Abstract
Structurally unrelated antibiotics MLSB (macrolide-lincosamide-streptogramin B) compromised with clinically resistant pathogens because of the cross-resistance resulting from the structural modification of rRNA A2058. The structure–activity relationships of a novel 3-O-descladinose azithromycin chemotype conjugating with nucleobases were fully explored with the aid of engineered E. coli SQ110DTC and SQ110LPTD. The conjugates of macrolides with nucleobases, especially adenine, displayed antibacterial superiority over telithromycin, azithromycin and clindamycin against rRNA A2058/2059-mutated engineered E. coli strains at the cost of lowering permeability and increasing vulnerability to efflux proteins against clinical isolates.
1. Introduction
Public health has been threatened due to the dramatically surging incidence of resistance in clinical isolates. More than half of the marketed antibacterial drugs serve as protein synthesis inhibitors by targeting 30S or 50S subunits of bacterial ribosomes [1]. Among them, macrolides are widely prescribed to treat community- or hospital-acquired bacterial pneumonia.
Recently, high-resolution crystal structures of 70S ribosome complexed with erythromycin derivatives revealed the convincible model for a prevailing resistance against structurally unrelated MLSB (macrolide-lincosamide-streptogramin B) antibiotics [2]. The methylated A2058 or A2058 mutation would disrupt the water-mediated hydrogen bonding to the 5-O-desosamine of macrolides, and the greatly decreasing affinity could not maintain the tight binding of the antibiotics when nascent peptides egress, leading to high-level resistance.
The second-generation semi-synthetic macrolides, clarithromycin and 15-membered azithromycin (Figure 1), failed to afford additional contacts with the ribosome and thus showed no efficacy against erythromycin-resistant bacteria. The third-generation ketolide (replacing 3-O-cladinose with 3-keto), telithromycin (Figure 1), succeeded in partially restoring the activities by supplying a secondary π–π interaction between the A752–A2609 base pair and the newly introduced imidazolyl pyridyl group [3,4]. The extended side chain of another ketolide, solithromycin, interacts with the same base pair [5]. However, the affinities of telithromycin or solithromycin to the A2058-methylated or mutated ribosome are too low to allow the usage of these drugs for treating high-level MLSB-resistant infections [2].
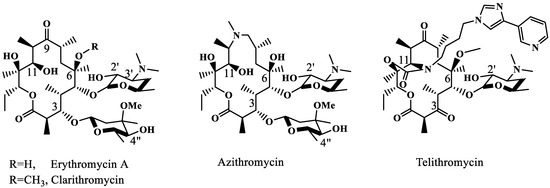
Figure 1.
Structures of the 1st-, 2nd- and 3rd-generation erythromycin.
To strengthen the affinities to the modified ribosome, a variety of mono/bi/fused-heteroaromatic groups were installed onto the end of the linkers derivatized from the 6-OH, 9-keto, 11-OH and 4″-OH of macrolides [6,7]. It was reported that the antibacterial activities of the novel macrolides that were constructed by a total synthesis route still heavily depend on the pendant side chains capable of binding the ribosome [8]. Carbamolides (3-O-carbamoyl), acylides (3-O-acyl) and alkylides (3-O-alkyl) showed a new mode of action, namely π–π interacting with the base pair C2610–G2505 [9,10,11,12]. In 2019, we attempted to introduce some heteroaromatic groups to 3-OH clarithromycin bridged by three-to eight-atom-long spacers. The molecular docking suggested that the most potent compound possessing a three-atom-long spacer shared the same mode of action as that of the known macrolides [13].
Apart from the chemotypes mentioned above, some conjugates have been investigated to achieve dual modes of action [14,15,16]. Quinolones linking to 4″-OH of macrolides have been extensively investigated since 2010, which was well summarized in a recent review [7]. The potent hybrids proved to be non-inhibitors in gyrase-based supercoiling assays [17,18]. In 2020, we reported that quinolones attaching to 6-OH of azithromycin could show moderate activities against E. coli DNA gyrase [19].
In continuation of our work, we report here a series of 3-O-descladinose azithromycin derivatives substituting 3-O-cladinose by seven- to seventeen-atom-length spacers anchored with various nucleobases (adenine, guanine, cytosine, thymine and uracil). There are two reasons to further this work. First, thymine and adenine were linked to 9- or 11-positions of some 3-O-cladinose-containing erythromycin derivatives, but the resulting activities, for unknown reasons, are poor [20,21]. The 3-O-derivatives of macrolides are not expected to activate the expression of inducible resistance genes, as the presence of 3-O-cladinose is required for efficient induction [22], but they failed to afford potency against A2058/2059-methylated or mutated strains. As purines and pyrimidines might form Watson–Crick interactions with rRNA bases, it would be interesting to know whether nucleobases attached at the C3-position of the macrolactone ring of macrolides could restore the activities against the ribosome-mutated strains. Secondly, the modes of action of ribosome-targeting inhibitors are less investigated in the early stage of new macrolides discovery [23] because it is difficult to isolate resistance mutants with alterations in the drug target sites because of the redundancy of rRNA genes in bacteria. To determine the biological function of nucleobases when attached to macrolides, we mutated ribosomal RNA A2058 or A2059 of a public strain E. coli SQ110 that has been engineered with only one chromosomal rrn allele [24].
2. Results and Discussion
The derivatization of the five nucleobases followed a modified procedure [25]. The nucleobases 3, 4, 8, 12 and 14 were obtained through three steps: a substitution reaction with methyl bromoacetate, protection and saponification, which are outlined in Scheme 1, Scheme 2, Scheme 3 and Scheme 4. To prevent the functional groups at the nucleobases, uracil, adenine, guanine and cytosine were protected by Boc, di-Boc, Bn and Cbz, respectively [26]. The nucleobases 5 and 10 were obtained through the reduction of 2 and 7 to alcohols by using NaBH4 and subsequent oxidization to aldehydes with Dess–Martin reagent.
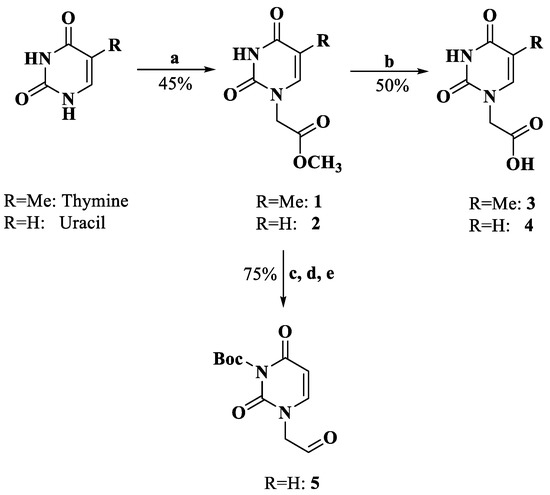
Scheme 1.
Synthesis of compounds 3–5. Reagents and conditions: a. DMF, K2CO3, BrCH2COOMe, rt; b. H2O, 2M NaOH; 4M HCl, 0 °C; c. CH2Cl2, DMAP, di-tert-butyl dicarbonate, rt; d. THF, MeOH, NaBH4, rt; e. CH2Cl2, Dess–Martin periodinane, rt.
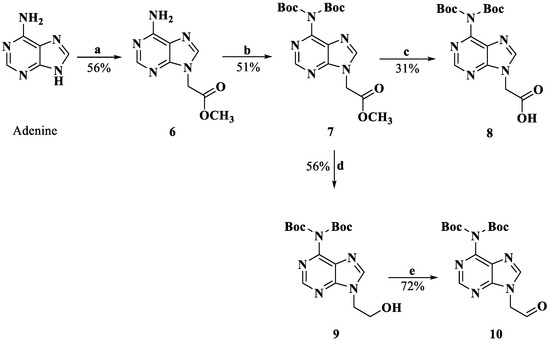
Scheme 2.
Synthesis of compounds 8 and 10. Reagents and conditions: a. DMF, K2CO3, BrCH2COOMe, rt; b. CH2Cl2, DMAP, di-tert-butyl dicarbonate, rt; c. MeOH, 2M NaOH; 4M HCl, 0 °C; d. THF, MeOH, NaBH4, rt; e. CH2Cl2, Dess–Martin periodinane, rt.
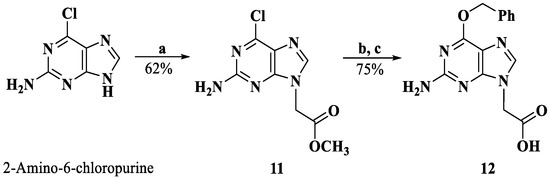
Scheme 3.
Synthesis of compound 12. Reagents and conditions: a. DMF, K2CO3, BrCH2COOMe, rt; b. DMF, NaH, benzyl alcohol, rt; c. MeOH, 2M NaOH; 4M HCl, 0 °C.
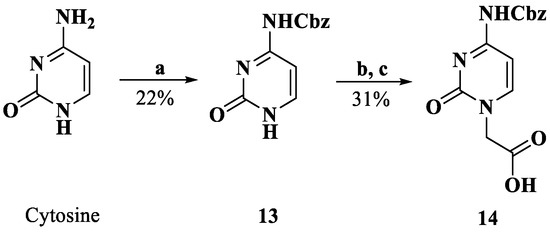
Scheme 4.
Synthesis of compound 14. Reagents and conditions: a. Pyridine, Cbz-Cl, 0 °C; b. DMF, K2CO3, BrCH2COOMe, rt; c. H2O, 2M NaOH; 4M HCl, 0 °C.
Intermediate 16 was prepared from azithromycin in three steps [27]. The general procedure for the synthesis of 26–35 (conjugating with nucleobases A/T/U/G/C) is listed in Scheme 5: introduction of various diamines to the 3-O position, amidization catalyzed by DCC/HOBT, the removal of 2′-acetyl group and the deprotection of the nucleobases [28,29]. Compounds 26T, 28T and 27U–32U were directly obtained without deprotection. Compounds 29A, 30A and 32A–34A were synthesized by removing the Boc groups under acidic conditions [30]. In the presence of Pd/C, a hydrogenolysis reaction was carried out to produce the target compounds of 29G–30G, 32G and 29C–30C [31,32]. 11,12-di-OH 35A was yielded from 11,12-cyclic carbonate 32A through a treatment with LiOH·H2O.
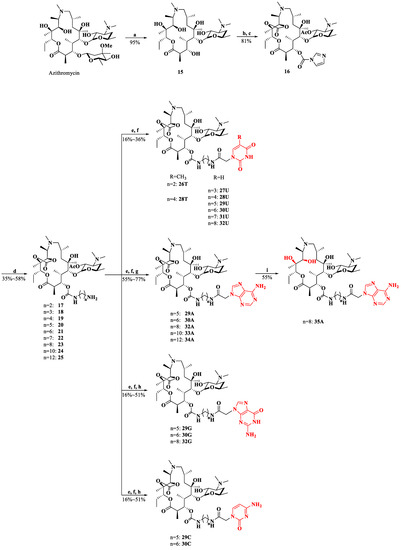
Scheme 5.
Synthesis of compounds 26–35. Reagents and conditions: a. EtOH, HCl, 40 °C; b. CH2Cl2, acetic anhydride, rt; c. CH2Cl2, 4-dimethylaminopyridine, N,N′-carbonyldiimdazole, Ar, rt; d. DMF, 1,2-diaminoethane/1,3-diaminopropane/1,4-diaminobutane/1,5-diaminopentane/1,6-diaminohexane/1,7-diaminoheptane/1,8-diaminooctane/1,10-diaminodecane/1,12-diaminododecane, rt. e. CH2Cl2, compounds 3, 4, 8, 12 or 14, 1-hydroxybenzotriazole, dicyclohexylcarbodiimide; f. MeOH, 60 °C; g. EtOH, HCl; h. MeOH, H2, Pd/C; i. THF:H2O = 1:1, LiOH·H2O.
In addition, a more flexible secondary amine (substituting for an amide) side chain spanning from macrolides and nucleobases was designed as a comparator (Scheme 6). In the presence of ZnCl2 and NaBH(OAc)3, the reaction of 23 with aldehydes 5 or 10 followed by deacetylation yielded the intermediates 36U and 36A [33]. The Boc group was then removed to produce the target compounds 37U and 37A. All target compounds 26–35 and 37 were structurally validated by using 1H NMR, 13C NMR and HRMS.
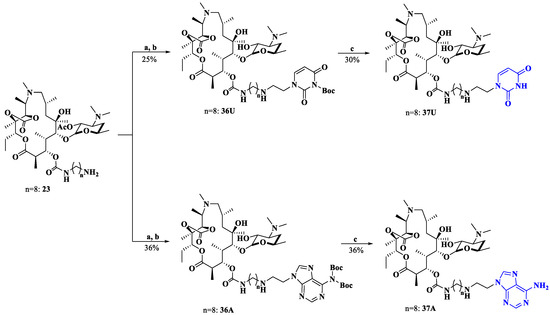
Scheme 6.
Synthesis of compounds 37U and 37A. Reagents and conditions: a. CH2Cl2, compound 5 or 10, ZnCl2, NaBH(OAc)3; b. MeOH, 60 °C; c. EtOH, HCl.
Initially, the MICs of compounds 26–32 were determined against a panel of clinical isolates. The data in Table 1 suggest that the influence of the bases on the activities followed the order of A > G > U > C (29A, 29G, 29U, 29C; 30A, 30G, 30U, 30C), and the activity increased when prolonging the length of the linker (27U–32U).

Table 1.
In vitro antibacterial activity of 26–32 against erythromycin-susceptible and -resistant clinical isolates.
Unexpectedly, the activities of compounds 26–32 were inferior to azithromycin against susceptible and efflux-encoded strains. To further determine whether conjugates 26–32 were vulnerable to outer membrane barriers and efflux pumps, we used the engineered E. coli stains SQ110DTC and SQ110LPTD to examine the MICs of compounds 30U, 30A, 30G, 30C, 32U and 32A (Table 2). SQ110 was rendered hypersusceptible to macrolides by deleting the gene encoding the multidrug efflux transporter TolC (strain SQ110DTC) or mutating the lptD gene, which renders the outer membrane more permeable (strain SQ110LPTD) [24]. The decreasing MICs against the SQ110 strains in comparison with the reference E. coli strain ATCC25922 indicated that the low activities of compounds 26–32 could be attributed to their poor permeability and vulnerability to efflux. Among them, compounds 30A and 32A, which have adenine groups, were the most active, which was in agreement with the SARs against the clinical isolates, as shown in Table 1. The presence of the uracil base was inferior to the adenine (30U vs. 30A, 32U vs. 32A). None of the compounds were active against the A2058/A2059-mutated SQ110 stains (Table 2).
