. [Google Scholar] [CrossRef]
- Igoe, N.; Bayle, E.D.; Fedorov, O.; Tallant, C.; Savitsky, P.; Rogers, C.; Owen, D.R.; Deb, G.; Somervaille, T.C.P.; Andrews, D.M.; et al. Design of a biased potent small molecule inhibitor of the bromodomain and PHD finger-containing (Brpf) proteins suitable for cellular and in vivo studies. J. Med. Chem. 2017, 60, 668–680. [Google Scholar] [CrossRef]
- Choudhary, C.; Brandts, C.; Schwable, J.; Tickenbrock, L.; Sargin, B.; Ueker, A.; Böhmer, F.D.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood 2007, 110, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Sly, L.M.; Argiropoulos, B.; Kuchenbauer, F.; Lai, C.; Weng, A.; Leung, M.; Lin, G.; Brookes, C.; Fung, S.; et al. Modeling the functional heterogeneity of leukemia stem cells: Role of STAT5 in leukemia stem cell self-renewal. Blood 2009, 114, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Brachet-Botineau, M.; Deynoux, M.; Vallet, N.; Polomski, M.; Juen, L.; Hérault, O.; Mazurier, F.; Viaud-Massuard, M.C.; Prié, G.; Gouilleux, F. A novel inhibitor of Stat5 signaling overcomes chemotherapy resistance in myeloid leukemia cells. Cancers 2019, 11, 2043. [Google Scholar] [CrossRef] [PubMed]
- Awadasseid, A.; Ma, X.; Wu, Y.; Zhang, W. G-quadruplex stabilization via small-molecules as a potential anti-cancer strategy. Biomed. Pharmacother. 2021, 139, 111550–111558. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Local, A.; Zhang, H.; Benbatoul, K.D.; Folger, P.; Sheng, X.; Tsai, C.Y.; Howell, S.B.; Rice, W.G. APTO-253 stabilizes G-quadruplex DNA, inhibits MYC expression, and induces DNA damage in acute myeloid leukemia cells. Mol. Cancer Ther. 2018, 17, 1177–1186. [Google Scholar] [CrossRef]
- Gueddouda, N.M.; Hurtado, M.R.; Moreau, S.; Ronga, L.; Das, R.N.; Savrimoutou, S.; Rubio, S.; Marchand, A.; Mendoza, O.; Marchivie, M.; et al. Design, synthesis, and evaluation of 2,9-bis[(substituted-aminomethyl)phenyl]-1,10-phenanthroline derivatives as G-quadruplex ligands. ChemMedChem 2017, 12, 146–160. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Shima, H.; Yamagata, K.; Aikawa, Y.; Shino, M.; Koseki, H.; Shimada, H.; Kitabayashi, I. Bromodomain-PHD finger protein 1 is critical for leukemogenesis associated with MOZ-TIF2 fusion. Int. J. Hematol. 2014, 99, 21–31. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512.e9. [Google Scholar] [CrossRef]
