

The Structural Basis of African Swine Fever Virus pS273R Protease Binding to E64 through Molecular Dynamics Simulations

Abstract
:1. Introduction
2. Results
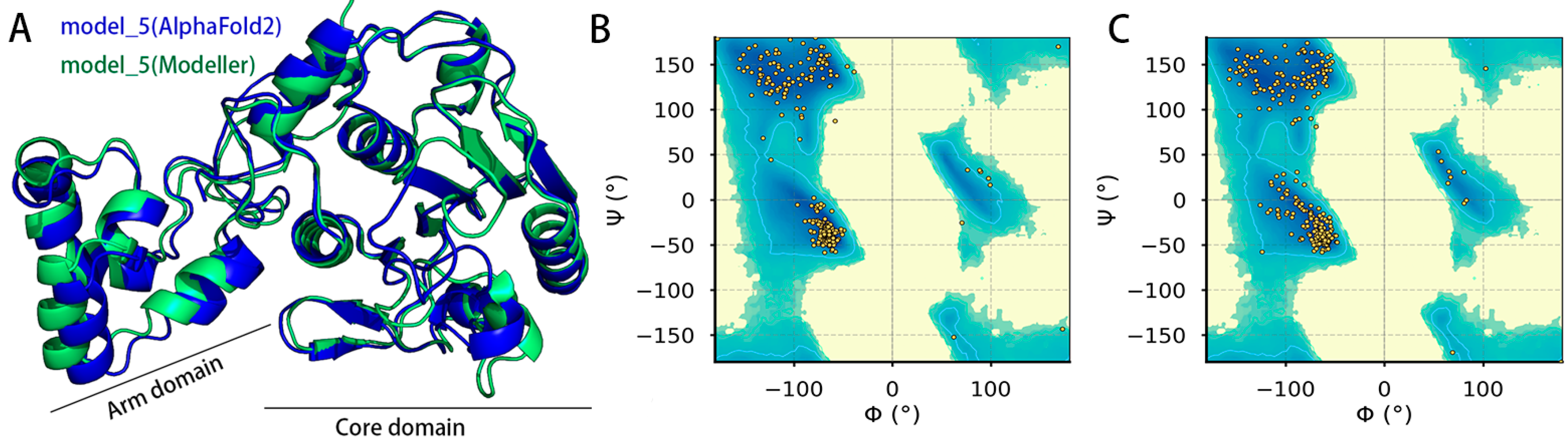
2.1. Model Evaluation of ASFV pS273R Modeling Structure
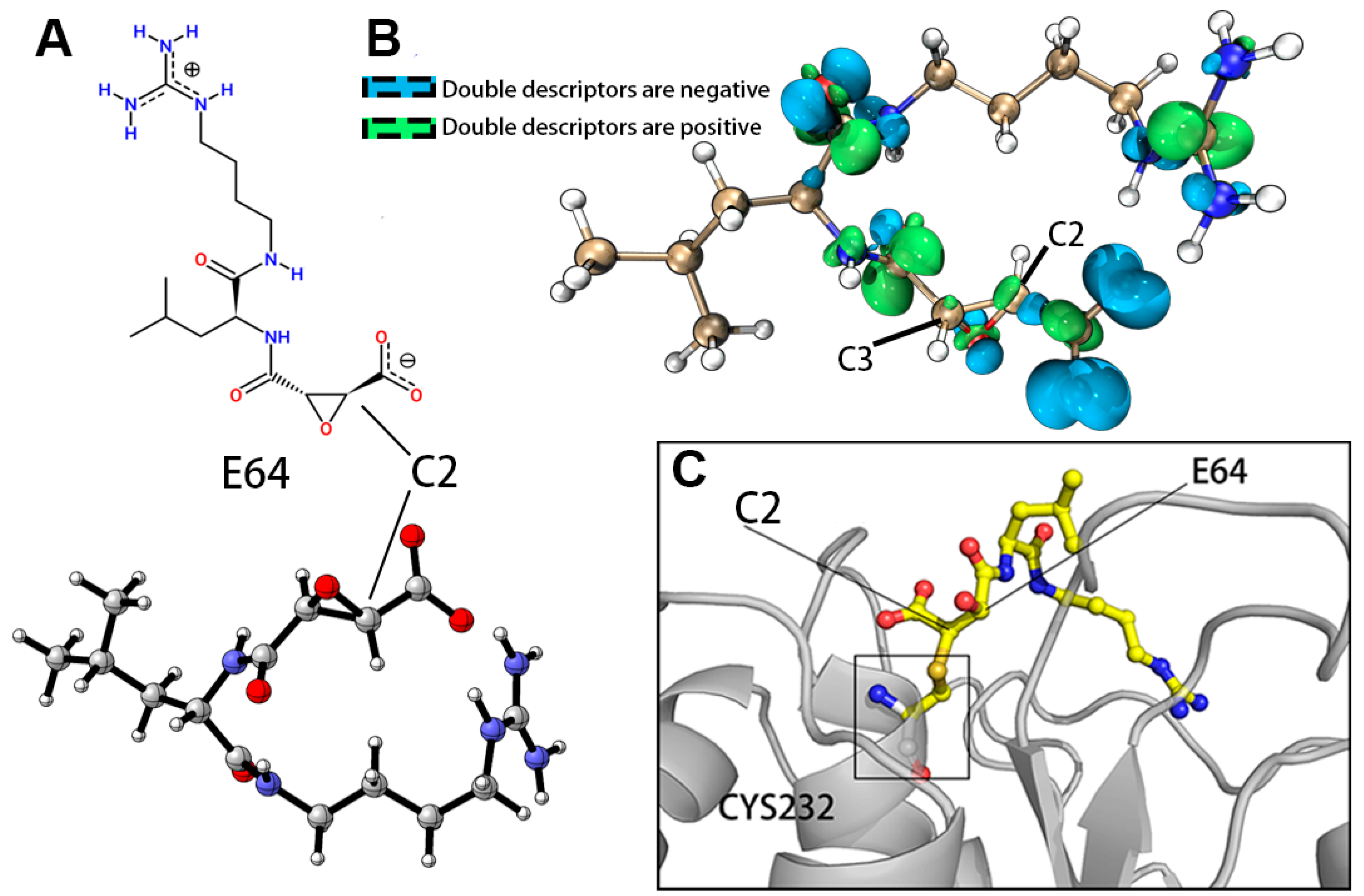
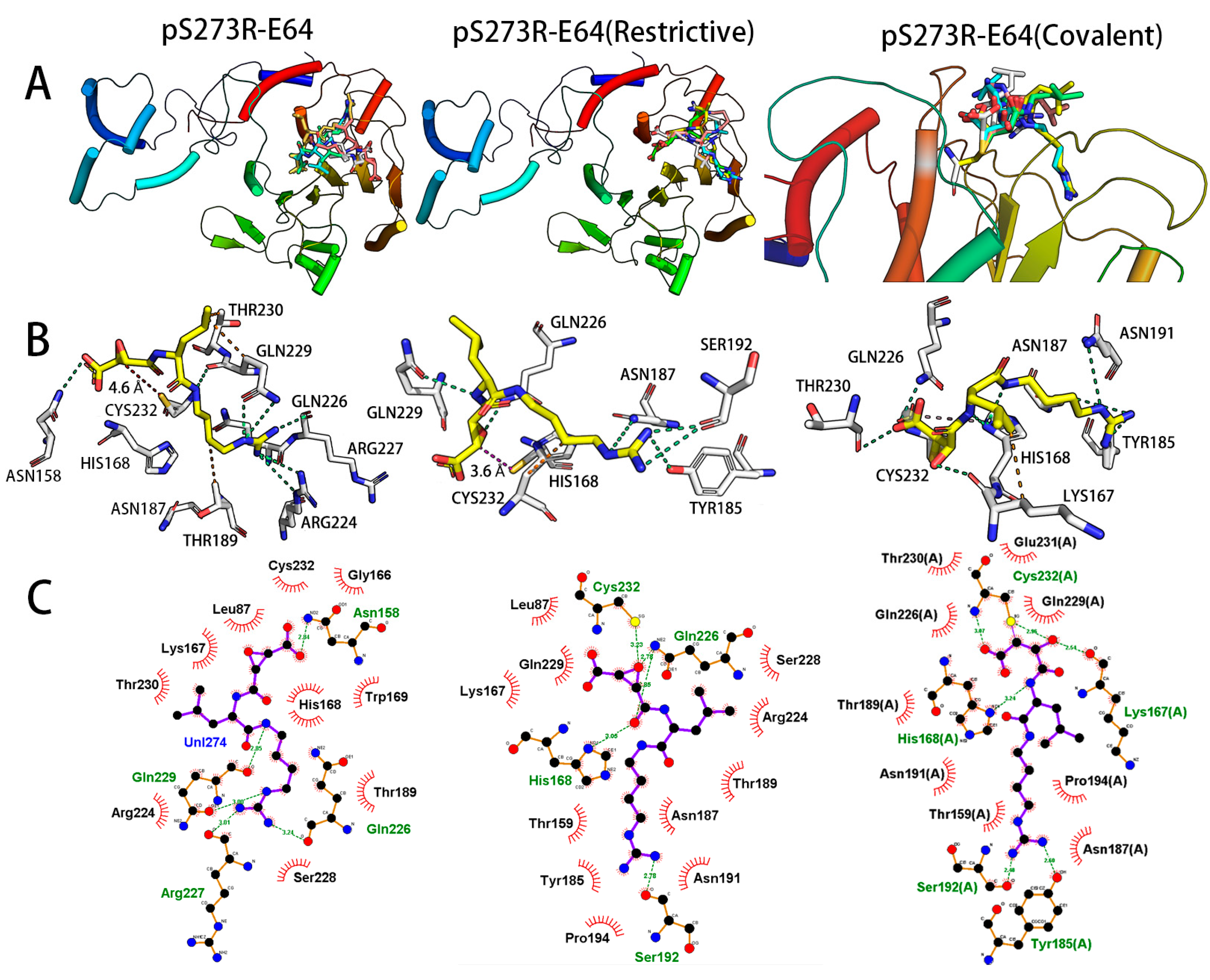
2.2. Molecular Docking Analysis of the ASFV pS273R and E64
2.3. Molecular Dynamics Simulation and Affinity Calculation of pS273R and E64 Complex
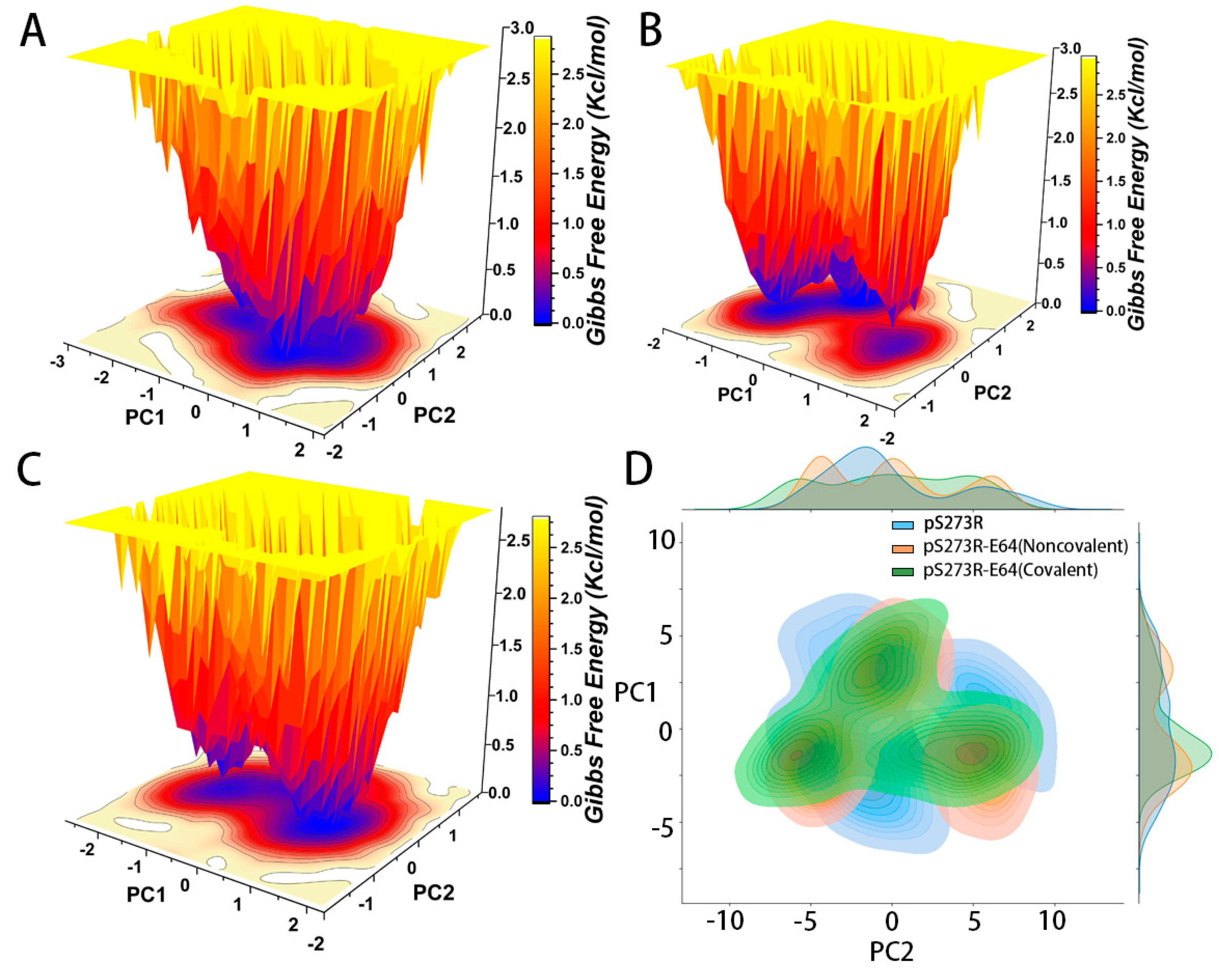
2.4. The Analysis of pS273R and pS273R−E64 Complexes in Different Bonding States Based on Gibbs Free Energy Landscape and Principal Component Analysis
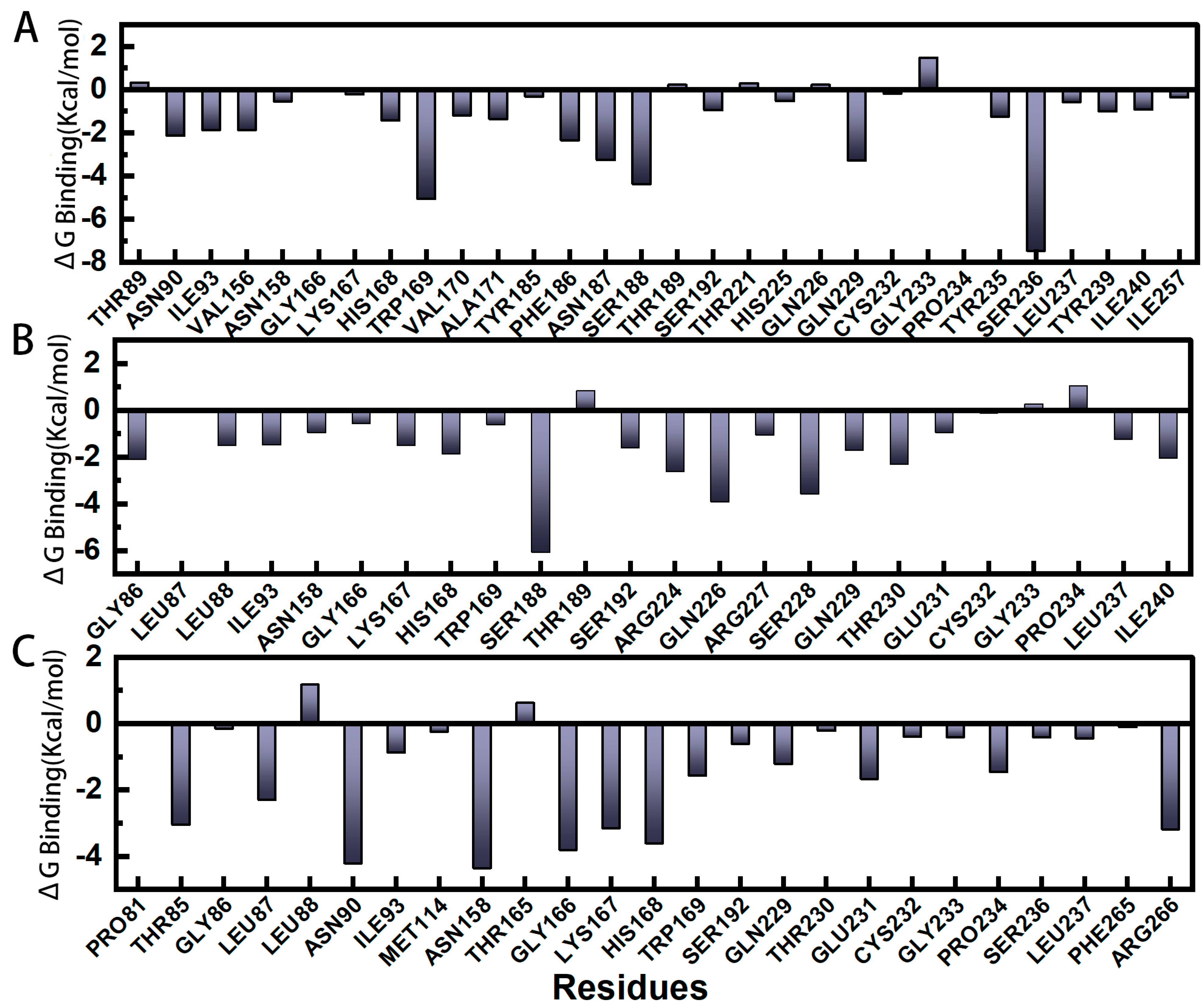
2.5. Gibbs Binding Free Energy Calculation and Decomposition of pS273R−E64 Complex Based on Molecular Mechanics Poisson–Boltzmann Surface Area
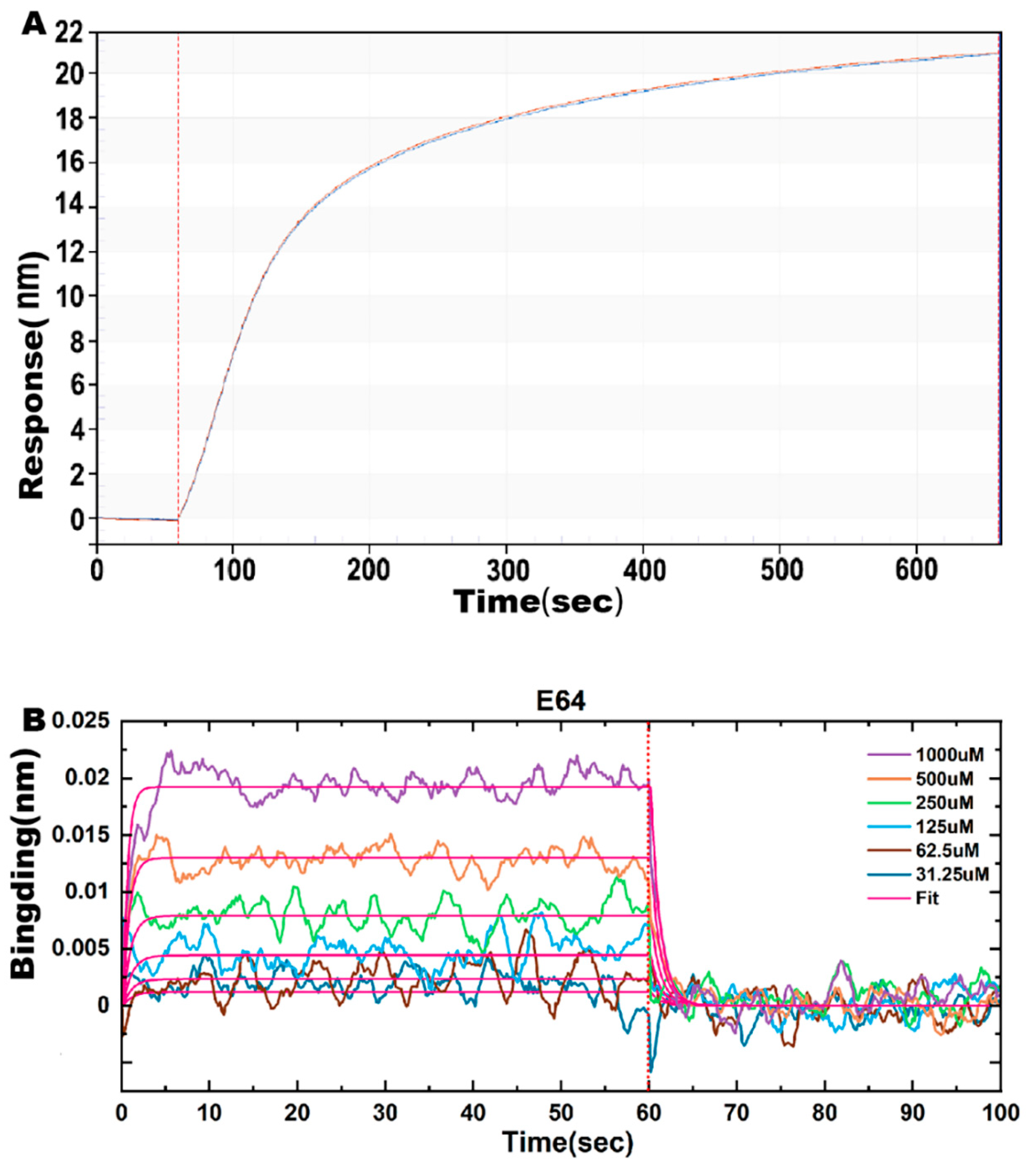
2.6. Effect of E64 on ASFV pS273R Proteinase Affinity
3. Discussion
4. Materials and Methods
4.1. Homology Modeling and Structural Evaluation
4.2. Molecular Optimization and Molecular Docking of E64
4.3. Molecular Dynamics Simulations
4.4. Affinity Assay of ASFV pS273R and E64
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Costard, S.; Mur, L.; Lubroth, J.; Sanchez-Vizcaino, J.M.; Pfeiffer, D.U. Epidemiology of African swine fever virus. Virus Res. 2013, 173, 191–197. [Google Scholar] [CrossRef]
- Gogin, A.; Gerasimov, V.; Malogolovkin, A.; Kolbasov, D. African swine fever in the North Caucasus region and the Russian Federation in years 2007–2012. Virus Res 2013, 173, 198–203. [Google Scholar] [CrossRef]
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef]