Table 2.
In vitro antibacterial activity of 30 and 32 against some standard, engineered and mutant E. coli strains.
It was reported that menadione (VK3) could inhibit the efflux pumps and alter the fluidity of the bacterial membrane of S. aureus [34]. Thus, compound 32A, in combination with the usage of menadione (VK3) at concentrations of MIC/4 or MIC/8, exhibited better activities against S. aureus than 3-O-descladinose-3-OH azithromycin while much lower activities of 32A than azithromycin (Table 3), which suggested that the introduction of nucleobases is detrimental to the resulting activities of the conjugates against the clinical isolates.
Table 3.
In vitro antibacterial activity of 32A alone or in combination with menadione against S. aureus.
With the most potent compound, 32A, in hand, the analogs of 32A were then screened, as shown in Table 4. The varying lengths of the linker (32A, 33A, 34A) revealed that the homolog of 33A was more potent than 32A and 34A. Replacing the base A with G/U (32A vs. 32G; 37A vs. 37U) and removing cyclic carbonation at 11,12-OH (32A vs. 35A) were not favorable for activities. The conversion of a rigid linker (amide functionality) to a flexible one (amine) resulted in two- to four-fold higher activities (32A vs. 37A).
Table 4.
In vitro antibacterial activity of 32G, 32A–35A and 37 against some engineered and mutant E. coli strains.
The molecular docking studies of the ligands (37A and 32A) against the G2099A mutant Haloarcula Marismortui 50S ribosomal subunit (PDB ID: 1YHQ [35]) were performed utilizing Autodock VINA in YASARA (YASARA Biosciences GmbH, Vienna, VIE, AUT) [36,37]. The suggested binding modes of 37A and 32A (Figure 2) indicated that hydrogen bonds were formed between the adenines of the conjugates and the surrounded ribosomal nucleobases (Tables S1 and S2), which accounted for the better activities of 37A and 32A than azithromycin against A2058/2059-mutated SQ110 strains. Meanwhile, higher binding affinities were determined for 37A than 32A (Table 5), which was in agreement with the higher activities of 37A than those of 32A (Table 4).
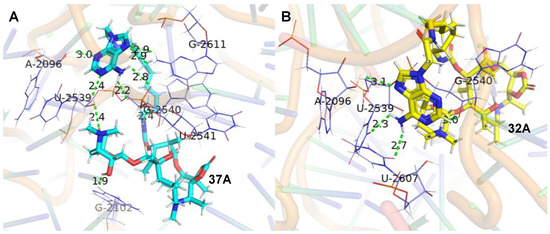
Figure 2.
Interactions of the ligands with 50S ribosomal subunit (PDB ID: 1YHQ). (A). Interaction between 37A (cyan) and 50S ribosomal subunit. (B). Interaction between 32A (yellow) and 50S ribosomal subunit. The green dashed lines represent conventional hydrogen bonds. The labeled bases are colored purple and shown in licorice by PyMOL [38].
Table 5.
The binding free energies (kcal/mol) and predicted KD values (nM) between the 50S ribosomal subunit (PDB ID: 1YHQ) and the ligands.
The adenine, 3′-N(CH3)2 of the desosamine, the carbamoyl group in 37A formed six conventional hydrogen bonds with both sugar and phosphate groups in the RNA backbone linked to A2096/ U2539/ G2540/ U2541 (Table S1). The adenine and 2′-OH of the desosamine in 37A formed three conventional hydrogen bonds with the 2-NH2 of G2611 and O6 of G2102. The adenine and 3′-N(CH3)2 of the desosamine in 32A formed two conventional hydrogen bonds with the sugar group in the RNA backbone linked to A2096 and G2540. The adenine in 32A formed two conventional hydrogen bonds with the O2 of U2607 and U2539 (Table S2). Therefore, it revealed that the 37A bound tightly to the backbone of the G2099A large ribosomal subunit. The reliability of the models constructed from protein–ligand docking was verified by using molecular dynamics (MD) simulations. Both methods yielded similar complex structures. Quantitatively, the RMSD values of the structures from the MD and docking are small (Figure S2), which indicates that the structural variation is minor. Interestingly, we found that after the molecular dynamic simulation, the 6-NH2 of compound 37A’s adenine formed two hydrogen bonds with the sugar in the RNA backbone linked to G2609. The -NH- of the linker chains formed two hydrogen bonds with the phosphate groups of G2540, and the -C=O- of the linker chains and the backbone of macrolides formed two hydrogen bonds with 6-NH2 of A2099 (Figure 3A, Table S3). The 6-OH of compound 32A formed a hydrogen bond with the sugar in the RNA backbone linked to U2541. The 2′-OH formed two hydrogen bonds with the 4-NH2 and N3 of C2104 and a hydrogen bond with 1-NH of G2102. Moreover, the -N-CH3 and O6 of the backbone of macrolides formed a hydrogen bond with the O2 of C2104 and 2-NH2 of G2102, respectively. The 3′-N(CH3)2 of the desosamine, 6-NH2 and N7 of adenine formed a hydrogen bond with the phosphate groups, O1 and the sugar in the RNA backbone linked to G2539, respectively. The -C=O- of the backbone of macrolides formed a hydrogen bond with 3-NH of U2620, and the N3 of adenine formed a hydrogen bond with the 2-NH2 of G2611 (Figure 3B, Table S4).
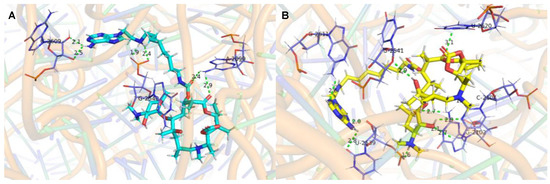
Figure 3.
Interactions of the ligands with 50S ribosomal subunit (PDB ID: 1YHQ) after molecular dynamic simulation. (A). Interaction between 37A (cyan) and 50S ribosomal subunit. (B). Interaction between 32A (yellow) and 50S ribosomal subunit. The green dashed lines represent conventional hydrogen bond. The labeled bases are colored purple and shown in licorice by PyMOL.
3. Materials and Methods
3.1. Synthetic Procedures
All of the solvents and reagents were obtained from commercial sources (Innochem, Beijing, China; Bidepharm, Shanghai, China; J&K scientific, Beijing, China) and used without further purification unless otherwise noted. The reactions were monitored by using thin-layer chromatography (TLC, Qingdao Ocean Chemical Co., Ltd., Qingdao, China) with silica gel HSGF254 precoated plates (0.2 mm), and the compounds were visualized under UV light (λ = 254 or 365 nm) and/or stained with iodine. Column chromatography was performed on silica gel (100–200 mesh). 1H and 13C spectra were taken in CDCl3, MeOH-d4 or DMSO-d6 on Bruker Ascend 400 MHz or Ascend 700 MHz spectrometers (Bruker, Massachusetts, Germany) with tetramethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS) were obtained with an Agilent Q-TOF 6520 (Agilent Technologies Inc., Santa Clara, CA, USA). The spectra of compounds are shown in Table S5.
3.1.1. Methyl Thymin-1-ylacetate (1)
To a suspension of thymine (1.000 g, 7.93 mmol) and K2CO3 (1.100 g, 7.93 mmol) in dry DMF, methyl bromoacetate was added (0.85 mL, 7.93 mmol), and the mixture was stirred overnight. Then, the mixture was filtered, the solid residue was cooled to 0 °C and then treated with water and 4M HCl. The precipitate was collected by filtration and washed with water to afford compound 1 (0.700 g, 3.54 mmol, 44.6%). HRMS (ESI) (M + H)+ m/z 199.0717, calcd for C8H11N2O4+ 199.0713. 1H NMR (DMSO-d6, 400 MHz) δ: 10.13 (s, 1 H, -NH-), 7.51 (s, 1 H, H-6), 4.49 (s, 2 H, -CH2-), 3.69 (s, 3 H, -O-CH3), 1.76 (s, 3 H, 5-CH3).
3.1.2. Thymin-1-ylacetic Acid (3)
Compound 1 (0.700 g, 3.54 mmol) was treated with water (3 mL) and 2M NaOH (3 mL). The mixture was boiled for 10 min, and then cooled to 0 °C and treated with 4M HCl to adjust the pH to 3. After stirring for 30 min, the solid was filtered and washed with water to afford compound 3 (0.325 g, 1.77 mmol, 50.0%). HRMS (ESI) (M − H)− m/z 183.0414, calcd for C7H7N2O4− 183.0411. 1H NMR (DMSO-d6, 400 MHz) δ: 13.08 (s, 1 H, -COOH), 11.34 (s, 1 H, -NH-), 7.49 (s, 1 H, H-6), 4.36 (s, 2 H, -CH2-), 1.75 (s, 3 H, 5-CH3).
3.1.3. Uracil-1-ylacetic Acid (4)
To a suspension of uracil (1.000 g, 8.92 mmol) and K2CO3 (1.356 g, 8.92 mmol) in dry DMF, methyl bromoacetate was added (0.96 mL, 8.92 mmol), and the mixture was stirred overnight. The mixture was filtered, and the solid residue was cooled to 0 ºC and treated with water and 4M HCl. The precipitate was collected by filtration and washed with water. The solid was treated with water (3 mL) and 2M NaOH (3 mL), and then the mixture was boiled for 10 min. The mixture was cooled to 0 °C and treated with 4M HCl to adjust the pH to 3. After stirring for 30 min, compound 4 (0.287 g, 1.69 mmol, 18.9%) was filtered and washed with water. HRMS (ESI) (M + H)+ m/z 171.0398, calcd for C6H7N2O4+ 171.0400. 1H NMR (DMSO-d6, 400 MHz) δ: 13.12 (s, 1 H, -COOH), 11.34 (s, 1 H, -NH-), 7.61 (d, J = 8.0 Hz, 1 H, H-6), 5.59 (d, J = 7.6 Hz, 1 H, H-5), 4.41 (s, 2 H, -CH2-).
3.1.4. (N3-(tert-butyloxy carbonyl)uracil-1-yl)acetaldehyde (5)
Compound 2 (2.313 g, 12.57 mmol) was dissolved in dry CH2Cl2, and then DMAP (0.566 g, 12.57 mmol) and di-tert-butyl dicarbonate (1.000 g, 15.08 mmol) were added to the solution over a period of 15 min at 0 °C. The cooling was discontinued, and the solution was stirred for 8 h. Subsequently, the mixture was treated with saturated NH4Cl repeatedly. After stirring for 30 min, the phases were separated, and the organic phase was washed with water and brine. The organic layer was concentrated and purified through column chromatography (CH2Cl2/EtOH = 10:0.1). Then, the resulting compound (0.400 g, 1.41 mmol) was dissolved in dry THF/MeOH (10:1), and NaBH4 (0.106 g, 2.82 mmol) was added to the solution. After stirring for 1 h at room temperature, the mixture was washed with water and brine, and the organic layer was concentrated. The resulting compound (0.218 g, 0.85 mmol) and Dess–Martin periodinane (0.396 g, 0.93 mmol) were dissolved in dry CH2Cl2. The solution was stirred for 1 h at room temperature and then washed with water and brine. The organic layer was concentrated and purified through column chromatography (CH2Cl2/EtOH = 10:0.3) to afford compound 5 (0.161 g, 0.64 mmol, 75.3%). 1H NMR (MeOD-d4, 400 MHz) δ: 9.72 (s, 1 H, -CHO), 7.03 (d, J = 8.0 Hz, 1 H, H-6), 5.83 (d, J = 8.0 Hz, 1 H, H-5), 4.64 (d, J = 4.8 Hz, 2 H, -CH2-), 1.63 (s, 9 H, -Boc).
3.1.5. Methyl Adenin-9-ylacetate (6)
To a suspension of adenine (1.000 g, 7.40 mmol) and K2CO3 (1.227 g, 8.88 mmol) in dry DMF, methyl bromoacetate was added (1.60 mL, 14.80 mmol). After stirring at room temperature for 5 h, the mixture was filtered, and the solid residue was treated with ethyl acetate, resulting in recrystallization. The solid was filtered off and followed by recrystallization using MeOH to yield 0.807 g (4.18 mmol, 56.5%) of white product. HRMS (ESI) (M + H)+ m/z 194.0670, calcd for C7H8N5O2+ 194.0673. 1H NMR (DMSO-d6, 400 MHz) δ: 8.13 (s, 1 H, H-2), 8.12 (s, 1 H, H-8), 7.27 (s, 2 H, -NH2), 5.09 (s, 2 H, -CH2-), 3.70 (s, 3 H, -O-CH3).
3.1.6. Methyl (N6-(di-tert-butyloxy carbonyl)adenin-9-yl)acetate (7)
Compound 6 (0.807 g, 4.18 mmol) was dissolved in dry CH2Cl2, and then DMAP (1.532 g, 12.54 mmol) and di-tert-butyl dicarbonate (2.737 g, 12.54 mmol) were added to the solution over a period of 15 min at 0 °C. The cooling was discontinued, and the solution was stirred overnight. Subsequently, the mixture was treated with saturated NH4Cl repeatedly. After stirring for 30 min, the phases were separated, and the organic phase was washed with water and brine. The organic layer was concentrated and purified through column chromatography (CH2Cl2/EtOH = 10:0.15) to obtain compound 7 (0.795 g, 2.15 mmol, 51.4%). 1H NMR (CDCl3, 400 MHz) δ: 8.86 (s, 1 H, H-2), 8.16 (s, 1 H, H-8), 5.08 (s, 2 H, -CH2-), 3.81 (s, 3 H, -CH3), 1.45 (s, 18 H, -N(Boc)2).
3.1.7. (N6-(di-tert-butyloxy carbonyl)adenin-9-yl)acetic Acid (8)
Compound 7 (0.795 g, 2.15 mmol) was mixed with MeOH, and 2M NaOH (5 mL) was added. After stirring for 30 min, CH2Cl2 and water were added to the solution for extraction. The water phase was adjusted to pH 3.0 with 4M HCl at 0 °C. Target compound 8 (0.507 g, 1.29 mmol, 30.9%) was filtered and washed with water. HRMS (ESI) (M + H)+ m/z 394.1713, calcd for C7H8N5O2+ 394.1721. 1H NMR (CDCl3, 400 MHz) δ: 8.77 (s, 1 H, H-2), 8.29 (s, 1 H, H-8), 4.86 (s, 2 H, -CH2-), 1.42 (s, 18 H, -N(Boc)2).