- Dorrance, A.M.; Liu, S.; Yuan, W.; Becknell, B.; Arnoczky, K.J.; Guimond, M.; Strout, M.P.; Feng, L.; Nakamura, T.; Yu, L.; et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J. Clin. Investig. 2006, 116, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Camós, M.; Esteve, J.; Jares, P.; Colomer, D.; Rozman, M.; Villamor, N.; Costa, D.; Carrió, A.; Nomdedéu, J.; Montserrat, E.; et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(P11;P13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of Hox gene expression. Cancer Res. 2006, 66, 6947–6954. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.J.; Lalonde, M.E.; Côté, J.; Yang, X.J.; Kutateladze, T.G. Crosstalk between epigenetic readers regulates the MOZ/MORF HAT complexes. Epigenetics 2014, 9, 186–193. [Google Scholar] [CrossRef]
- Ghazy, E.; Zeyen, P.; Herp, D.; Hügle, M.; Schmidtkunz, K.; Erdmann, F.; Robaa, D.; Schmidt, M.; Morales, E.R.; Romier, C.; et al. Design, synthesis, and biological evaluation of dual targeting inhibitors of histone deacetylase 6/8 and bromodomain BRPF1. Eur. J. Med. Chem. 2020, 200, 112338–112357. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Kolbinger, F.R.; Zeyen, P.; Ghazy, E.; Herp, D.; Schmidtkunz, K.; Melesina, J.; Shaik, T.B.; Erdmann, F.; Schmidt, M.; et al. Structure-based design and biological characterization of selective histone deacetylase 8 (HDAC8) inhibitors with anti-neuroblastoma activity. J. Med. Chem. 2017, 60, 10188–10204. [Google Scholar] [CrossRef]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Wen, Z.H.; Wan, K.; Yuan, D.; Zeng, X.; Liang, G.; Zhu, J.; Xu, B.; Luo, H. A novel synthesized 3′ 5′-diprenylated chalcone mediates the proliferation of human leukemia cells by regulating apoptosis and autophagy pathways. Biomed. Pharmacother. 2018, 106, 794–804. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, Z.; Ge, W.; Kuang, B.; Xu, J.; Yang, J.; Chen, Y.; Zhang, Q. Synthesis and biological evaluation of dithiocarbamate esters of parthenolide as potential anti-acute myelogenous leukaemia agents. J. Enzyme Inhib. Med. Chem. 2018, 33, 1376–1391. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.; Liu, X.; Gao, Y.; Hu, J.; Chen, H. Discovery of novel negletein derivatives as potent anticancer agents for acute myeloid leukemia. Chem. Biol. Drug Des. 2018, 91, 924–932. [Google Scholar] [CrossRef]
- de Almeida, P.A.; de Souza, L.F.S.; Maioral, M.F.; Duarte, B.F.; Walter, L.O.; Pirath, Í.M.S.; Speer, D.B.; Sens, L.; Tizziani, T.; de Oliveira, A.S.; et al. Cell cycle arrest and apoptosis induction by a new 2,4- dinitrobenzenesulfonamide derivative in acute leukemia cells. J. Pharm. Pharm. Sci. 2021, 24, 23–36. [Google Scholar] [CrossRef]
- Yoshida, C.; Higashi, T.; Hachiro, Y.; Fujita, Y.; Yagi, T.; Takechi, A.; Nakata, C.; Miyashita, K.; Kitada, N.; Saito, R.; et al. Synthesis of polyenylpyrrole derivatives with selective growth inhibitory activity against T-Cell acute lymphoblastic leukemia cells. Bioorg. Med. Chem. Lett. 2021, 37, 127837–127840. [Google Scholar] [CrossRef] [PubMed]