- Tao, D.; Sun, D.; Liu, Y.; Wei, S.; Yang, Z.; An, T.; Shan, F.; Chen, Z.; Liu, J. One year of African swine fever outbreak in China. Acta Trop. 2020, 211, 105602. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, H.; Yang, C.; Gao, Y.; Yu, X.; Chen, X.; Cui, R.; Zheng, L.; Li, S.; Li, X.; et al. Structure of the error-prone DNA ligase of African swine fever virus identifies critical active site residues. Nat. Commun. 2019, 10, 387. [Google Scholar] [CrossRef]
- Jori, F.; Vial, L.; Penrith, M.L.; Perez-Sanchez, R.; Etter, E.; Albina, E.; Michaud, V.; Roger, F. Review of the sylvatic cycle of African swine fever in sub-Saharan Africa and the Indian ocean. Virus Res. 2013, 173, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Vinuela, E. African swine fever virus. Curr. Top. Microbiol. Immunol. 1985, 116, 151–170. [Google Scholar] [PubMed]
- Arabyan, E.; Kotsynyan, A.; Hakobyan, A.; Zakaryan, H. Antiviral agents against African swine fever virus. Virus Res. 2019, 270, 197669. [Google Scholar] [CrossRef]
- Gallardo, C.; Blanco, E.; Rodríguez, J.M.; Carrascosa, A.L.; Sanchez-Vizcaino, J.M. Antigenic properties and diagnostic potential of African swine fever virus protein pp62 expressed in insect cells. J. Clin. Microbiol. 2006, 44, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Cabot, B.; Esteban, J.I.; Martell, M.; Genesca, J.; Vargas, V.; Esteban, R.; Guardia, J.; Gomez, J. Structure of replicating hepatitis C virus (HCV) quasispecies in the liver may not be reflected by analysis of circulating HCV virions. J. Virol. 1997, 71, 1732–1734. [Google Scholar] [CrossRef] [Green Version]
- Alejo, A.; Andrés, G.; Salas, M.L. African Swine Fever virus proteinase is essential for core maturation and infectivity. J. Virol. 2003, 77, 5571–5577. [Google Scholar] [CrossRef]