3.1.8. (N6-(di-tert-butyloxy carbonyl)adenin-9-yl)ethanol (9)
Compound 7 (0.500 g, 1.23 mmol) was dissolved in dry THF/MeOH (10:1), and NaBH4 (0.093 g, 2.45 mmol) was added to the solution. The mixture was stirred for 1 h at room temperature and then washed with water and brine. The organic layer was concentrated and purified through column chromatography (CH2Cl2/EtOH = 10:0.5) to afford compound 9 (0.260 g, 0.69 mmol, 56.0%). 1H NMR (CDCl3, 400 MHz) δ: 8.84 (s, 1 H, H-2), 8.14 (s, 1 H, H-8), 4.41 (t, J = 4.8 Hz, 2 H, -CH2-), 4.01 (t, J = 4.8 Hz, 2 H, -CH2-OH), 3.57 (s, 1 H, -OH), 1.46 (s, 18 H, -N(Boc)2).
3.1.9. (N6-(di-tert-butyloxy carbonyl)adenin-9-yl)acetaldehyde (10)
Compound 9 (0.260 g, 0.69 mmol) and Dess–Martin periodinane (0.349 g, 0.83 mmol) were dissolved in dry CH2Cl2. The solution was stirred for 1 h at room temperature, then washed with water and brine. The organic layer was concentrated and purified through column chromatography (CH2Cl2/EtOH = 10:0.3) to afford compound 10 (0.189 g, 0.50 mmol, 72.0%). HRMS (ESI) (M + H)+ m/z 378.1768, calcd for C17H24N5O5+ 378.1772. 1H NMR (CDCl3, 400 MHz) δ: 9.86 (s, 1 H, -CHO), 8.87 (s, 1 H, H-2), 8.11 (s, 1 H, H-8), 5.22 (s, 2 H, -CH2-), 1.48 (s, 18 H, -N(Boc)2).
3.1.10. Methyl (2-Amino-6-chloropurin-9-yl)acetate (11)
To a suspension of 2-amino-6-chloropurine (1.000 g, 5.91 mmol) and K2CO3 (2.450 g, 17.73 mmol) in DMF, methyl bromoacetate was added (0.76 mL, 7.09 mmol), and the mixture was stirred for 5 h. Water was added, the suspension was filtered, and the solid was dried to afford intermediate 11 (0.880 g, 3.65 mmol, 62%).
3.1.11. (2-Amino-6-(benzyloxy)purin-9-yl)acetic acid (12)
NaH (0.730 g, 18.25 mmol) was dissolved in benzyl alcohol (10 mL) for 2 h at 0 °C, a solution of compound 11 (0.880 g, 3.65 mmol) in DMF was slowly added, and the resultant suspension was stirred overnight at room temperature. Then, 1M NaOH was added, and the solution was washed with ethyl acetate. The water phase was acidified to pH 3 with 4M HCl and extracted with ethyl acetate repeatedly. The combined organic phases were washed with brine and concentrated. The residue was recrystallized from EtOH (yield 74.8%). 1H NMR (DMSO-d6, 400 MHz) δ: 7.84 (s, 1 H, H-8), 7.52–7.35 (m, 5 H, -phenyl), 6.50 (s, 2 H, -NH2), 5.50 (s, 2 H, -O-CH2-), 4.82 (s, 2 H, -CH2-).
3.1.12. N4-(Benzyloxycarbonyl)cytosine (13)
Cbz-Cl (2.54 mL, 18.0 mmol) was added dropwise over a suspension of cytosine (1.000 g, 9.0 mmol) in pyridine (10 mL) at 0 °C. The mixture was stirred overnight. Water was added, and the pH was adjusted to 1 with 4M HCl. The resultant white precipitate was filtered off, washed with water, and dried to afford 13 (0.510 g, 2.08 mmol, 22%). 1H NMR (DMSO-d6, 400 MHz) δ: 7.79 (d, J = 7.2 Hz, 1 H, H-6), 7.40–7.34 (m, 5 H, -phenyl), 6.91 (d, J = 7.2 Hz, 1 H, H-5), 5.17 (s, 2 H, -O-CH2-).
3.1.13. N4-(Benzyloxycarbonyl)cytosin-1-yl)acetic Acid (14)
To a suspension of compound 13 (0.510 g, 2.08 mmol) and K2CO3 (0.288 g, 2.08 mmol) in dry DMF, methyl bromoacetate was added (0.23 mL, 2.08 mmol), and the mixture was stirred overnight. The mixture was filtered, and the solid residue was treated with water and 4M HCl at 0 °C. The precipitate was collected through filtration and treated with water (3 mL) and 2M NaOH (3 mL). After stirring for 30 min, the solution was cooled to 0 ºC and treated with 4M HCl to adjust the pH to 3. The mixture was filtered and washed with water to obtain a white solid, 14 (0.198 g, 0.65 mmol, 31.4%). HRMS (ESI) (M + H)+ m/z 304.0954, calcd for C14H14N3O5+ 304.0928. 1H NMR (DMSO-d6, 400 MHz) δ: 8.04 (d, J = 7.2 Hz, 1 H, H-6), 7.41–7.35 (m, 5 H, -phenyl), 7.02 (d, J = 6.8 Hz, 1 H, H-5), 5.20 (s, 1 H, -O-CH2-), 4.53 (s, 2 H, -CH2-).
3.1.14. 3-O-descladinosyl-3-OH azithromycin (15)
To a solution of azithromycin (20.000 g, 26.7 mmol) in ethanol (35 mL), 1 M hydrochloric acid was slowly dripped into the reaction system. After stirring for 1 h at 40 °C, the pH was adjusted to 10 with NH3·H2O, and then CH2Cl2 was added for extraction. The organic phase was washed with water and brine and dried to afford compound 15 (15.500 g, 26.2 mmol, 98.1%) as a white solid. HRMS (ESI) (M + H)+ m/z 591.4194, calcd for C30H59N2O9+ 591.4215. 1H NMR (MeOD-d4, 400 MHz) δ: 4.92 (dd, J = 10.8, 2.4 Hz, 1 H, H-13), 4.68 (d, J = 7.2 Hz, 1 H, H-1′), 3.63–3.60 (m, 2 H, H-3, H-11), 3.57–3.51 (m, 2 H, H-5, H-5′), 3.26 (dd, J = 10.0, 7.6 Hz, 1 H, H-2′), 2.84 (q, J = 10.0, 7.2 Hz, 1 H, H-10), 2.68–2.57 (m, 2 H, H-2, H-3′), 2.50–2.46 (m, 1 H, H-9a), 2.35 (s, 3 H, N-CH3), 2.33 (s, 6 H, -N(CH3)2), 2.24–2.22 (m, 1 H, H-4), 2.17–2.11 (m, 1 H, H-9b), 1.90–1.84 (m, 2 H, H-8, H-14eq), 1.74–1.65 (m, 2 H, H-4′a, H-7a), 1.58–1.42 (m, 2 H, H-7b, H-14ax), 1.25 (s, 3 H, 6-CH3), 1.24–1.20 (m, 7 H, H-4′b, 2-CH3, 4-CH3), 1.11 (d, J = 6.8 Hz, 3 H, 5′-CH3), 1.08 (s, 3 H, 12-CH3), 1.00 (d, J = 7.6 Hz, 3 H, 10-CH3), 0.92 (d, J = 7.2 Hz, 3 H, 8-CH3), 0.88 (t, J = 7.2 Hz, 3 H, 15-CH3).
3.1.15. 2′-O-acetyl-3-O-descladinosyl-3-O-imidazolylcarbonyl azithromycin-11,12-cyclic carbonate (16)
To a stirred solution of compound 15 (15.500 g, 26.2 mmol) in CH2Cl2 (150 mL), acetic anhydride was added (3.22 mL, 34.1 mmol). The mixture was stirred at room temperature for 1 h and quenched by the addition of saturated NaHCO3. The remaining acetic anhydride was removed by washing thoroughly with saturated NaHCO3. Then, the organic phase was washed with water and brine and concentrated to yield 2′-O-acetyl-3-O-descladinosylazithromycin (15.900 g, 25.1 mmol, 95.7%) as a white solid.
The resulting compound (15.900 g, 25.1 mmol) was dissolved in dry CH2Cl2 (10 mL), then 4-dimethylaminopyridine (6.131 g, 50.2 mmol) and N, N-carbonyl diimidazole (12.176 g, 75.3 mmol) were added to this solution under Ar. The reaction was stirred at room temperature for 12 h and was washed with saturated NH4Cl solution, water and brine. The organic phase was evaporated under reduced pressure, and then an appropriate amount of ethyl acetate was added. The mixture was stirred in an ice bath for 2 h and filtered to afford a white solid, 16 (15.928 g, 21.2 mmol, 84.6%). HRMS (ESI) (M + H)+ m/z 753.4280, calcd for C37H61N4O12+ 753.4278. 1H NMR (CDCl3, 400 MHz) δ: 8.22 (s, 1 H, H-imidazole), 7.51 (t, J = 1.5 Hz, 1 H, H-imidazole), 7.14 (dd, J = 1.7, 0.9 Hz, 1 H, H-imidazole), 6.62 (s, 1 H, 6-OH), 5.67 (dd, J = 9.2, 3.3 Hz, 1 H, H-3), 5.07 (dd, J = 10.8, 2.4 Hz, 1 H, H-13), 4.69 (dd, J = 10.6, 7.5 Hz, 1 H, H-2′), 4.59 (s, 1 H, H-11), 4.02 (d, J = 7.5 Hz, 1 H, H-1′), 3.60 (d, J = 2.7 Hz, 1 H, H-5), 3.21–3.12 (m, 1 H, H-2), 2.86 (q, J = 6.6 Hz, 1 H, H-10), 2.69–2.60 (m, 1 H, H-5′), 2.40–2.34 (m, 1 H, H-9a), 2.34–2.29 (m, 1 H, H-3′), 2.23–2.30 (m, 4 H, N-CH3, H-4), 2.20 (s, 6 H, -N(CH3)2), 2.09 (s, 3 H, 2′-O-CO-CH3), 2.07–2.03 (m, 1 H, H-9b), 1.94–1.82 (m, 2 H, H-7a, H-14eq), 1.79–1.67 (m, 1 H, H-8), 1.64–1.56 (m, 1 H, H-14ax), 1.55–1.48 (m, 2 H, H-7b, H-4′a), 1.43 (s, 3 H, 12-CH3), 1.31–1.26 (m, 1 H, H-4′b), 1.25 (d, J = 7.1 Hz, 3 H, 5′-CH3), 1.22 (s, 3 H, 6-CH3), 1.11 (d, J = 6.1 Hz, 3 H, 10-CH3), 1.09–1.04 (m, 6 H, 2-CH3, 4-CH3), 0.94–0.88 (m, 6 H, 8-CH3, 15-CH3).
3.1.16. General Procedures for the Synthesis of Intermediates 17–25
Compound 16 (1 eq) was dissolved in DMF, then a diamine (5 eq) was added to this solution. The reaction was stirred at room temperature for 2 h. Water was added, and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with water and brine. The organic layer was concentrated and purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.4:0.05) to obtain intermediates 17–25.
3.1.17. General Procedures for the Synthesis of Target Compounds 26T, 28T, 27U–32U
Compounds 17–23 (1 eq), compound 3 or 4 (1.2 eq) and 1-hydroxybenzotriazole (HOBT, 1.1 eq) were dissolved in CH2Cl2, and then the solution was cooled to 0 °C and dicyclohexylcarbodiimide (DCC, 1.2 eq) was added. The ice bath was removed after 1.5 h, and the stirring was continued for another 2.5 h at rt. The precipitated DCU was removed through filtration and washed with CH2Cl2. Then, the combined filtrate was washed with water and brine and then concentrated. The resulting product was dissolved in methanol and stirred at 60 °C for 2 h. The methanol was evaporated, and the crude product was purified through column chromatography to afford the target compounds, 26T, 28T and 27U–32U.
3-O-descladinosyl-3-O-(2-(2-(thymin-1-yl)acetamido)ethyl)carbamoyl azithromycin-11,12-cyclic carbonate (26T)
Compound 17 (0.277 g, 0.37 mmol), compound 3 (0.103 g, 0.45 mmol), DCC (0.092 g, 0.45 mmol) and HOBT (0.055 g, 0.40 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.5:0.05) to afford compound 26T (80.2 mg, 0.09 mmol, 24.3%). Melting point: 145.3–146.8 °C. HRMS (ESI) (M + H)+ m/z 869.4871, calcd for C41H69N6O14+ 869.4866. 1H NMR (CDCl3, 400 MHz) δ: 7.46 (s, 1 H, -NH-thymine), 7.05 (s, 1 H, H-thymine), 6.30 (s, 1 H, -OH), 5.87 (s, 1 H, -O-CONH-), 5.12 (d, J = 8.8 Hz, 1 H, H-3), 5.06 (dd, J = 10.0, 2.4 Hz, 1 H, H-13), 4.65 (s, 1 H, H-11), 4.29 (s, 2 H, -CH2-T), 4.18 (d, J = 7.2 Hz, 1 H, H-1′), 3.57 (s, 1 H, H-5), 3.46–3.31 (m, 6 H, H-5′, -CONHCH2CH2-, H-2′), 2.88–2.82 (m, 2 H, H-10, H-2), 2.71–2.69 (m, 1 H, H-3′), 2.40–2.37 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.18–2.16 (m, 1 H, H-4), 2.07–2.01 (m, 1 H, H-9b), 1.92 (s, 4 H, thymine-CH3, H-8), 1.86–1.82 (m, 1 H, H-14eq), 1.73–1.70 (m, 1 H, H-4′a), 1.62–1.54 (m, 2 H, H-14ax, H-7a), 1.43 (s, 3 H, 12-CH3), 1.39–1.36 (m, 2 H, H-7b, H-4′b), 1.29 (s, 3 H, 6-CH3), 1.23 (d, J = 6.0 Hz, 3 H, 5′-CH3), 1.17 (d, J = 6.8 Hz, 3 H, 2-CH3), 1.08–1.05 (m, 6 H, 10-CH3, 4-CH3), 0.94–0.89 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.4, 167.4, 164.5, 157.3, 153.4, 151.8, 140.9, 111.1, 104.2, 88.9, 86.0, 85.0, 79.1, 76.1, 73.2, 71.0, 69.1, 67.7, 65.3, 61.6, 51.4, 43.3, 42.2, 40.9, 39.9, 36.6, 35.0, 29.0, 26.2, 25.6, 21.9, 21.4, 21.1, 15.1, 13.9, 12.4, 10.3, 9.6, 5.3.