- Cury, N.M.; Capitão, R.M.; do Canto Borges de Almeida, R.; Artico, L.L.; Ronchi Corrêa, J.; Dos Santos, E.F.S.; Yunes, J.A.; Correia, C.R.D. Synthesis and evaluation of 2-carboxy indole derivatives as potent and selective antileukemic agents. Eur. J. Med. Chem. 2019, 181, 111570–111587. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kril, L.M.; Yu, T.; Zhang, W.; Frasinyuk, M.S.; Bondarenko, S.P.; Kondratyuk, K.M.; Hausman, E.; Martin, Z.M.; Wyrebek, P.P.; et al. Semisynthetic aurones inhibit tubulin polymerization at the colchicine-binding site and repress PC-3 tumor xenografts in nude Mice and Myc-induced T-ALL in zebrafish. Sci. Rep. 2019, 9, 6439–6453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shao, X.; Zhong, T.; Wu, Y.; Xu, A.; Sun, X.; Gao, H.; Liu, Y.; Lan, T.; Tong, Y.; et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat. Chem. Biol. 2021, 17, 567–575. [Google Scholar] [CrossRef]
- Jin, T.; Wang, P.; Long, X.; Jiang, K.; Song, P.; Wu, W.; Xu, G.; Zhou, Y.; Li, J.; Liu, T. Design, synthesis, and biological evaluation of orally bioavailable CHK1 inhibitors active against acute myeloid leukemia. ChemMedChem 2021, 16, 1477–1487. [Google Scholar] [CrossRef]
- Gębarowski, T.; Wiatrak, B.; Gębczak, K.; Tylińska, B.; Gąsiorowski, K. Effect of new olivacine derivatives on P53 protein level. Pharmacol. Rep. 2020, 72, 214–224. [Google Scholar] [CrossRef]
- Simonetti, G.; Boga, C.; Durante, J.; Micheletti, G.; Telese, D.; Caruana, P.; Di Rorà, A.G.L.; Mantellini, F.; Bruno, S.; Martinelli, G.; et al. Synthesis of novel tryptamine derivatives and their biological activity as antitumor agents. Molecules 2021, 26, 683. [Google Scholar] [CrossRef]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef]
- Takahashi, S. Kinase inhibitors and interferons as other myeloid differentiation inducers in leukemia therapy. Acta Haematol. 2022, 145, 113–121. [Google Scholar] [CrossRef]
- Jasztold-Howorko, R.; Tylińska, B.; Bbiaduń, O.; Barowski, T.G.; Gasiorowski, K. New pyridocarbazole derivatives. synthesis and their in vitro anticancer activity. Acta Pol. Pharm.-Drug Res. 2013, 70, 823–832. [Google Scholar]
- Hassin, O.; Oren, M. Drugging P53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2022; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Boyson, C.A.; Di Liberto, M.; Huang, X.; Hannah, J.; Dorn, D.C.; Moore, M.A.S.; Chen-Kiang, S.; Zhou, P. CDK4/6 inhibitor PD 0332991 sensitizes acute myeloid leukemia to cytarabine-mediated cytotoxicity. Cancer Res. 2015, 75, 1838–1845. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Molecule. | Target(s) | Disease(s) | IC50 /GI50 | Reference | SAR/Docking Studies |
|---|---|---|---|---|---|