- Andrés, G.; Alejo, A.; Simón-Mateo, C.; Salas, M.L. African swine fever virus protease, a new viral member of the SUMO-1-specific protease family. J. Biol. Chem. 2001, 276, 780–787. [Google Scholar] [CrossRef]
- Andrés, G.; Alejo, A.; Salas, J.; Salas, M.L. African swine fever virus polyproteins pp220 and pp62 assemble into the core shell. J. Virol. 2002, 76, 12473–12482. [Google Scholar] [CrossRef]
- Li, G.; Liu, X.; Yang, M.; Zhang, G.; Wang, Z.; Guo, K.; Gao, Y.; Jiao, P.; Sun, J.; Chen, C.; et al. Crystal Structure of African Swine Fever Virus pS273R Protease and Implications for Inhibitor Design. J. Virol. 2020, 94, e02125-19. [Google Scholar] [CrossRef]
- Liu, B.; Cui, Y.; Lu, G.; Wei, S.; Yang, Z.; Du, F.; An, T.; Liu, J.; Shen, G.; Chen, Z. Small molecule inhibitor E-64 exhibiting the activity against African swine fever virus pS273R. Bioorganic Med. Chem. 2021, 35, 116055. [Google Scholar] [CrossRef]
- Wang, J.; Ji, M.; Yuan, B.; Luo, A.; Jiang, Z.; Zhu, T.; Liu, Y.; Kamau, P.M.; Jin, L.; Lai, R. Peptide OPTX-1 From Ornithodoros papillipes Tick Inhibits the pS273R Protease of African Swine Fever Virus. Front. Microbiol. 2021, 12, 778309. [Google Scholar] [CrossRef]
- Reverter, D.; Lima, C.D. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure 2004, 12, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Mizoue, K.; Kitamura, K.; Tse, W.C.; Huber, C.P.; Ishida, T. Structural basis of inhibition of cysteine proteases by E-64 and its derivatives. Biopolymers 1999, 51, 99–107. [Google Scholar] [CrossRef]
- Blass, G.; Levchenko, V.; Ilatovskaya, D.V.; Staruschenko, A. Chronic cathepsin inhibition by E-64 in Dahl salt-sensitive rats. Physiol. Rep. 2016, 4, e12950. [Google Scholar] [CrossRef]