3-O-descladinosyl-3-O-(4-(2-(thymin-1-yl)acetamido)butyl)carbamoyl azithromycin-11,12-cyclic carbonate (28T)
Compound 19 (0.321 g, 0.36 mmol), compound 3 (0.080 g, 0.43 mmol), DCC (0.089 g, 0.43 mmol) and HOBT (0.054 g, 0.39 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.5:0.05) to afford compound 28T (100.5 mg, 0.11 mmol, 30.6%). Melting point: 139.5–141.2 °C. HRMS (ESI) (M + H)+ m/z 897.5169, calcd for C43H73N6O14+ 897.5179. 1H NMR (CDCl3, 400 MHz) δ: 7.10 (s, 1 H, H-thymine), 7.00 (s, 1 H, -NH-thymine), 6.15 (s, 1 H, -OH), 5.65 (s, 1 H, -O-CONH-), 5.12 (d, J = 10.0 Hz, 1 H, H-3), 5.06 (dd, J = 10.4, 2.4 Hz, 1 H, H-13), 4.61 (s, 1 H, H-11), 4.31 (s, 2 H, -CH2-T), 4.13 (d, J = 7.2 Hz, 1 H, H-1′), 3.52 (s, 1 H, H-5), 3.44–3.09 (m, 6 H, H-5′, -CONH-CH2CH2CH2CH2-, H-2′), 2.88–2.83 (m, 2 H, H-10, H-2), 2.71–2.69 (m, 1 H, H-3′), 2.41–2.36 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.17−2.15 (m, 1 H, H-4), 2.06–1.99 (m, 1 H, H-9b), 1.92–1.82 (m, 5 H, thymine-CH3, H-8, H-14eq), 1.78–1.75 (m, 1 H, H-4′a), 1.56–1.43 (m, 6 H, H-14ax, H-7a, -CONH-CH2CH2CH2-CH2-), 1.43 (s, 3 H, 12-CH3), 1.34–1.28 (m, 2 H, H-7b, H-4′b), 1.24–1.19 (m, 6 H, 6-CH3, 5′-CH3), 1.18 (d, J = 6.8 Hz, 3 H, 2-CH3), 1.08–1.05 (m, 6 H, 10-CH3, 4-CH3), 0.93−0.88 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.4, 166.8, 156.7, 153.4, 152.1, 141.0, 111.2, 103.7, 88.5, 86.3, 84.9, 78.9, 75.9, 73.0, 71.0, 69.0, 68.0, 65.5, 61.9, 51.6, 43.3, 42.0, 40.4, 39.8, 39.2, 36.0, 35.0, 29.7, 27.1, 26.1, 25.8, 21.8, 21.4, 21.1, 15.6, 13.6, 12.4, 10.2, 9.5, 5.0.
3-O-descladinosyl-3-O-(3-(2-(uracil-1-yl)acetamido)propyl)carbamoyl azithromycin-11, 12-cyclic carbonate (27U)
Compound 18 (0.355 g, 0.50 mmol), compound 4 (0.100 g, 0.60 mmol), DCC (0.123 g, 0.60 mmol) and HOBT (0.074 g, 0.55 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.5:0.05) to afford compound 27U (80.5 g, 0.09 mmol, 18.0%). Melting point: 139.6–140.6 °C. HRMS (ESI) (M + H)+ m/z 869.4864, calcd for C41H69N6O14+ 869.4866. 1H NMR (CDCl3, 400 MHz) δ: 7.31 (s, 1 H, -NH-uracil), 7.25 (d, J = 7.6 Hz, 1 H, H-uracil), 6.15 (s, 1 H, -OH), 5.89 (s, 1 H, -O-CONH-), 5.71 (d, J = 7.6 Hz, 1 H, H-uracil), 5.11 (d, J = 10.4 Hz, 1 H, H-3), 5.04 (dd, J = 10.4, 2.4 Hz, 1 H, H-13), 4.58 (s, 1 H, H-11), 4.48–4.27 (m, 2 H, -CH2-U), 4.11 (d, J = 7.6 Hz, 1 H, H-1′), 3.51 (s, 1 H, H-5), 3.48–3.09 (m, 6 H, H-5′, -CONH-CH2CH2CH2-, H-2′), 2.89–2.81 (m, 2 H, H-10, H-2), 2.73–2.67 (m, 1 H, H-3′), 2.37–2.36 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.16–2.14 (m, 1 H, H-4), 2.05−2.01 (m, 1 H, H-9b), 1.89−1.82 (m, 2 H, H-8, H-14eq), 1.74−1.70 (m, 1 H, H-4′a), 1.69–1.54 (m, 4 H, H-14ax, H-7a, -CONHCH2CH2CH2-), 1.43 (s, 3 H, 12-CH3), 1.34–1.31 (m, 2 H, H-7b, H-4′b), 1.24–1.21 (m, 6 H, 6-CH3, 2-CH3), 1.16 (d, J = 7.2 Hz, 3 H, 5′-CH3), 1.08–1.04 (m, 6 H, 10-CH3, 4-CH3), 0.93–0.88 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.6, 167.0, 164.5, 157.0, 153.4, 151.9, 145.2, 103.8, 102.6, 88.2, 86.2, 85.0, 78.8, 75.9, 73.1, 71.0, 69.1, 67.9, 64.9, 61.8, 51.5, 43.3, 42.0, 39.7, 37.7, 36.5, 36.1, 35.0, 29.5, 28.9, 26.0, 25.8, 21.7, 21.4, 21.1, 15.5, 13.6, 10.2, 9.6, 5.1.
3-O-descladinosyl-3-O-(4-(2-(uracil-1-yl)acetamido)butyl)carbamoyl azithromycin-11,12-cyclic carbonate (28U)
Compound 19 (0.250 g, 0.34 mmol), compound 4 (0.070 g, 0.41 mmol), DCC (0.084 g, 0.41 mmol) and HOBT (0.051 g, 0.37 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.5:0.05) to afford compound 28U (52.7 mg, 0.06 mmol, 17.5%). Melting point: 136.9–138.3 °C. HRMS (ESI) (M + H)+ m/z 883.5014, calcd for C42H71N6O14+ 883.5023. 1H NMR (CDCl3, 400 MHz) δ: 7.30 (d, J = 8.0 Hz, 1 H, H-uracil), 7.17 (s, 1 H, -NH-uracil), 6.23 (s, 1 H, -OH), 5.72 (d, J = 8.0 Hz, 1 H, H-uracil), 5.68 (s, 1 H, -O-CONH-), 5.11 (d, J = 10.4 Hz, 1 H, H-3), 5.06 (dd, J = 10.4, 2.4 Hz, 1 H, H-13), 4.59 (s, 1 H, H-11), 4.38 (s, 2 H, -CH2-U), 4.12 (d, J = 7.2 Hz, 1 H, H-1′), 3.51 (s, 1 H, H-5), 3.40–3.04 (m, 6 H, H-5′, -CONH-CH2CH2CH2CH2-, H-2′), 2.90–2.84 (m, 2 H, H-10, H-2), 2.64–2.58 (m, 1 H, H-3′), 2.39–2.31 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.17–2.14 (m, 1 H, H-4), 2.06–2.00 (m, 1 H, H-9b), 1.89–1.82 (m, 2 H, H-8, H-14eq), 1.72–1.69 (m, 1 H, H-4′a), 1.60–1.57 (m, 6 H, H-14ax, H-7a, -CONHCH2CH2CH2CH2-), 1.43 (s, 3 H, 12-CH3), 1.33−1.31 (m, 2 H, H-7b, H-4′b), 1.23–1.17 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.08–1.05 (m, 6 H, 10-CH3, 4-CH3), 0.93–0.88 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.6, 166.7, 164.5, 156.8, 153.4, 152.0, 145.3, 103.7, 102.6, 88.1, 86.3, 84.9, 78.8, 75.9, 73.1, 71.0, 69.1, 68.0, 65.5, 61.8, 51.5, 43.3, 42.0, 40.4, 39.2, 36.0, 35.0, 29.3, 27.2, 26.1, 25.8, 21.7, 21.4, 21.1, 15.6, 13.6, 10.2, 9.5, 5.0.
3-O-descladinosyl-3-O-(5-(2-(uracil-1-yl)acetamido)pentyl)carbamoyl azithromycin-11,12-cyclic carbonate (29U)
Compound 20 (0.285 g, 0.38 mmol), compound 4 (0.078 g, 0.45 mmol), DCC (0.094 g, 0.45 mmol) and HOBT (0.057 g, 0.42 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.45:0.05) to afford compound 29U (60.5 mg, 0.07 mmol, 18.4%). Melting point: 137.8–139.9 °C. HRMS (ESI) (M + H)+ m/z 897.5181, calcd for C43H73N6O14+ 897.5179. 1H NMR (CDCl3, 400 MHz) δ: 7.32 (d, J = 8.0 Hz, 1 H, H-uracil), 7.00 (s, 1 H, -NH-uracil), 6.17 (s, 1 H, -OH), 5.71 (d, J = 8.0 Hz, 1 H, H-uracil, -O-CONH-), 5.12 (d, J = 10.4 Hz, 1 H, H-3), 5.05 (dd, J = 10.8, 2.4 Hz, 1 H, H-13), 4.58 (s, 1 H, H-11), 4.43 (s, 2 H, -CH2-U), 4.13 (d, J = 7.2 Hz, 1 H, H-1′), 3.51 (s, 1 H, H-5), 3.42–3.19 (m, 6 H, H-5′, -CONH-CH2(CH2)3CH2-, H-2′), 2.90–2.80 (m, 2 H, H-10, H-2), 2.73–2.67 (m, 1 H, H-3′), 2.40–2.35 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.16–2.14 (m, 1 H, H-4), 2.04–1.99 (m, 1 H, H-9b), 1.88–1.84 (m, 2 H, H-8, H-14eq), 1.76–1.72 (m, 1 H, H-4′a), 1.61–1.50 (m, 6 H, H-14ax, H-7a, -CONHCH2CH2CH2CH2CH2-), 1.42 (s, 3 H, 12-CH3), 1.37–1.28 (m, 4 H, -CONH-CH2CH2CH2CH2CH2-, H-7b, H-4′b), 1.23–1.17 (m, 9 H, 6-CH3, 5′-CH3, 2-CH3), 1.08–1.05 (m, 6 H, 10-CH3, 4-CH3), 0.92–0.88 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.6, 166.6, 164.4, 156.8, 153.4, 151.6, 145.5, 103.6, 102.3, 88.0, 86.4, 84.8, 78.9, 76.0, 73.1, 71.0, 69.1, 68.1, 65.6, 61.9, 51.0, 43.2, 42.0, 38.7, 35.9, 35.0, 29.8, 29.5, 28.8, 26.0, 25.8, 25.3, 21.7, 21.4, 21.1, 15.7, 13.5, 9.4, 5.0.
3-O-descladinosyl-3-O-(6-(2-(uracil-1-yl)acetamido)hexyl)carbamoyl azithromycin-11,12-cyclic carbonate (30U)
Compound 21 (0.316 g, 0.42 mmol), compound 4 (0.085 g, 0.50 mmol), DCC (0.104 g, 0.50 mmol) and HOBT (0.063 g, 0.46 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.45:0.05) to afford compound 30U (61.3 mg, 0.07 mmol, 16.0%). Melting point: 137.5–139.6 °C. HRMS (ESI) (M + H)+ m/z 911.5326, calcd for C44H75N6O14+ 911.5336. 1H NMR (CDCl3, 400 MHz) δ: 7.33 (d, J = 8.0 Hz, 1 H, H-uracil), 7.06 (s, 1 H, -NH-uracil), 6.19 (s, 1 H, -OH), 5.71 (d, J = 8.0 Hz, 1 H, H-uracil), 5.63 (s, 1 H, -O-CONH-), 5.11 (d, J = 10.4 Hz, 1 H, H-3), 5.05 (dd, J = 10.8, 2.4 Hz, 1 H, H-13), 4.60 (s, 1 H, H-11), 4.45 (s, 2 H, -CH2-U), 4.12 (d, J = 7.2 Hz, 1 H, H-1′), 3.54 (s, 1 H, H-5), 3.44–3.04 (m, 6 H, H-5′, -CONH-CH2(CH2)4CH2-, H-2′), 2.90–2.84 (m, 2 H, H-10, H-2), 2.64–2.58 (m, 1 H, H-3′), 2.39–2.31 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.17–2.14 (m, 1 H, H-4), 2.06–2.00 (m, 1 H, H-9b), 1.89–1.82 (m, 2 H, H-8, H-14eq), 1.72–1.69 (m, 1 H, H-4′a), 1.60–1.57 (m, 6 H, H-14ax, H-7a, -CONHCH2CH2(CH2)2CH2CH2-), 1.43–1.31 (m, 9 H, 12-CH3, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2-), 1.23–1.17 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.08–1.05 (m, 6 H, 10-CH3, 4-CH3), 0.93–0.88 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.6, 166.7, 164.5, 156.8, 153.4, 152.0, 145.3, 103.7, 102.6, 88.1, 86.3, 84.9, 78.8, 75.9, 73.1, 71.0, 69.1, 68.0, 65.5, 61.8, 51.5, 43.3, 42.0, 40.4, 39.2, 36.0, 35.0, 29.3, 27.2, 26.1, 25.8, 21.7, 21.4, 21.1, 15.6, 13.6, 10.2, 9.5, 5.0.