| 5-(4-fluorophenyl)-N-phenyloxazol-2-amine (compound 7c) | FLT3, FLT3-ITD | AML | MV4-11: 95.51 ± 1.16 nM MOLM-13: 61.9 ± 2.45 nM | [54] | Yes |
| 4-((6,7-dimethoxyquinoline- 4-yl)oxy)aniline derivatives (compound 12c) | FLT3 | AML | MV4-11: 8.29 ± 0.24 µM HL-60: 14.16 ± 0.22 µM | [55] | Yes |
| 4-((6,7-dimethoxyquinoline- 4-yl)oxy)aniline derivatives (compound 12g) | FLT3 | AML | MV4-11: 5.80 ± 0.42 µM HL-60: 8.91 ± 0.66 µM | [55] | Yes |
| 4-azaaryl-N-phenylpyrimidin-2-amine derivative (compound 12b) | FLT3, FLT3-ITD and their mutants | AML | MV4-11: 0.074 ± 0.010 µM MOLM-13 0.023 ± 0.001 µM | [56] | Yes |
| 4-azaaryl-N-phenylpyrimidin-2-amine derivative (compound 12r) | FLT3, FLT3-ITD and their mutants | AML | MV4-11: 0.017 ± 0.012 µM MOLM-13: 0.0004 ± 0.0002 µM | [56] | Yes |
| 3-aminoisoquinoline analogs (HSW630-1) | FLT3 | AML | MV4-11: 0.15 µM MOLM-14: 0.15 µM | [57] | Yes |
| 3-amino and 1-aminoisoquinoline benzamides (HSN286) | FLT3, SRC kinases | AML | MV4-11: 0.492 nM MOLM-14: 0.721 nM | [58] | Yes |
| N-(3,4-dimethoxybenzyl)-1-phenyl-1H-benzimidazol-5-amine derivative (compound HP1328) | FLT3, FLT3-ITD, c-KIT | AML | Ba/F3 FLT3-ITD: 75.4 ± 3.2 nM MV4-11: 165.0 ± 17.5 nM MOLM-13: 66.8 ± 4.8 nM | [59] | Yes |
| pyrimidine-4,6-diamine derivative (compound 13a) | FLT3 | AML | Ba/F3 FLT3-ITD: 131.2 nM MV4-11: 9.9 nM MOLM-14: 24.4 nM MOLM-14D835Y:1842 nM MOLM-14F691L: 1345 nM | [60] | Yes |
| amino acid-substituted sunitinib analogue released active compound candidates (20a) | FLT3-ITD | AML | MV4-11: 2.2 ± 0.3 µM | [61] | Yes |
| phenylethenylquinazoline derivatives (compound III) | FLT3-ITD, FLT3D835Y, FLT3-ITDD835Y | AML | MV4-11: 0.03 ± 0.00 µM | [62] | Yes |
| bis(1H-indol-2-yl)methanones, detivatives (compound 16) | FLT3, FLT3-ITD, FLT3D835Y, RET, ZAK | AML | MV4-11: <0.001 µM MOLM-13: <0.001 µM EOL-1: <0.001 µM | [63] | Yes |
| (Z)-N-(5-((5-Fluoro-2-oxoindolin-3-ylidene)methyl)-4-methyl-1H-pyrrol-3-yl)-3-(pyrrolidin-1-yl)propanamide (compound 17) | FLT3-ITD and its mutants | AML | MV4-11: 23.5 ± 1.2 nM MOLM-13: 35.5 ± 2.1 nM Ba/F3 FLT3-ITD: 12.7 ± 0.1 nM Ba/F3 FLT3-ITDD835Y: 36.7 ± 0.7 nM Ba/F3 FLT3-ITDD835V: 26.8 ± 1.5 nM Ba/F3 FLT3-ITDF691L: 43.6 ± 3.1 nM | [64] | Yes |
| imidazo[1,2-a]pyridine-thiophene derivative (compound 5o) | FLT3 and its mutants | AML | MOLM-14: 0.52 ± 0.062 µM MOLM-14 FLT3-ITDD835Y: 0.53 ± 0.022 µM MOLM-14 FLT3-ITDF691L: 0.57 ± 0.058 µM | [65] | Yes |
| 2-Aminopyrimidine derivative (compound 30) | FLT3, TRKA, Aurora A | AML | MV4-11: 3.20 ± 0.77 nM Ba/F3 FLT3-ITD: 23.32 ± 8.27 nM Ba/F3 FLT3D835V: 1.41 ± 0.11 nM Ba/F3 FLT3D835F: 5.02 ± 0.87 nM Ba/F3 FLT3F691L: 28.84 ± 5.01 nM Ba/F3 FLT3-ITDD835Y: 19.23 ± 10.46 nM Ba/F3 FLT3-ITDF691L: 99.62 ± 3.22 nM | [66] | Yes |