- Barrett, A.J.; Kembhavi, A.A.; Brown, M.A.; Kirschke, H.; Knight, C.G.; Tamai, M.; Hanada, K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 1982, 201, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Yamamoto, D.; Matsumoto, K.; Inoue, M.; Ishida, T.; Mizuno, H.; Sumiya, S.; Kitamura, K. Crystal structure of papain-E64-c complex. Binding diversity of E64-c to papain S2 and S3 subsites. Biochem. J. 1992, 287 Pt 3, 797–803. [Google Scholar] [CrossRef]
- Gomes, M.T.; Teixeira, R.D.; Lopes, M.T.; Nagem, R.A.; Salas, C.E. X-ray crystal structure of CMS1MS2: A high proteolytic activity cysteine proteinase from Carica candamarcensis. Amino Acids 2012, 43, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Arafet, K.; Ferrer, S.; Marti, S.; Moliner, V. Quantum mechanics/molecular mechanics studies of the mechanism of falcipain-2 inhibition by the epoxysuccinate E64. Biochemistry 2014, 53, 3336–3346. [Google Scholar] [CrossRef]
- Mladenovic, M.; Ansorg, K.; Fink, R.F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic insights into the inhibition of cysteine proteases: First QM/MM calculations clarifying the stereoselectivity of epoxide-based inhibitors. J. Phys. Chem. B 2008, 112, 11798–11808. [Google Scholar] [CrossRef]
- Yongqing, T.; Wilmann, P.G.; Pan, J.; West, M.L.; Brown, T.J.; Mynott, T.; Pike, R.N.; Wijeyewickrema, L.C. Determination of the crystal structure and substrate specificity of ananain. Biochimie 2019, 166, 194–202. [Google Scholar] [CrossRef]
- Arafet Cruz, K. Computational Studies of the Mechanism of Catalysis and Inhibition of Cysteine Proteases. Ph.D. Thesis, Universitat Jaume I, Castello, Spain, 2017. [Google Scholar]