3-O-descladinosyl-3-O-(7-(2-(uracil-1-yl)acetamido)heptyl)carbamoyl azithromycin-11, 12-cyclic carbonate (31U)
Compound 22 (0.362 g, 0.47 mmol), compound 4 (0.095 g, 0.56 mmol), DCC (0.095 g, 0.56 mmol) and HOBT (0.070 g, 0.52 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.4:0.05) to afford compound 31U (100.2 mg, 0.11 mmol, 23.0%). Melting point: 120.5–121.9 °C. HRMS (ESI) (M + H)+ m/z 925.5489, calcd for C45H77N6O14+ 925.5492. 1H NMR (CDCl3, 400 MHz) δ: 7.34 (d, J = 8.0 Hz, 1 H, H-uracil), 6.97 (s, 1 H, -NH-uracil), 6.08 (s, 1 H, -H), 5.71 (d, J = 8.0 Hz, 1 H, H-uracil), 5.58 (s, 1 H, -O-CONH-), 5.11 (d, J = 10.4 Hz, 1 H, H-3), 5.05 (dd, J = 10.8, 2.4 Hz, 1 H, H-13), 4.58 (s, 1 H, H-11), 4.45 (q, J = 16.0, 12.0 Hz, 2 H, -CH2-U), 4.13 (d, J = 7.2 Hz, 1 H, H-1′), 3.52 (s, 1 H, H-5), 3.43–3.03 (m, 6 H, H-5′, -CONH-CH2(CH2)5CH2-, H-2′), 2.90–2.81 (m, 2 H, H-10, H-2), 2.64–2.58 (m, 1 H, H-3′), 2.37–2.31 (m, 7 H, -N(CH3)2, H-9a), 2.24 (s, 3 H, N-CH3), 2.17–2.11 (m, 1 H, H-4), 2.05–1.99 (m, 1 H, H-9b), 1.88–1.80 (m, 2 H, H-8, H-14eq), 1.71–1.67 (m, 1 H, H-4′a), 1.61–1.50 (m, 6 H, H-14ax, H-7a, -CONHCH2CH2(CH2)3CH2CH2-), 1.42 (s, 3 H, 12-CH3), 1.32-1.26 (m, 8 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2-), 1.22–1.18 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.07–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.93–0.90 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.7, 166.7, 164.8, 156.6, 153.4, 151.6, 145.6, 103.6, 102.2, 87.8, 86.4, 84.8, 78.8, 75.9, 73.1, 70.9, 69.2, 68.1, 65.6, 61.9, 51.0, 43.3, 41.9, 40.6, 39.9, 39.3, 36.0, 35.0, 29.6, 29.0, 28.9, 28.1, 26.2, 26.0, 25.8, 21.7, 21.4, 21.2, 15.7, 13.5, 10.1, 9.4, 5.0.
3-O-descladinosyl-3-O-(8-(2-(uracil-1-yl)acetamido)octyl)carbamoyl azithromycin-11,12-cyclic carbonate (32U)
Compound 23 (0.366 g, 0.47 mmol), compound 4 (0.095 g, 0.56 mmol), DCC (0.095 g, 0.56 mmol) and HOBT (0.070 g, 0.52 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.4:0.05) to afford compound 32U (83.5 mg, 0.09 mmol, 18.9%). Melting point: 121.9–123.6 °C. HRMS (ESI) (M + H)+ m/z 939,5642, calcd for C46H79N6O14+ 939.5649. 1H NMR (CDCl3, 400 MHz) δ: 7.34 (d, J = 8.0 Hz, 1 H, H-uracil), 6.87 (s, 1 H, -NH-uracil), 6.11 (s, 1 H, -OH), 5.71 (d, J = 8.0 Hz, 1 H, H-uracil), 5.47 (s, 1 H, -O-CONH-), 5.11 (d, J = 10.4 Hz, 1 H, H-3), 5.06 (dd, J = 10.8, 2.4 Hz, 1 H, H-13), 4.60 (s, 1 H, H-11), 4.41 (q, J = 22.4, 15.6 Hz, 2 H, -CH2-U), 4.12 (d, J = 7.2 Hz, 1 H, H-1′), 3.54 (s, 1 H, H-5), 3.43–3.03 (m, 6 H, H-5′, -CONH-CH2(CH2)6CH2-, H-2′), 2.90–2.81 (m, 2 H, H-10, H-2), 2.64–2.58 (m, 1 H, H-3′), 2.39–2.37 (m, 7 H, -N(CH3)2, H-9a), 2.24 (s, 3 H, N-CH3), 2.16-2.14 (m, 1 H, H-4), 2.05–1.99 (m, 1 H, H-9b), 1.89–1.82 (m, 2 H, H-8, H-14eq), 1.69–1.66 (m, 1 H, H-4′a), 1.62–1.48 (m, 6 H, H-14ax, H-7a, -CONHCH2CH2(CH2)4CH2CH2-), 1.42 (s, 3 H, 12-CH3), 1.29–1.25 (m, 10 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-), 1.22–1.17 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.08-1.06 (m, 6 H, 10-CH3, 4-CH3), 0.92–0.88 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.6, 166.6, 164.6, 156.5, 153.4, 151.5, 145.5, 103.6, 102.2, 87.9, 86.4, 84.9, 78.8, 77.3, 75.9, 73.1, 70.9, 69.2, 68.1, 65.6, 61.9, 50.8, 43.3, 42.0, 40.9, 40.0, 39.6, 36.0, 35.0, 29.9, 29.1, 29.0, 28.7, 26.4, 26.3, 26.0, 25.8, 21.7, 21.4, 21.2, 15.6, 13.5, 10.1, 9.4, 5.0.
3.1.18. General Procedures for the Synthesis of Target Compounds 29A–30A, 32A–34A
Compound 20–21 or 23–25 (1 eq), compound 8 (1.2 eq) and HOBT (1.1 eq) were dissolved in CH2Cl2, then the solution was cooled to 0 °C and DCC (1.2 eq) was added. The ice bath was removed after 1.5 h, and the stirring was continued for another 2.5 h at rt. The precipitated DCU was removed through filtration and washed with DCM. Then, the combined filtrate was washed with water and brine and concentrated. The resulting mixture was dissolved in methanol and stirred at 60 °C for 2 h. The methanol was concentrated, and the crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.5:0.05). The resulting compound was dissolved in EtOH/HCl (8:2 mL), and the solution was stirred for 30 min. Then, the pH was adjusted to 10 with NH3·H2O, and then CH2Cl2 was added for extraction. The organic phase was washed with water and brine once, and the crude product was purified through column chromatography to afford compounds 29A–30A and 32A–34A.
3-O-descladinosyl-3-O-(5-(2-(adenin-9-yl)acetamido)pentyl)carbamoyl azithromycin-11, 12-cyclic carbonate (29A)
Compound 20 (0.425 g, 0.57 mmol), compound 8 (0.267 g, 0.68 mmol), DCC (0.115 g, 0.68 mmol) and HOBT (0.085 g, 0.63 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.25:0.05) to afford compound 29A (95.8 mg, 0.11 mmol, 55.0%). Melting point: 127.4–130.4 °C. HRMS (ESI) (M + H)+ m/z 920.5424, calcd for C44H74N9O12+ 920.5451. 1H NMR (CDCl3, 400 MHz) δ: 8.34 (s, 1 H, H-adenine), 7.96 (s, 1 H, H-adenine), 6.16 (s, 1 H, -OH), 5.83 (s, 1 H, -NH2-), 5.45 (s, 1 H, -O-CONH-), 5.14 (d, J = 10.0 Hz, 1 H, H-3), 5.07–4.87 (m, 1 H, H-13, -CH2-A), 4.58 (s, 1 H, H-11), 4.15 (d, J = 7.2 Hz, 1 H, H-1′), 3.56 (s, 1 H, H-5), 3.45–3.06 (m, 6 H, H-5′, -CONH-CH2(CH2)3CH2-, H-2′), 2.88–2.81 (m, 2 H, H-10, H-2), 2.73–2.68 (m, 1 H, H-3′), 2.45–2.34 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.18–2.16 (m, 1 H, H-4), 2.04–1.99 (m, 1 H, H-9b), 1.90–1.83 (m, 2 H, H-8, H-14eq), 1.77–1.73 (m, 1 H, H-4′a), 1.61–1.48 (m, 6 H, H-14ax, H-7a, -CONHCH2CH2CH2CH2CH2-), 1.41 (s, 3 H, 12-CH3), 1.37–1.28 (m, 4 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2-), 1.25–1.18 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.07–1.04 (m, 6 H, 10-CH3, 4-CH3), 0.93–0.87 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.4, 156.6, 155.6, 153.1, 141.5, 119.4, 103.7, 86.3, 84.9, 78.9, 75.9, 73.0, 70.7, 69.0, 65.8, 61.8, 46.7, 43.4, 40.3, 39.6, 36.0, 29.5, 28.4, 26.1, 25.6, 23.6, 21.7, 21.4, 21.1, 15.6, 13.6, 10.1, 5.1.
3-O-descladinosyl-3-O-(6-(2-(adenin-9-yl)acetamido)hexyl)carbamoyl azithromycin-11, 12-cyclic carbonate (30A)
Compound 21 (0.250 g, 0.33 mmol), compound 8 (0.157 g, 0.40 mmol), DCC (0.067 g, 0.40 mmol) and HOBT (0.049 g, 0.36 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.3:0.05) to afford compound 30A (58.5 mg, 0.06 mmol, 54.6%). Melting point: 128.1–129.9 °C. HRMS (ESI) (M + H)+ m/z 934.5600, calcd for C45H76N9O12+ 934.5608. 1H NMR (CDCl3, 400 MHz) δ: 8.33 (s, 1 H, H-adenine), 7.95 (s, 1 H, H-adenine), 7.03 (t, J = 5.6 Hz, 1 H, -CONH-CH2-A), 6.13–6.07 (m, 3 H, -NH2-, -OH), 5.28 (s, 1 H, -O-CONH-), 5.15 (d, J = 10.0 Hz, 1 H, H-3), 5.06 (dd, J = 10.8, 2.0 Hz, 1 H, H-13), 4.90 (q, 2 H, -CH2-A), 4.60 (s, 1 H, H-11), 4.12 (d, J = 7.2 Hz, 1 H, H-1′), 3.56 (s, 1 H, H-5), 3.42–3.01 (m, 6 H, H-5′, -CONH-CH2(CH2)4CH2-, H-2′), 2.89–2.82 (m, 2 H, H-10, H-2), 2.49–2.43 (m, 1 H, H-3′), 2.38–2.30 (m, 7 H, -N(CH3)2, H-9a), 2.24 (s, 3 H, N-CH3), 2.18–2.16 (m, 1 H, H-4), 2.04–1.99 (m, 1 H, H-9b), 1.89–1.82 (m, 2 H, H-8, H-14eq), 1.68–1.55 (m, 3 H, H-4′a, H-14ax, H-7a), 1.46–1.42 (m, 7 H, -CONHCH2CH2(CH2)2CH2CH2-, 12-CH3), 1.27–1.18 (m, 13 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2-, 6-CH3, 2-CH3, 5′-CH3), 1.08–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.92-0.90 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.5, 166.2, 156.6, 155.7, 153.4, 153.1, 150.0, 141.3, 119.3, 103.8, 88.0, 86.3, 84.9, 78.9, 77.3, 75.9, 73.0, 70.8, 69.4, 68.0, 61.8, 47.0, 43.3, 42.1, 40.4, 39.2, 36.0, 34.9, 30.0, 28.9, 28.8, 26.1, 25.8, 25.7, 25.6, 21.7, 21.4, 21.2, 15.6, 10.1, 5.1.
3-O-descladinosyl-3-O-(8-(2-(adenin-9-yl)acetamido)octyl)carbamoyl azithromycin-11, 12-cyclic carbonate (32A)
Compound 23 (0.315 g, 0.40 mmol), compound 8 (0.188 g, 0.48 mmol), DCC (0.081 g, 0.48 mmol) and HOBT (0.059 g, 0.44 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.3:0.05) to afford compound 32A (97.8 mg, 0.10 mmol, 76.9%). Melting point: 125.7–127.2 °C. HRMS (ESI) (M + H)+ m/z 962.5928, calcd for C47H80N9O12+ 962.5921. 1H NMR (MeOD-d4, 400 MHz) δ: 8.19 (s, 1 H, H-adenine), 8.10 (s, 1 H, H-adenine), 5.11–5.04 (m, 2 H, H-3, H-13), 4.93 (q, 2 H, -CH2-A), 4.60 (s, 1 H, H-11), 4.20 (d, J = 7.2 Hz, 1 H, H-1′), 3.66–3.19 (m, 6 H, H-5′, -CONH-CH2(CH2)6CH2-, H-2′, H-5), 3.04–2.95 (m, 3 H, -CONHCH2-, H-10, H-2), 2.60–2.55 (m, 1 H, H-3′), 2.40–2.33 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.20–2.13 (m, 2 H, H-4, H-9b), 1.84–1.78 (m, 2 H, H-8, H-14eq), 1.72–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.52–1.50 (m, 4 H, -CONHCH2CH2(CH2)4CH2CH2-), 1.46 (s, 3 H, 12-CH3), 1.42–1.25 (m, 10 H, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-, H-7b, H-4′b), 1.25–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94–0.89 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 100 MHz) δ: 172.6, 164.7, 154.9, 153.5, 151.1, 150.1, 147.3, 139.8, 116.0, 99.1, 84.1, 82.7, 81.1, 76.0, 73.5, 71.1, 68.7, 66.3, 64.9, 62.1, 59.0, 42.8, 40.8, 38.6, 38.2, 37.2, 36.9, 33.5, 31.5, 28.5, 27.2, 26.6, 26.5, 24.1, 23.4, 22.5, 18.9, 17.9, 17.7, 12.3, 9.8, 6.8, 5.9.
3-O-descladinosyl-3-O-(10-(2-(adenin-9-yl)acetamido)decyl)carbamoyl azithromycin-11, 12-cyclic carbonate (33A)
Compound 24 (0.302 g, 0.37 mmol), compound 8 (0.175 g, 0.44 mmol), DCC (0.092 g, 0.44 mmol) and HOBT (0.055 g, 0.41 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.2:0.05) to afford compound 33A (100.8 mg, 0.10 mmol, 59.8%). Melting point: 127.5–128.9 °C. HRMS (ESI) (M + H)+ m/z 990.6198, calcd for C49H84N9O12+ 990.6234. 1H NMR (MeOD-d4, 400 MHz) δ: 8.19 (s, 1 H, H-adenine), 8.10 (s, 1 H, H-adenine), 5.10–5.04 (m, 2 H, H-3, H-13), 4.93 (s, 2 H, -CH2-A), 4.59 (s, 1 H, H-11), 4.20 (d, J = 7.2 Hz, 1 H, H-1′), 3.66 (s, 2 H, H-5), 3.44–3.40 (m, 1 H, H-5′), 3.27–3.19 (m, 4 H, H-2′, -CONH-CH2(CH2)8CH2-), 3.05–2.91 (m, 3 H, H-2, H-10, -CONH-CH2(CH2)9-), 2.62–2.56 (m, 1 H, H-3′), 2.40–2.33 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.20–2.12 (m, 2 H, H-4, H-9b), 1.84-1.78 (m, 2 H, H-8, H-14eq), 1.72–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.51–1.50 (m, 4 H, -CONHCH2CH2(CH2)6CH2CH2-), 1.46 (s, 3 H, 12-CH3), 1.37–1.31 (m, 14 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2CH2CH2-), 1.25–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94–0.89 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 176 MHz) δ: 174.9, 167.0, 157.3, 155.9, 153.5, 152.4, 149.6, 142.2, 118.3, 101.5, 86.5, 85.0, 83.4, 78.3, 75.9, 73.4, 71.0, 68.6, 67.2, 64.4, 61.4, 45.1, 43.1, 40.9, 40.5, 39.6, 39.3, 35.9, 33.8, 30.8, 29.6, 29.2, 29.0, 28.9,26.5, 25.8, 24.8, 21.3, 20.3, 20.1, 14.7, 12.1, 9.2, 8.2, 3.8.