| 2-Aminopyrimidine derivative (compound 36) | FLT3, TRKA, Aurora A | AML | MV4-11: 0.75 ± 0.11 nM Ba/F3 FLT3-ITD: 0.84 ± 0.33 nM Ba/F3 FLT3D835V: 1.29 ± 0.10 nM Ba/F3 FLT3D835F: 0.16 ± 0.03 nM Ba/F3 FLT3F691L: 3.56 ± 0.48 nM Ba/F3 FLT3-ITDD835Y: 1.71 ± 0.54 nM Ba/F3 FLT3-ITDF691L: 14.50 ± 1.02 nM | [66] | Yes |
| 1H-pyrazole-3-carboxamide derivatives (compound 8t) | FLT3, CDKs, KDR/VEGFR2, ERK7, FLT1, FLT4, GSK3 | AML, T-ALL | MV4-11: 1.22 nM HL-60 1.15 µM CCRF-CEM: 0.22 µM MOLT-4 0.08 µM | [67] | Yes |
| Molecule | Target(s) | Disease(s) | IC50 /GI50 | Cell Cycle Arrest (Phase, Model, Dose, Time Point) | Reference(s) | SAR/Docking Studies |
|---|---|---|---|---|---|---|
| N-(4-(6-Acetamidopyrimidin-4-yloxy)phenyl)-2-(2-(trifluoromethyl)phenyl)acetamide (CHMFL-FLT3-335, compound 27) | FLT3-ITD, PDGFRβ, HPK1, CSF1R, PDGFRα, c-KIT | AML | MV4-11: 0.284 ± 0.018 µM MOLM-13: 0.466 ± 0.026 µM MOLM-14: 0.343 ± 0.025 µM | G0/G1, MV4-11: 0.1–3 µM, 24 h MOLM-13: 0.1–3 µM, 12 h MOLM-14: 0.3–3 µM, 24 h | [68] | Yes |
| pyrrolo[2,3-d]pyrimidine derivatives (compound 9u) | FLT3-ITD, PDGFRα, ABL1 and their mutants, HCK, LCK, RET, LYN, MET, MER, TIE2 | AML | MV4-11: 0.089 ± 0.001 nM MOLM-13: 0.022 ± 0.003 nM Ba/F3 FLT3-ITD: 0.92 ± 0.01 nM Ba/F3 FLT3-ITDD835Y: 20.71 ± 2.32 nM Ba/F3 FLT3-ITDF691L: 12.99 ± 0.87 nM |
G0/G1
MV4-11: 1–30 nM, 24 h MOLM-13: 3–30 nM, 24 h | [69] | Yes |
| 8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (HSD1169, compound 10) | FLT3, FLT3-ITD and its mutants, LRRK2, MELK, DYRK1B, CLK1, TRKC, MNK2, TOPK, ROCK2, MSK2, p70S6K, PKG1a, CDK2/Cyclin A1 | AML | MV4-11: 5.4 nM MOLM-14: 4.9 nM MOLM-13 FLT3-ITDD835Y: 5.1 nM | G1, MV4-11: 62.5 nM, 48–72 h | [70,71] | Yes |
| 8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine derivative (compound 49) | FLT3, FLT3-ITD and its mutants, LRRK2, MELK, DYRK1B, CLK1, TRKC | AML | MV4-11: 0.070 µM MOLM-13: 0.068 µM MOLM-14: 0.026 µM MOLM14 FLT3D835Y: 0.036 µM MOLM14 FLT3F691L: 0.053 µM Ba/F3 FLT3-ITD: 0.062 µM | G1, MV4-11: 20–500 nM, 24 h | [71] | Yes |
| O-methylated flavonol (compound 11) | FLT3, other kinases | AML, ALL | MOLM-13: 2.65 ± 0.28 μM MV4-11: 1.99 ± 0.25 μM HL-60: 12 ± 4.39 µM MOLT-4: 7.95 ± 1.9 µM | G0/G1, MOLM-13: 3 μM, 16–72 h MV4-11: 3 μM, 16–72 h | [72] | Yes |
| Flavonoid derivatives (compound 31) | FLT3, FLT3D835Y, FLT3-ITD | AML | MOLM-13: 2.6 ± 0.20 μM MV4-11: 2.6 ± 0.46 μM | G0/G1 MOLM-13: 3 μM, 72 h MV4-11: 3–30 μM, 72 h | [73] | Yes |
| Flavonoid derivatives (compound 32) | FLT3, FLT3D835Y, FLT3-ITD | AML | MOLM-13: 32 5.9 ± 0.58 μM MV4-11: 7.9 ± 0.20 μM | G0/G1 MV4-11: 3–10 μM, 72 h | [73] | Yes |