- da Silva, J.A.V.; Nepovimova, E.; Ramalho, T.C.; Kuca, K.; Celmar Costa França, T. Molecular modeling studies on the interactions of 7-methoxytacrine-4-pyridinealdoxime, 4-PA, 2-PAM, and obidoxime with VX-inhibited human acetylcholinesterase: A near attack conformation approach. J. Enzym. Inhib. Med. Chem. 2019, 34, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Lima, C.D. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 2000, 5, 865–876. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein–ligand binding free energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- Wang, K.K.; Po-Wai, Y. Calpain inhibition: An overview of its therapeutic potential. Trends Pharmacol. Sci. 1994, 15, 412–419. [Google Scholar] [CrossRef]
- Rasnick, D. Small synthetic inhibitors of cysteine proteases. Perspect. Drug Discov. Des. 1996, 6, 47–63. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Katane, M.; Osaka, N.; Matsuda, S.; Maeda, K.; Kawata, T.; Saitoh, Y.; Sekine, M.; Furuchi, T.; Doi, I.; Hirono, S. Identification of novel D-amino acid oxidase inhibitors by in silico screening and their functional characterization in vitro. J. Med. Chem. 2013, 56, 1894–1907. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Sim, V. D-amino acid-based peptide inhibitors as early or preventative therapy in Alzheimer disease. Prion 2014, 8, 119–124. [Google Scholar] [CrossRef]
- Nyamai, D.W.; Tastan Bishop, Ö. Identification of selective novel hits against plasmodium falciparum prolyl tRNA synthetase active site and a predicted allosteric site using in silico approaches. Int. J. Mol. Sci. 2020, 21, 3803. [Google Scholar] [CrossRef]
- Jonniya, N.A.; Sk, M.F.; Kar, P. Characterizing an allosteric inhibitor-induced inactive state in with-no-lysine kinase 1 using Gaussian accelerated molecular dynamics simulations. Phys. Chem. Chem. Phys. 2021, 23, 7343–7358. [Google Scholar] [CrossRef]