3-O-descladinosyl-3-O-(12-(2-(adenin-9-yl)acetamido)dodecyl)carbamoyl azithromycin-11, 12-cyclic carbonate (34A)
Compound 25 (0.250 g, 0.30 mmol), compound 8 (0.141 g, 0.36 mmol), DCC (0.074 g, 0.36 mmol) and HOBT (0.045 g, 0.33 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O=10:0.2:0.05) to afford compound 34A (86.4 mg, 0.08 mmol, 56.7%). Melting point: 126.9–127.7 °C. HRMS (ESI) (M + H)+ m/z 1018.6531, calcd for C51H88N9O12+ 1018.6547. 1H NMR (MeOD-d4, 400 MHz) δ: 8.19 (s, 1 H, H-adenine), 8.10 (s, 1 H, H-adenine), 5.10–5.04 (m, 2 H, H-3, H-13), 4.93 (s, 2 H, -CH2-A), 4.59 (s, 1 H, H-11), 4.20 (d, J = 7.2 Hz, 1 H, H-1′), 3.66 (s, 2 H, H-5), 3.44–3.40 (m, 1 H, H-5′), 3.28–3.19 (m, 4 H, H-2′, -CONH-CH2(CH2)10CH2-), 3.05–2.91 (m, 3 H, H-2, H-10, -CONH-CH2(CH2)11-), 2.62–2.56 (m, 1 H, H-3′), 2.40–2.36 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.20–2.12 (m, 2 H, H-4, H-9b), 1.87–1.78 (m, 2 H, H-8, H-14eq), 1.72–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.53-1.50 (m, 4 H, -CONHCH2CH2(CH2)8CH2CH2-), 1.46 (s, 3 H, 12-CH3), 1.37–1.26 (m, 18 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2-), 1.20–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94-0.89 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 176 MHz) δ: 174.9, 167.0, 157.3, 155.9, 153.5, 152.4, 149.6, 142.2, 118.3, 101.5, 86.5, 85.0, 83.4, 78.3, 75.9, 73.4, 71.0, 68.6, 67.2, 64.4, 61.4, 45.1, 43.1, 40.9, 40.5, 39.6, 39.3, 35.9, 33.8, 30.8, 29.6, 29.3, 29.0, 28.9, 26.5, 25.7, 24.8, 21.3, 20.3, 20.1,14.7, 12.1, 9.2, 8.3, 3.8.
3.1.19. 3-O-descladinosyl-3-O-(8-(2-(adenin-9-yl)acetamido)octyl)carbamoyl azithromycin (35A)
Compound 32A (0.164 g, 0.17 mmol) was dissolved in THF/H2O (1:1), and LiOH·H2O (0.036 g, 0.85 mmol) was added to the solution. The mixture was stirred for 5 h at room temperature and then extracted with a mixture of water and CH2Cl2. The organic phase was concentrated and purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.3:0.05) to afford compound 35A (87.8 mg, 0.09 mmol, 55.2%). Melting point: 125.7–127.2 °C. HRMS (ESI) (M + H)+ m/z 936.6102, calcd for C46H82N9O11+ 936.6128. 1H NMR (MeOD-d4, 400 MHz) δ: 8.19 (s, 1 H, H-adenine), 8.11 (s, 1 H, H-adenine), 5.11–5.04 (m, 2 H, H-3, H-13), 4.93 (q, 2 H, -CH2-A), 4.60 (s, 1 H, H-11), 4.20 (d, J = 7.2 Hz, 1 H, H-1′), 3.66–3.19 (m, 6 H, H-5′, -CONH-CH2(CH2)6CH2-, H-5, H-2′), 3.04–2.95 (m, 3 H, H-10, H-2, -CONHCH2-), 2.60–2.55 (m, 1 H, H-3′), 2.40–2.33 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.20–2.13 (m, 2 H, H-4, H-9b), 1.84–1.78 (m, 2 H, H-8, H-14eq), 1.72–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.52–1.50 (m, 4 H, -CONHCH2CH2(CH2)4CH2CH2-), 1.46 (s, 3 H, 12-CH3), 1.42–1.25 (m, 10 H, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-, H-7b, H-4′b), 1.25–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94-0.89 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 176 MHz) δ: 175.7, 167.1, 157.4, 155.9, 152.4, 149.6, 142.2, 118.3, 101.3, 83.3, 78.7, 77.4, 75.8, 74.1, 73.4, 71.1, 69.7, 68.5, 64.4, 62.4, 43.1, 40.5, 39.6, 39.3, 35.6, 30.9, 29.6, 28.9, 28.8, 26.4, 26.1, 25.1, 20.5, 20.3, 20.1, 16.1, 14.6, 9.8, 8.2, 5.9.
3.1.20. General Procedures for the Synthesis of Target Compounds 29G–30G, 32G and 29C–30C
Compound 20–21 or 23 (1 eq), compound 12 or 14 (1.2 eq) and HOBT (1.1 eq) were dissolved in CH2Cl2, then the solution was cooled to 0 °C and DCC (1.2 eq) was added. The ice bath was removed after 1.5 h, and the stirring was continued for another 2.5 h at rt. The precipitated DCU was removed through filtration and washed with DCM. Then, the combined filtrate was washed with water and brine and then concentrated. The resulting mixture was dissolved in methanol and stirred under reflux for 2 h. The methanol was concentrated, and the crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.5:0.05). The resulting compound was dissolved in MeOH, then Pd/C (15% wt) was added under H2. After stirring for 24 h at room temperature, the Pd/C was filtered. The filtrate was concentrated and purified through column chromatography to afford compounds 29G–30G, 32G and 29C–30C.
3-O-descladinosyl-3-O-(5-(2-(guanin-9-yl)acetamido)pentyl)carbamoyl azithromycin -11, 12-cyclic carbonate (29G)
Compound 20 (0.365 g, 0.49 mmol), compound 12 (0.176 g, 0.59 mmol), DCC (0.100 g, 0.59 mmol) and HOBT (0.073 g, 0.54 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.3:0.05) to afford compound 29G (42.6 mg, 0.05 mmol, 31.3%). Melting point: 138.7–140.0 °C. HRMS (ESI) (M + H)+ m/z 936.5394, calcd for C44H74N9O13+ 936.5401. 1H NMR (DMSO-d6, 400 MHz) δ: 10.58 (s, 1 H, -NH-guanine), 8.16 (s, 1 H, -CONH-), 7.59 (s, 1 H, H-guanine),7.33(s, 1 H, -O-CONH-), 6.46 (s, 1 H, -NH2-), 5.30 (s, 1 H, -OH), 4.92–4.86 (m, 2 H, H-3, H-13), 4.61 (s, 2 H, -CH2-G), 4.43 (s, 1 H, H-11), 4.19 (d, J = 7.2 Hz, 1 H, H-1′), 3.40–3.02 (m, 7 H, H-5′, -CONH-CH2(CH2)3CH2-, H-5, H-2′), 2.92–2.82 (m, 2 H, H-10, H-2), 2.28–2.25 (m, 1 H, H-3′), 2.18–2.10 (m, 1 H, H-9a), 2.10 (s, 3 H, N-CH3), 2.06–2.04 (m, 1 H, H-4), 1.78–1.61 (m, 4 H, H-8, H-14eq, H-14ax, H-7a), 1.43–1.40 (m, 7 H, -CONHCH2CH2CH2CH2CH2-, 12-CH3), 1.38–1.24 (m, 5 H, H-7b, H-4′b, H-4′a, -CONHCH2CH2CH2CH2CH2-), 1.11–1.05 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.00–0.96 (m, 6 H, 10-CH3, 4-CH3), 0.86–0.82 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (DMSO-d6, 100 MHz) δ: 175.0, 166.7, 157.3, 156.8, 154.1, 153.3, 151.9, 138.8, 116.6, 101.1, 86.0, 85.7, 83.2, 79.7, 77.5, 76.0, 73.1, 68.0, 66.8, 65.2, 60.5, 45.2, 42.9, 40.6, 40.5, 39.1, 35.9, 34.8, 30.7, 29.5, 29.1, 26.3, 25.4, 24.0, 22.0, 21.5, 21.3, 15.5, 14.1, 10.2, 9.6, 5.8.
3-O-descladinosyl-3-O-(6-(2-(guanin-9-yl)acetamido)hexyl)carbamoyl azithromycin-11, 12-cyclic carbonate (30G)
Compound 21 (0.408 g, 0.54 mmol), compound 12 (0.194 g, 0.65 mmol), DCC (0.110 g, 0.65 mmol) and HOBT (0.080 g, 0.59 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.3:0.05) to afford compound 30G (28.5 mg, 0.03 mmol, 15.8%). Melting point: 132.8–134.5 °C. HRMS (ESI) (M + H)+ m/z 950.5568, calcd for C45H76N9O13+ 950.5557. 1H NMR (DMSO-d6, 400 MHz) δ: 10.52 (s, 1 H, -NH-guanine), 8.11 (s, 1 H, -CONH-), 7.59 (s, 1 H, H-guanine),7.28 (s, 1 H, -O-CONH-), 6.40 (s, 1 H, -NH2-), 5.32 (s, 1 H, -OH), 4.92–4.86 (m, 2 H, H-3, H-13), 4.60 (s, 2 H, -CH2-G), 4.43 (s, 1 H, H-11), 4.12 (d, J = 7.2 Hz, 1 H, H-1′), 3.41–3.02 (m, 7 H, H-5′, -CONH-CH2(CH2)4CH2-, H-5, H-2′), 2.92–2.82 (m, 2 H, H-10, H-2), 2.25–2.17 (m, 7 H, -N(CH3)2, H-9a), 2.10 (s, 3 H, N-CH3), 2.04–2.01 (m, 1 H, H-4), 1.78–1.57 (m, 4 H, H-8, H-14eq, H-14ax, H-7a), 1.43 (s, 3 H, 12-CH3), 1.34–1.26 (m, 10 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2-), 1.08–1.04 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 0.99–0.95 (m, 6 H, 10-CH3, 4-CH3), 0.86–0.82 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (DMSO-d6, 100 MHz) δ: 175.0, 167.5, 158.0, 157.3, 154.0, 153.5, 152.1, 139.0, 115.8, 100.9, 86.4, 85.0, 83.2, 78.2, 76.0, 73.4, 70.0, 68.3, 67.2, 65.1, 61.4, 45.0, 43.1, 40.9, 40.4, 39.0, 35.9, 33.8, 30.3, 29.4, 28.9, 26.0, 25.9, 25.7, 24.8, 21.3, 20.3, 19.9, 14.7, 12.1, 9.2, 8.2, 3.8.
3-O-descladinosyl-3-O-(8-(2-(guanin-9-yl)acetamido)octyl)carbamoyl azithromycin-11, 12-cyclic carbonate (32G)
Compound 23 (0.343 g, 0.44 mmol), compound 12 (0.261 g, 0.52 mmol), DCC (0.109 g, 0.52 mmol) and HOBT (0.065 g, 0.48 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.25:0.05) to afford compound 32G (62.7 mg, 0.06 mmol, 14.6%). Melting point: 131.6-133.0 °C. HRMS (ESI) (M + H)+ m/z 978.5842, calcd for C47H80N9O13+ 978.5870. 1H NMR (MeOD-d4, 400 MHz) δ: 7.70 (s, 1 H, H-guanine), 5.10–5.04 (m, 2 H, H-3, H-13), 4.74 (s, 2 H, -CH2-G), 4.43 (s, 1 H, H-11), 4.59 (d, J = 7.2 Hz, 1 H, H-1′), 3.66 (s, 1 H, H-5), 3.44–3.40 (m, 1 H, H-5′), 3.28–3.18 (m, 4 H, H-2′, -CONH-CH2(CH2)6CH2-), 3.06–2.91 (m, 3 H, H-10, H-2, -CONHCH2-), 2.67-2.60 (m, 1 H, H-3′), 2.40–2.36 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.21–2.12 (m, 2 H, H-4, H-9a), 1.84–1.78 (m, 2 H, H-8, H-14eq), 1.74-1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.51-1.49 (m, 4 H, -CONH-CH2CH2(CH2)4CH2CH2-), 1.47 (s, 3 H, 12-CH3), 1.32–1.24 (m, 10 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-), 1.20–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09-1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94–0.89 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 176 MHz) δ: 175.0, 167.4, 158.0, 157.3, 154.0, 153.5, 152.1, 139.0, 115.8, 101.4, 86.5, 85.0, 83.4, 78.3, 75.9, 73.4, 70.9, 68.6, 67.2, 64.5, 61.4, 45.0, 43.1, 40.9, 40.5, 39.5, 39.2, 35.9, 33.8, 30.8, 29.6, 28.9, 26.5, 25.8, 24.8, 21.3, 20.3, 20.0, 14.7, 12.1, 9.2, 8.2, 3.8.