| 5,7,4′-trihydroxy-6-methoxyflavone (compound 40) | FLT3, FLT3D835Y, FLT3-ITD | AML | MOLM-13: 40 7.0 ± 0.71 μM MV4-11: 6.8 ± 0.66 μM | G0/G1 MOLM-13: 3 μM, 72 h MV4-11: 3–10 μM, 72 h | [73] | Yes |
| Fradcarbazole A derivative (compound 6) | FLT3, c-KIT, CDK2 | AML | MV4-11: 0.32 ± 0.03 µM | G0/G1, MV4-11: 0.15–0.6 μM, 24 h | [74] | No |
| 3-amine-pyrazole-5-benzimidazole compounds (67) | FLT3-ITD and its mutants | AML | MOLM-13: 9.85 ± 1.03 nM MV4-11: 2.93 ± 0.31 nM Ba/F3 FLT3-ITD: 7.60 ± 0.58 nM Ba/F3 FLT3-ITDF691L: 8.30 ± 0.51 nM Ba/F3 TEL-FLT3D835V, Ba/F3 FLT3-ITDY842H, Ba/F3 FLT3-ITDD835V: <1.50 nM | G1, MV4-11: 10–100 nM, 24 h | [75] | Yes |
| 6-(pyrimidin-4-yl)- 1H-pyrazolo[4,3-b]pyridine derivative (compound 23k) | FLT3, CDK4 | AML | MV4-11: 70 ± 8 nM | G0/G1, MV4-11: 200 nM, 24 h | [76] | Yes |
| 1-H-pyrazole-3-carboxamide derivatives (compound 50) | FLT3, CDK2,4,6 | AML | MV4-11: 0.008 ± 0.001 µM | G0/G1, MV4-11: 0.02–0.2 μM, 24 h | [77] | Yes |
| pyrido-dipyrimidines (compound 20) | topoisomerase II, FLT3 | AML | HL-60: 0.48 ± 0.08 µM | G2/M, HL-60: 2.26 μM, 24 h | [78] | Yes |
| N-phenyl-4-(thiazol-5-yl)pyrimidin-2-amines and 4-(indol-3-yl)-N-phenylpyrimidin-2-amines (16a) | FLT3, MNK2 | AML | MV4-11: 0.60 ± 0.10 µM | G1, MV4-11: 4.8 μM, 48 h | [79] | Yes |
| Molecule | Target(s) | Disease(s) | IC50 /GI50 | Reference | SAR/Docking Studies |
|---|---|---|---|---|---|
| Indole scaffold derivative (FD223, compound 13) | PI3Kδ | AML | HL-60: 2.25 μM, MOLM-16: 0.87 μM EOL-1: 2.82 μM KG-1: 5.82 μM | [92] | Yes |
| PPARα/γ ligand derivative (compound 17f) | STAT5 | AML | KG1a: 2.638 ± 0.51 μM MV4-11: 3.549 ± 0.47 μM | [93] | Yes |
| 17f analogs (compounds 7a and 7a’) | STAT5 | AML | KG-1a: 7a, 7.8±0.9 μM 7a’, 6.9±0.8 μM MOLM-13: 7a, 5.4±0.5 μM 7a’, 4.7±1.0 μM | [94] | No |
| 2,9-bis[4-(pyridinylalkylaminomethyl)phenyl]-1,10-phenanthroline derivatives (compound 1g–i) | G- quadruplexes | AML | MV4-11: 1g, 2.1 ± 0.5 µM 1h, 1.3 ± 0.3 µM 1i, 1.6 ± 0.4 µM U937: 1 g, 2.0 ± 0.8 µM 1h, 2.0 ± 0.7 µM 1i, 3.0 ± 0.9 µM HL-60: 1 g, 3.0 ± 1.0 µM 1h, 8.0 ± 0.9 µM 1i, 3.0 ± 0.8 µM | [95] | FRET melting experiments and native electrospray mass spectrometry |
| biimidazole derivative (BIM-2) | G- quadruplexes | AML | U937: 9.2 μM | [96] | Yes |
| Quinizarin quaternary ammonium salt 3 | unknown | AML, T-ALL | HL-60: 1.40 ± 0.81 μM MOLT-4: 2.61 ± 0.15 μM Jurkat: 2.80 ± 0.22 μM | [97] | Yes |
| 3,5-dimethylisoxazole derivatives (compound 58) | BRD4 | AML | MV4-11: 0.15 ± 0.02 µM HL-60: 1.21 ± 0.02 µM | [98] | Yes |
| 3-Hydroxyisoindolin-1-one derivate (compound 10e) | BRD4 | AML | MV4-11: 0.420 ± 0.011 µM HL-60: 0.365 ± 0.018 µM | [99] | Yes |
| Quinolin-2-one Hit (compound 13-d) | BRPF1 | AML | OCI-AML2: 1.3 µM NOMO-1: 4.6 µM THP-1: 5.7 µM KG-1: 7.0 µM MV4-11: 9.9 µM | [100] | Yes |