- Bronswijk-Deddens, L. Epitope Binning of Human Monoclonal Antibodies in Classical Sandwich and In-Tandem Orientation Using the Octet System Based on Biolayer Interferometry. Methods Mol. Biol. 2018, 1785, 207–220. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein structure modeling with MODELLER. In Structural Proteomics; Springer: Berlin/Heidelberg, Germany, 2008; pp. 145–159. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T. ERRAT: An empirical atom-based method for validating protein structures. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Guo, S.S.; Liu, J.; Zhou, X.G.; Zhang, G.J. DeepUMQA: Ultrafast Shape Recognition-based Protein Model Quality Assessment using Deep Learning. Bioinformatics 2022. online ahead of print. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Moellmann, J.; Grimme, S. DFT-D3 study of some molecular crystals. J. Phys. Chem. C 2014, 118, 7615–7621. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Houk, K. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; Rong, C.-Y.; Lu, T.; Liu, S.-B. Hirshfeld charge as a quantitative measure of electrophilicity and nucleophilicity: Nitrogen-containing systems. Acta Phys. Chim. Sin. 2014, 30, 2055–2062. [Google Scholar]
- Fuentealba, P.; Pérez, P.; Contreras, R. On the condensed Fukui function. J. Chem. Phys. 2000, 113, 2544–2551. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Gohlke, H.E. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Cryst. 2002, 40, 82–92. [Google Scholar]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA methods in virtual screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Quality Assessment Method | Model_5 (Modeller) | Model_5 (AlphaFold2) | |
|---|---|---|---|
| Ramachandran plot | Favored regions | 96.8% | 94.8% |
| Allowed regions | 3.2% | 5.2% | |
| SAVES v6.0 | ERRAT | 78.87 | 98.49 |
| VERIFY(3D-1D score) | 89.74% | 97.80% | |
| DeepUMQA v3.0 | Global lDDT | 73.81 | 85.87 |