3-O-descladinosyl-3-O-(5-(2-(cytosin-1-yl)acetamido)pentyl)carbamoyl azithromycin -11,12-cyclic carbonate (29C)
Compound 20 (0.406 g, 0.55 mmol), compound 14 (0.200 g, 0.66 mmol), DCC (0.112 g, 0.66 mmol) and HOBT (0.082 g, 0.61 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.25:0.05) to afford compound 29C (82.3 mg, 0.10 mmol, 51.2%). Melting point: 135.2-137.0 °C. HRMS (ESI) (M + H)+ m/z 896.5369, calcd for C43H74N7O13+ 896.5339. 1H NMR (CDCl3, 400 MHz) δ: 7.60 (s, 1 H, -CONH-), 7.37 (d, J = 7.2 Hz, 1 H, H-cytosine), 6.22 (s, 1 H, -OH), 5.84 (d, J = 7.2 Hz, 2 H, H-cytosine, -O-CONH-), 5.10 (d, J =10.4 Hz, 1 H, H-3), 5.04 (dd, J = 10.8, 2.0 Hz, 1 H, H-13), 4.59 (s, 1 H, H-11), 4.46–4.35 (m, 2 H, -CH2-cytosine), 4.14 (d, J = 7.2 Hz, 1 H, H-1′), 3.56–2.98 (m, 6 H, H-5′, -CONH-CH2(CH2)3CH2-, H-5, H-2′), 2.88–2.83 (m, 3 H, H-10, H-2, -CONH-CH2-), 2.51–2.50 (m, 1 H, H-3′), 2.38–2.29 (m, 7 H, H-9a, -N(CH3)2), 2.25 (s, 3 H, N-CH3), 2.12–2.00 (m, 2 H, H-4, H-9b), 1.89–1.81 (m, 2 H, H-14eq, H-7a), 1.68–1.49 (m, 7 H, H-4′a, -CO-NH-CH2CH2CH2CH2CH2-, H-8, H-14ax), 1.42 (s, 3 H, 12-CH3), 1.25–1.16 (m, 13 H, H-7b, H-4′b, -CO-NH-CH2CH2CH2CH2CH2-, 6-CH3, 5′-CH3, 2-CH3), 1.07–1.04 (m, 6 H, 10-CH3, 4-CH3), 0.92–0.87 (m, 6 H, 8-CH3, 15- CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.8, 167.9, 166.4, 155.6, 153.4, 146.4, 103.0, 95.2, 86.7, 86.3, 84.9, 78.5, 75.9, 73.2, 70.8, 69.1, 68.0, 65.6, 61.8, 53.0, 43.3, 41.9, 40.6, 40.4, 39.3, 36.0, 34.9, 29.5, 29.1, 28.6, 26.0, 25.8, 23.7, 21.7, 21.4, 21.2, 15.6, 13.5, 10.2, 9.4, 5.0.
3-O-descladinosyl-3-O-(6-(2-(cytosin-1-yl)acetamido)hexyl)carbamoyl azithromycin-11, 12-cyclic carbonate (30C)
Compound 21 (0.376 g, 0.50 mmol), compound 14 (0.182 g, 0.60 mmol), DCC (0.102 g, 0.60 mmol) and HOBT (0.074 g, 0.55 mmol) were dissolved in DCM according to the general procedure. The crude product was purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.25:0.05) to afford compound 30C (65.6 mg, 0.07 mmol, 46.1%). Melting point: 131.9-133.4 °C. HRMS (ESI) (M + H)3+ m/z 304.1881, calcd for C44H76N7O133+ 304.1880. 1H NMR (CDCl3, 400 MHz) δ: 7.40 (d, J = 7.2 Hz, 1 H, H-cytosine), 6.16 (s, 1 H, -OH), 5.87 (d, J = 6.8 Hz, 1 H, H-cytosine), 5.69 (s, 1 H, -O-CONH-), 5.11 (d, J =10.4 Hz, 1 H, H-3), 5.05 (dd, J = 10.8, 2.0 Hz, 1 H, H-13), 4.60 (s, 1 H, H-11), 4.49-4.38 (m, 2 H, -CH2-cytosine), 4.17 (d, J = 7.2 Hz, 1 H, H-1′), 3.56–3.02 (m, 6 H, H-5′, -CONH-CH2(CH2)4CH2-, H-5, H-2′), 2.88–2.83 (m, 3 H, H-10, H-2, -CONH-CH2-), 2.70 (m, 1 H, H-3′), 2.38–2.32 (m, 7 H, -N(CH3)2, H-9a), 2.25 (s, 3 H, N-CH3), 2.14–2.12 (m, 1 H, H-4), 2.06–2.00 (m, 1 H, H-9b), 1.89–1.87 (m, 2 H, H-8, H-14eq), 1.73–1.70 (m, 1 H, H-4′a), 1.61–1.58 (m, 2 H, H-14ax, H-7a), 1.52–1.48 (m, 4 H, -CONHCH2CH2(CH2)2CH2CH2-), 1.42 (s, 3 H, 12-CH3), 1.31–1.17 (m, 15 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2-, 6-CH3, 2-CH3, 5′-CH3), 1.08–1.05 (m, 6 H, 10-CH3, 4-CH3), 0.92–0.88 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (CDCl3, 100 MHz) δ: 174.8, 167.7, 166.2, 156.5, 153.4, 146.2, 103.2, 95.2, 87.1, 86.3, 84.9, 78.5, 75.9, 73.1, 70.8, 69.1, 68.0, 65.7, 61.8, 53.3, 43.3, 42.0, 40.5, 40.4, 40.3, 39.1, 36.0, 34.9, 29.8, 26.0, 25.9, 25.8, 21.7, 21.4, 21.2, 15.6, 13.5, 10.2, 9.4, 5.0.
3.1.21. General Procedures for the Synthesis of Intermediates 36U and 36A
Compound 23 (1 eq) was dissolved in CH2Cl2, then NaBH(OAc)3 (2 eq), ZnCl2 (1.2 eq) and compound 5 or 10 (2 eq) were added to the solution. After stirring for 2 h at room temperature, the mixture was washed with water and brine and then concentrated. The resulting mixture was dissolved in methanol and stirred at 60 °C for 2 h. The methanol was concentrated, and the crude product was purified through column chromatography (CH2Cl2/EtOH/NH3·H2O = 10:0.3:0.05) to afford intermediates 36U and 36A.
3-O-descladinosyl-3-O-(8-((2-(N6-(di-tert-butyloxycarbonyl)adenin-9-yl)ethyl)amino)octyl)carbamoylazithromycin-11,12-cyclic carbonate (36A)
Compound 36A (100 mg, 0.09 mmol, 36.0%) was prepared from compound 23 (0.200 g, 0.25 mmol), compound 10 (0.189 g, 0.50 mmol), NaBH(OAc)3 (0.108 g, 0.50 mmol) and ZnCl2 (0.034 g, 0.30 mmol) according to the general procedure. HRMS (ESI) (M + H)+ m/z 1148.7133, calcd for C57H98N9O15+ 1148.7177. 1H NMR (MeOD-d4, 400 MHz) δ: 8.84 (s, 1 H, H-adenine), 8.54 (s, 1 H, H-adenine), 5.10–5.04 (m, 2 H, H-3, H-13), 4.60 (s, 1 H, H-11), 4.48 (t, J = 6.4 Hz, 2 H, -CH2-A), 4.20 (d, J = 7.2 Hz, 1 H, H-1′), 3.66 (s, 1 H, H-5), 3.45–3.40 (m, 1 H, H-5′), 3.28–3.23 (m, 2 H, -CONH-CH2-, H-2′), 3.11 (t, J = 6.4 Hz, 2 H, -CH2-CH2-A),3.05–2.90 (m, 3 H, H-2, H-10, -CONH-CH2-), 2.60-2.57 (m, 3 H, -CH2-NH-CH2-, H-3′), 2.37-2.34 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.20–2.10 (m, 2 H, H-4, H-9b), 1.90–1.78 (m, 2 H, H-8, H-14eq), 1.69–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.51–1.49 (m, 2 H, -CONHCH2CH2-), 1.46 (s, 3 H, 12-CH3), 1.39 (m, 20 H, -CH2CH2-NH-, -(Boc)2),1.31–1.30 (m, 10 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-), 1.20–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09-1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94–0.89 (m, 6 H, 8-CH3, 15-CH3).
3.1.22. General Procedures for the Synthesis of Target Compounds 37U and 37A
Intermediate 36U or 36A was dissolved in EtOH/HCl (8:2 mL) and the solution was stirred for 30 min. Then, the pH was adjusted to 10 with NH3·H2O, and then CH2Cl2 was added for extraction. The organic phase was washed with water and brine once and then purified through column chromatography (CH2Cl2/MeOH/NH3·H2O = 10:0.7:0.05) to afford compounds 37U and 37A.
3-O-descladinosyl-3-O-(8-((2-(uracil-1-yl)ethyl)amino)octyl)carbamoyl azithromycin-11,12-cyclic carbonate (37U)
Compound 37U (22.0 mg, 0.02 mmol, 29.7%) was prepared from compound 36U (80 mg, 0.08 mmol) according to the general procedure. Melting point: 129.5-130.6 °C. HRMS (ESI) (M + H)+ m/z 925.5859, calcd for C46H81N6O13+ 925.5856. 1H NMR (MeOD-d4, 400 MHz) δ: 7.55 (d, J = 7.6 Hz, 1 H, H-uracil), 5.63 (d, J = 8.0 Hz, 1 H, H-uracil), 5.10–5.04 (m, 2 H, H-3, H-13), 4.60 (s, 1 H, H-11), 4.20 (d, J = 7.6 Hz, 1 H, H-1′), 3.85 (t, J = 6.4 Hz, 2 H, -CH2-U), 3.66 (s, 1 H, H-5), 3.44–3.39 (m, 1 H, H-5′), 3.27–3.18 (m, 2 H, -CONH-CH2-, H-2′), 3.06–2.91 (m, 3 H, -CONH-CH2-, H-10, H-2), 2.86 (t, J = 6.4 Hz, 2 H, -CH2-CH2-U), 2.60–2.57 (m, 3 H, -CH2-NH-CH2-, H-3′), 2.40–2.34 (m, 7 H, -N(CH3)2, H-9a), 2.24 (s, 3 H, N-CH3), 2.18–2.13 (m, 2 H, H-4, H-9b), 1.88–1.78 (m, 2 H, H-8, H-14eq), 1.72–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.52–1.47 (m, 7 H, 12-CH3, -CONHCH2CH2(CH2)4CH2CH2-), 1.42–1.23 (m, 10 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-), 1.20–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09–1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94–0.90 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 176 MHz) δ: 174.9, 165.5, 157.3, 153.5, 151.6, 146.1, 101.4, 100.7, 86.5, 85.0, 83.4, 78.3, 75.9, 73.4, 71.1, 68.6, 67.2, 64.4, 61.4, 49.1, 43.1, 40.9, 40.5, 39.6, 35.9, 33.8, 30.9, 29.6, 29.2, 29.1, 29.0, 26.9, 26.5, 25.7, 24.8, 21.3, 20.3, 20.0, 14.7, 12.1, 9.2, 8.2, 3.7.
3-O-descladinosyl-3-O-(8-((2-(adenin-9-yl)ethyl)amino)octyl)carbamoyl azithromycin -11, 12-cyclic carbonate (37A)
Compound 37A (30.8 mg, 0.03 mmol, 36.1%) was prepared from compound 36A (100 mg, 0.09 mmol) according to the general procedure. Melting point: 125.7-127.1 °C. HRMS (ESI) (M + H)+ m/z 948.6117, calcd for C47H82N9O11+ 948.6128. 1H NMR (MeOD-d4, 400 MHz) δ: 8.20 (s, 1 H, H-adenine), 8.12 (s, 1 H, H-adenine), 5.10–5.04 (m, 2 H, H-3, H-13), 4.59 (s, 1 H, H-11), 4.34 (t, J = 6.4 Hz, 2 H, -CH2-A), 4.20 (d, J = 7.2 Hz, 1 H, H-1′), 3.66 (s, 1 H, H-5), 3.45–3.40 (m, 1 H, H-5′), 3.26–3.23 (m, 2 H, -CONH-CH2-, H-2′), 3.06-3.03 (m, 5 H, -CH2-CH2-A, H-2, H-10, -CONH-CH2-), 2.60–2.56 (m, 3 H, -CH2-NH-CH2-, H-3′), 2.40–2.34 (m, 7 H, -N(CH3)2, H-9a), 2.23 (s, 3 H, N-CH3), 2.21–2.11 (m, 2 H, H-4, H-9b), 1.89–1.78 (m, 2 H, H-8, H-14eq), 1.71–1.63 (m, 3 H, H-4′a, H-14ax, H-7a), 1.49–1.43 (m, 7 H, -CONHCH2CH2(CH2)4CH2CH2-, 12-CH3), 1.29–1.25 (m, 10 H, H-7b, H-4′b, -CONHCH2CH2CH2CH2CH2CH2CH2CH2-), 1.20–1.16 (m, 9 H, 6-CH3, 2-CH3, 5′-CH3), 1.09-1.06 (m, 6 H, 10-CH3, 4-CH3), 0.94-0.89 (m, 6 H, 8-CH3, 15-CH3). 13C NMR (MeOD-d4, 176 MHz) δ: 174.9, 157.3, 155.9, 153.5, 152.3, 149.5, 141.6, 118.7, 101.4, 86.5, 85.0, 83.4, 78.3, 75.9, 73.4, 71.0, 68.6, 67.2, 64.4, 61.4, 48.9, 48.2, 43.2, 43.1, 40.9, 40.5, 39.6, 35.9, 33.8, 30.8, 29.6, 29.1, 29.0, 28.9, 26.8, 26.5, 24.8, 21.3, 20.3, 20.0, 14.7, 12.1, 9.2, 8.3, 3.8.
3.2. Antibacterial Assay
Minimum inhibitory concentrations (MICs) were determined using the microbroth dilution method, which was recommended by the Clinical and Laboratory Standards Institute. Standard ATCC strains and standardized antibiotics were used as quality controls in each experiment where stock solutions of the target compounds and clinical drugs were prepared to different concentrations of 256, 128, 64, 32, 16, 8, 4, 2, 1 and 0.25 μg/mL. The fresh bacterial strains purified through plate activation in advance were used for testing. The final concentration of the tested bacterial solution is 5 × 105 CFU/mL.
3.3. Docking Methods
The molecular docking studies of the ligands (37A and 32A) against target G2099A mutant 50S ribosomal subunit (PDB ID: 1YHQ) were performed utilizing Autodock VINA in YASARA (YASARA Biosciences GmbH, Vienna, VIE, AUT) [36,37]. To remove bumps and ascertain the covalent geometry of the ligands, the structures were all energy-minimized with the NOVA force field [39]. The dockings were undertaken by defining the simulation cell box of size 30 Å × 30 Å × 30 Å for the G2099A mutant 50S ribosomal subunit. Then, the docking studies on the aptamer were carried out through the built-in docking simulation macro ‘dock_run.mcr’ using an AMBER03 force field with 25 poses and 9 clusters for each situation [40].