| Molecule | Target(s) | Disease | IC50 /GI50 | Reference | SAR/Docking Studies |
|---|---|---|---|---|---|
| 3′, 5′-diprenylated chalcone | unknown | Erythroleukemia | HEL: 2.027 ± 0.523 µmol/L | [117] | No |
| dithiocarbamate esters of parthenolide (compound 7l) | unknown | AML | KG-1a: 0.7 ± 0.2 μM. HL-60: 1.7 ± 0.5 μM. | [118] | No |
| Neglectin derivative (compound 8) | unknown | AML | HL-60: 7.24 ± 0.15 µM | [119] | No |
| 2,4-dinitrobenzenesulfonamide derivative (S1) | unknown | T-ALL | Jurkat: 6.0 ± 0.4 μM | [120] | No |
| Octatetraenylpyrrole (compound 3s) | unknown | T-ALL | CCRF-CEM: 0.27 μM. | [121] | No |
| Molecule | Target(s) | Disease(s) | IC50 /GI50 | Reference | SAR/Docking Studies |
|---|---|---|---|---|---|
| 2-carbomethoxy-3-substituted indoles (compound 20) | Tubulin (suggested by cell phenotype) | ALL, AML | CEM: 0.22 µM RS4;11: 0.30 µM | [122] | No |
| Z)-2-((2-((1-ethyl-5-methoxy-1H-indol-3-yl)methylene)-3-oxo-2,3-dihydrobenzofuran-6-yl)oxy)acetonitrile (5a) | Tubulin | T-ALL, B-ALL | CRF-CEM: 244 nM DND41: 210 nM Jurkat: 273 nM HBP-ALL: 94 nM Loucy: 334 nM MOLT-4: 241 nM MOLT-16: 234 nM RPMI8402: 301 nM NALM-16: 272 nM REH: 287 nM | [123] | Yes |
| CDK2-PROTAC | CDK2 | AML | MV4-11: 100 nM < IC50 < 1 µM | [124] | Yes |
| Diaminopyrimidine derivatives (compound 13) | CHK1 | AML | MV4-11: 0.035 ± 0.007 μM | [125] | Yes |
| 1-substituted pyrido[4,3-b]carbazole derivatives of olivacine | TP53 restoration | T-ALL | CCFR/CEM: Compound 1: 0.442 ± 0.062 µM Compound 2: 0.520 ± 0.185 µM Compound 3: 0.359 ± 0.109 µM | [126] | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghelli Luserna di Rorà, A.; Jandoubi, M.; Martinelli, G.; Simonetti, G. Targeting Proliferation Signals and the Cell Cycle Machinery in Acute Leukemias: Novel Molecules on the Horizon. Molecules 2023, 28, 1224. https://doi.org/10.3390/molecules28031224
Ghelli Luserna di Rorà A, Jandoubi M, Martinelli G, Simonetti G. Targeting Proliferation Signals and the Cell Cycle Machinery in Acute Leukemias: Novel Molecules on the Horizon. Molecules. 2023; 28(3):1224. https://doi.org/10.3390/molecules28031224
Chicago/Turabian StyleGhelli Luserna di Rorà, Andrea, Mouna Jandoubi, Giovanni Martinelli, and Giorgia Simonetti. 2023. "Targeting Proliferation Signals and the Cell Cycle Machinery in Acute Leukemias: Novel Molecules on the Horizon" Molecules 28, no. 3: 1224. https://doi.org/10.3390/molecules28031224
APA StyleGhelli Luserna di Rorà, A., Jandoubi, M., Martinelli, G., & Simonetti, G. (2023). Targeting Proliferation Signals and the Cell Cycle Machinery in Acute Leukemias: Novel Molecules on the Horizon. Molecules, 28(3), 1224. https://doi.org/10.3390/molecules28031224