| Global lDDT (Refined) | 82.21 | 82.43 | |
| Atom | Hirshfeld Charges (e) | Condensed Fukui Functions (e) | Condensed Local Electrophilicity/Nucleophilicity Index (e*eV) | |||||
|---|---|---|---|---|---|---|---|---|
| q (N) | q (N + 1) | q (N − 1) | f- | f+ | f0 | Electrophilicity | Nucleophilicity | |
| C2 | 0.0233 | 0.0042 | 0.0512 | 0.0279 | 0.0191 | 0.0235 | 0.01034 | 0.10878 |
| C3 | 0.0209 | 0.0035 | 0.0403 | 0.0194 | 0.0174 | 0.0184 | 0.00938 | 0.07557 |
| Pose Number | Docking Score (Kcal/mol) | ||
|---|---|---|---|
| Molecular Docking (Common) | Molecular Docking (Restrictive) | Molecular Docking (Covalent) | |
| 1 | −7.85 | −6.91 | −3.8 |
| 2 | −7.49 | −6.62 | −3.5 |
| 3 | −7.47 | −6.44 | −3.0 |
| 4 | −7.35 | −6.35 | −3.1 |
| 5 | −7.08 | −6.30 | −2.4 |
| RMSD (Å) | Rg (Å) | SASA (Å2) | H-bone | ||
|---|---|---|---|---|---|
| pS273R | Trajectory 1 | 19.1 ± 0.23 | 20.17 ± 0.011 | 13372.7 ± 183.7 | - |
| Trajectory 2 | 16.7 ± 0.21 | 20.09 ± 0.010 | 13648.2 ± 227.5 | - | |
| Trajectory 3 | 16.7 ± 0.26 | 20.11 ± 0.010 | 13716.4 ± 206.3 | - | |
| pS273R−E64 (Noncovalent) | Trajectory 1 | 14.7 ± 0.16 | 20.00 ± 0.011 | 13874.2 ± 252.7 | 3.209 ± 1.142 |
| Trajectory 2 | 17.6 ± 0.12 | 20.29 ± 0.008 | 13970.8 ± 188.9 | 4.662 ± 1.012 | |
| Trajectory 3 | 18.6 ± 0.25 | 20.30 ± 0.016 | 14122.4 ± 227.1 | 2.886 ± 1.284 | |
| pS273R−E64 (Covalent) | Trajectory 1 | 14.9 ± 0.18 | 20.02 ± 0.010 | 13598.5 ± 171.9 | 6.087 ± 1.468 |
| Trajectory 2 | 13.2 ± 0.19 | 20.05 ± 0.010 | 13614.6 ± 191.9 | 4.776 ± 1.240 | |
| Trajectory 3 | 14.0 ± 0.23 | 20.08 ± 0.012 | 13655.6 ± 189.2 | 2.692 ± 1.036 |
| MM/PBSA (Kcal/mol) | Trajectory 1 | Trajectory 2 | Trajectory 3 |
|---|---|---|---|
| ΔE vdw | −45.84 ± 3.17 | −46.25 ± 3.02 | −49.17 ± 2.81 |