3.4. Molecular Dynamics
To justify whether the models constructed from protein–ligand docking were reliable, molecular dynamics (MD) simulations of the bindings between G2099A mutant 50S ribosomal subunit with the ligands (37A and 32A) were performed. The AMBER14 force field was utilized for the MD simulation, as implemented in the YASARA program. For the treatment of long-range coulomb forces beyond an 8 Å cutoff, the MD simulation used periodic boundary conditions and the particle-mesh Ewald method. NaCl was used at a concentration of 0.9%, and the density of HOH was 0.997 g/mL in the MD cell. No restraints were applied during the MD simulation using the settings employed in the second equilibration dynamics. Energies and coordinates every 100 ps were saved with a total simulation length of 40–50 ns under a constant temperature (298 K) and uncontrolled pressure in an NVT ensemble. The structural stability of the protein–ligand complex was examined by analyzing the average values of the potential energy with root mean square deviation (RMSD) throughout the trajectory. The RMSD profiles of all MD structures (Figure S1) show that the variation in the RMSD values tends to be stable (<1 Å) after 40–50 ns, indicating that equilibrium was reached, and the last MD structure was chosen for the analysis.
4. Conclusions
In summary, macrolides conjugating with nucleobases exhibited considerably low activities against clinical isolates due to poor accumulation in the cell, which was confirmed in the presence of menadione (VK3) that could inhibit the efflux pumps and alter the fluidity of the bacterial membrane of S. aureus. Thus, we turned to exploit an engineered E. coli SQ110 in combination with its rRNA-mutated strains to investigate the SARs of the ribosome-targeting inhibitors. Notably, 34A was the most potent against A2059G-mutated strains (34A 16 μg/mL vs. telithromycin 128 μg/mL, azithromycin > 256 μg/mL and clindamycin > 128 μg/mL). Compounds 37A and 37U showed weak activities against the A2058G-mutated strain (128 μg/mL vs. telithromycin > 128 μg/mL, azithromycin > 256 μg/mL and clindamycin > 128 μg/mL). Molecular docking suggested additional hydrogen bonding formation between the adenine of the conjugates and the surrounding ribosomal nucleobases.
The conjugates had superior profiles over a family of macrolide-lincosamide-streptogramin B against A2058/2059-mutated strains, which suggested that a suitable extended alkyl–aryl arm derived from the C-3 of macrolactone, perhaps supplied extra opportunities to combat high-level A2058 mutated resistance. Further research on novel conjugates to address the inefficiency of this work against clinical isolates is underway.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28031327/s1, Figure S1: The RMSD profiles of MD simulations of the G2099A mutant 50S ribosomal subunit with the ligands (37A and 32A). Figure S2: Superposition of two complexes including the last structures obtained from the MD and the complexes obtained from docking. Table S1: The interaction of hydrogen bonds between G2099A Large Ribosomal Subunits (PDB ID: 1YHQ) and 37A. Table S2: The interaction of hydrogen bonds between G2099A mutant 50S ribosomal subunit (PDB ID: 1YHQ) and 32A. Table S3: The interaction of hydrogen bonds between G2099A mutant 50S ribosomal subunit (PDB ID: 1YHQ) and 37A after molecular dynamic simulation. Table S4: The interaction of hydrogen bonds between G2099A mutant 50S ribosomal subunit (PDB ID: 1YHQ) and 32A after molecular dynamic simulation. Table S5: HRMS and NMR spectra checklist for the intermediates and target compounds (1–16 and 26–37). 1H and 13C NMR Spectra for the representative compounds.
Author Contributions
Conceptualization and methodology, J.L., X.L. and M.Y.; formal analysis and investigation, X.L., W.L. and B.F.; writing—original draft preparation, J.L. and X.L.; writing—review and editing, J.L., W.L. and M.Y.; project administration and funding acquisition, J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work is financially supported by the National Natural Science Foundation of China (21878022; 81673335; 32201053), the National Key R&D Program of China (2021YFC2103100, 2021YFF0600700) and the Beijing Institute of Technology Research Fund Program for Young Scholars (3100012222222).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data necessary for the understanding of the research are present in the manuscript. Additional data related to the molecular docking data and NMR spectra are available as Supplementary Material.
Acknowledgments
The authors highly appreciate the valuable help from Alexander S. Mankin and Nora C. Vazquez-Laslop at the University of Illinois in Chicago. We thank Xu-dong Qu for the support of YASARA in molecular docking and molecular dynamic simulation experiments. The authors also appreciate the spectral data support from the Analysis & Testing Center, Beijing Institute of Technology. The image of the graphical abstract was created with tools obtained from BioRen-der.com.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Svetlov, M.S.; Syroegin, E.A.; Aleksandrova, E.V.; Atkinson, G.C.; Gregory, S.T.; Mankin, A.S.; Polikanov, Y.S. Structure of Erm-modified 70S ribosome reveals the mechanism of macrolide resistance. Nat. Chem. Biol. 2021, 17, 412–420. [Google Scholar] [CrossRef]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed]
- Llano-Sotelo, B.; Dunkle, J.; Klepacki, D.; Zhang, W.; Fernandes, P.; Cate, J.H.D.; Mankin, A.S. Binding and Action of CEM-101, a New Fluoroketolide Antibiotic That Inhibits Protein Synthesis. Antimicrob. Agents Chemother. 2010, 54, 4961–4970. [Google Scholar] [CrossRef]
- Liang, J.-H.; Han, X. Structure-Activity Relationships and Mechanism of Action of Macrolides Derived from Erythromycin as Antibacterial Agents. Curr. Top. Med. Chem. 2013, 13, 3131–3164. [Google Scholar] [CrossRef]
- Janas, A.; Przybylski, P. 14-and 15-membered lactone macrolides and their analogues and hybrids: Structure, molecular mechanism of action and biological activity. Eur. J. Med. Chem. 2019, 182, 111662. [Google Scholar] [CrossRef]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Carlsen, P.N.; et al. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [Google Scholar] [CrossRef]
- Magee, T.V.; Han, S.; McCurdy, S.P.; Nguyen, T.T.; Granskog, K.; Marr, E.S.; Maguire, B.A.; Huband, M.D.; Chen, J.M.; Subashi, T.A.; et al. Novel 3-O-carbamoyl erythromycin A derivatives (carbamolides) with activity against resistant staphylococcal and streptococcal isolates. Bioorg. Med. Chem. Lett. 2013, 23, 1727–1731. [Google Scholar] [CrossRef]
- Liang, J.H.; Lv, W.; Li, X.L.; An, K.; Cushman, M.; Wang, H.; Xu, Y.C. Synthesis and antibacterial activity of 9-oxime ether non-ketolides, and novel binding mode of alkylides with bacterial rRNA. Bioorg. Med. Chem. Lett. 2013, 23, 1387–1393. [Google Scholar] [CrossRef]
- Han, X.; Lv, W.; Guo, S.Y.; Cushman, M.; Liang, J.H. Synthesis and structure-activity relationships of novel 9-oxime acylides with improved bactericidal activity. Bioorg. Med. Chem. 2015, 23, 6437–6453. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Asaka, T.; Kashimura, M.; Misawa, Y.; Suzuki, K.; Sato, M.; Kameo, K.; Morimoto, S.; Nishida, A. Synthesis and antibacterial activity of acylides (3-O-acyl-erythromycin derivatives): A novel class of macrolide antibiotics. J. Med. Chem. 2001, 44, 4027–4030. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Lv, W.; Guo, S.Y.; Li, Y.X.; Fan, B.Z.; Cushman, M.; Kong, F.S.; Zhang, J.; Liang, J.H. Synthesis and structure-bactericidal activity relationships of non-ketolides: 9-Oxime clarithromycin 11,12-cyclic carbonate featured with three-to eight-atom-length spacers at 3-OH. Eur. J. Med. Chem. 2019, 171, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.F.; Liu, K.; Jin, S.G.; Huigens, R.W. Design, synthesis and biological evaluation of a halogenated phenazine-erythromycin conjugate prodrug for antibacterial applications. Org. Biomol. Chem. 2021, 19, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Tevyashova, A.N.; Bychkova, E.N.; Korolev, A.M.; Isakova, E.B.; Mirchink, E.P.; Osterman, I.A.; Erdei, R.; Szucs, Z.; Bata, G. Synthesis and evaluation of biological activity for dual-acting antibiotics on the basis of azithromycin and glycopeptides. Bioorg. Med. Chem. Lett. 2019, 29, 276–280. [Google Scholar] [CrossRef]
- Krajacic, M.B.; Novak, P.; Cindric, M.; Brajsa, K.; Dumic, M.; Kujundzic, N. Azithromycin-sulfonamide conjugates as inhibitors of resistant Streptococcus pyogenes strains. Eur. J. Med. Chem. 2007, 42, 138–145. [Google Scholar] [CrossRef]
- Paljetak, H.C.; Verbanac, D.; Padovan, J.; Dominis-Kramaric, M.; Kelneric, Z.; Peric, M.; Banjanac, M.; Ergovic, G.; Simon, N.; Broskey, J.; et al. Macrolones Are a Novel Class of Macrolide Antibiotics Active against Key Resistant Respiratory Pathogens In Vitro and In Vivo. Antimicrob. Agents Chemother. 2016, 60, 5337–5348. [Google Scholar] [CrossRef]
- Pavlovic, D.; Mutak, S. Discovery of 4″-Ether Linked Azithromycin-Quinolone Hybrid Series: Influence of the Central Linker on the Antibacterial Activity. ACS Med. Chem. Lett. 2011, 2, 331–336. [Google Scholar] [CrossRef]
- Fan, B.-Z.; Hiasa, H.; Lv, W.; Brody, S.; Yang, Z.-Y.; Aldrich, C.; Cushman, M.; Liang, J.-H. Design, synthesis and structure-activity relationships of novel 15-membered macrolides: Quinolone/quinoline-containing sidechains tethered to the C-6 position of azithromycin acylides. Eur. J. Med. Chem. 2020, 193, 112222. [Google Scholar] [CrossRef]
- Costa, A.M.; Vilarrasa, J. Hybrids of macrolides and nucleobases or nucleosides. Tetrahedron Lett. 2000, 41, 3371–3375. [Google Scholar] [CrossRef]
- Esteban, J.; Costa, A.M.; Cruzado, M.C.; Faja, M.; Garcia, P.; Vilarrasa, J. Clarithromycin-adenine and related conjugates. Tetrahedron Lett. 2006, 47, 1919–1922. [Google Scholar] [CrossRef]
- Vazquez-Laslop, N.; Klepacki, D.; Mulhearn, D.C.; Ramu, H.; Krasnykh, O.; Franzblau, S.; Mankin, A.S. Role of antibiotic ligand in nascent peptide-dependent ribosome stalling. Proc. Natl. Acad. Sci. USA 2011, 108, 10496–10501. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.S.; Lee, M.; Jin, X.; Teijaro, C.N.; Wheeler, S.E.; Jacobson, M.A.; Buttaro, B.; Andrade, R.B. Synthesis, Biological Evaluation, and Computational Analysis of Biaryl Side-Chain Analogs of Solithromycin. Chemmedchem 2021, 16, 3368–3373. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.P.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, K.L.; Egholm, M.; Behrens, C.; Christensen, L.; Hansen, H.F.; Vulpius, T.; Petersen, K.H.; Berg, R.H.; Nielsen, P.E.; Buchardt, O. Synthesis of peptide nucleic-acid monomers containing the 4 natural nucleobases—Thymine, cytosine, adenine, and guanine and their oligomerization. J. Org. Chem. 1994, 59, 5767–5773. [Google Scholar] [CrossRef]
- Farèse, A.; Patino, N.; Condom, R.; Dalleu, S.; Guedj, R. Liquid phase synthesis of a peptidic nucleic acid dimer. Tetrahedron Lett. 1996, 37, 1413–1416. [Google Scholar] [CrossRef]
- Yan, M.; Ma, R.; Jia, L.; Venter, H.; Ma, S. Synthesis and antibacterial activity of novel 3-O-descladinosylazithromycin derivatives. Eur. J. Med. Chem. 2017, 127, 874–884. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Abdelbaky, A.S.; Prokhorov, I.A.; Gnuskova, E.V.; Esipova, O.V.; Kirillova, Y.G. Convenient and Efficient Syntheses of Peptide Nucleic Acid Purine Monomers. Curr. Org. Chem. 2019, 23, 2122–2130. [Google Scholar] [CrossRef]
- Ashworth, I.W.; Cox, B.G.; Meyrick, B. Kinetics and Mechanism of N-Boc Cleavage: Evidence of a Second-Order Dependence upon Acid Concentration. J. Org. Chem. 2010, 75, 8117–8125. [Google Scholar] [CrossRef]
- Bashir-Uddin Surfraz, M.; Akhtar, M.; Allemann, R.K. Bis-benzyl protected 6-amino cyclitols are poisonous to Pd/C catalysed hydrogenolysis of benzyl ethers. Tetrahedron Lett. 2004, 45, 1223–1226. [Google Scholar] [CrossRef]
- Sridhar, P.R.; Anjaneyulu, B.; Rao, B.U. Regioselective Anomeric O-Benzyl Deprotection in Carbohydrates. Eur. J. Org. Chem. 2021, 2021, 5665–5668. [Google Scholar] [CrossRef]
- Liu, X.-P.; Lv, W.; Zhao, F.; Ding, J.; Zhang, J.-R.; Xue, F.; Zhang, J.-Z.; Liu, L.-Y.; Cushman, M.; Li, Y.; et al. Design and synthesis of novel macrolones bridged with linkers from 11,12-positions of macrolides. Bioorg. Med. Chem. Lett. 2022, 68, 128761. [Google Scholar] [CrossRef]
- Tintino, S.R.; Oliveira-Tintino, C.D.M.; Campina, F.F.; Weslley Limaverde, P.; Pereira, P.S.; Siqueira-Junior, J.P.; Coutinho, H.D.M.; Quintans-Júnior, L.J.; da Silva, T.G.; Leal-Balbino, T.C.; et al. Vitamin K enhances the effect of antibiotics inhibiting the efflux pumps of Staphylococcus aureus strains. Med. Chem. Res. 2018, 27, 261–267. [Google Scholar] [CrossRef]
- Tu, D.; Blaha, G.; Moore, P.B.; Steitz, T.A. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 2005, 121, 257–270. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W.; Pymol. 2020. Available online: http://www.pymol.org/pymol (accessed on 30 October 2022).
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).