| ΔE elec | −17.66 ± 3.64 | −35.54 ± 6.28 | −25.40 ± 4.24 |
| ΔG pol | 22.93 ± 2.92 | 46.15 ± 5.73 | 38.32 ± 2.90 |
| ΔG nonpol | −33.66 ± 0.77 | −33.87 ± 1.35 | −35.97 ± 0.91 |
| ΔEDISPER | 55.45 ± 0.88 | 56.55 ± 1.43 | 58.82 ± 0.92 |
| ΔGGAS | −63.50 ± 2.34 | −81.78 ± 6.97 | −74.57 ± 2.49 |
| ΔGSOLV | 44.72 ± 2.96 | 68.82 ± 5.65 | 61.17 ± 3.32 |
| -TΔS | 7.16 ± 0.06 | 6.29 ± 0.06 | 4.64 ± 0.05 |
| ΔG Binding | −11.62 ± 2.87 | −6.67 ± 3.87 | −8.76 ± 2.69 |
| Variant | KD (M) | Kon (1/Ms) | Kdis (1/s) |
|---|---|---|---|
| E64 | 9.027 × 10−4 | 1.014 × 103 | 9.150 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, G.; Ou, K.; Jing, Y.; Zhang, H.; Feng, S.; Yang, Z.; Shen, G.; Liu, J.; Wu, C.; Wei, S. The Structural Basis of African Swine Fever Virus pS273R Protease Binding to E64 through Molecular Dynamics Simulations. Molecules 2023, 28, 1435. https://doi.org/10.3390/molecules28031435
Lu G, Ou K, Jing Y, Zhang H, Feng S, Yang Z, Shen G, Liu J, Wu C, Wei S. The Structural Basis of African Swine Fever Virus pS273R Protease Binding to E64 through Molecular Dynamics Simulations. Molecules. 2023; 28(3):1435. https://doi.org/10.3390/molecules28031435
Chicago/Turabian StyleLu, Gen, Kang Ou, Yiwen Jing, Huan Zhang, Shouhua Feng, Zuofeng Yang, Guoshun Shen, Jinling Liu, Changde Wu, and Shu Wei. 2023. "The Structural Basis of African Swine Fever Virus pS273R Protease Binding to E64 through Molecular Dynamics Simulations" Molecules 28, no. 3: 1435. https://doi.org/10.3390/molecules28031435
APA StyleLu, G., Ou, K., Jing, Y., Zhang, H., Feng, S., Yang, Z., Shen, G., Liu, J., Wu, C., & Wei, S. (2023). The Structural Basis of African Swine Fever Virus pS273R Protease Binding to E64 through Molecular Dynamics Simulations. Molecules, 28(3), 1435. https://doi.org/10.3390/molecules28031435