

Synthesis: Molecular Structure, Thermal-Calorimetric and Computational Analyses, of Three New Amine Borane Adducts

 , ,
, ,  ,
,  and
and 
Abstract
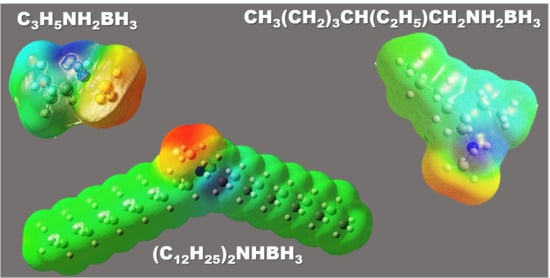
:
1. Introduction
2. Results and Discussion
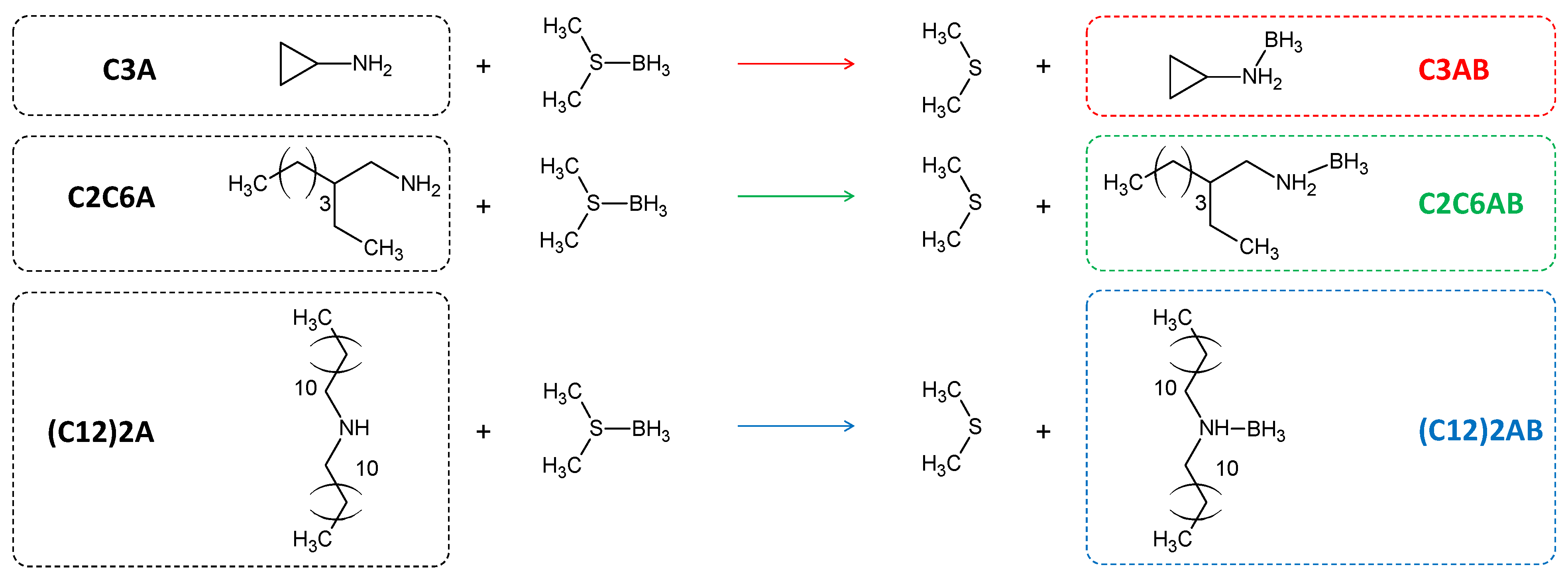
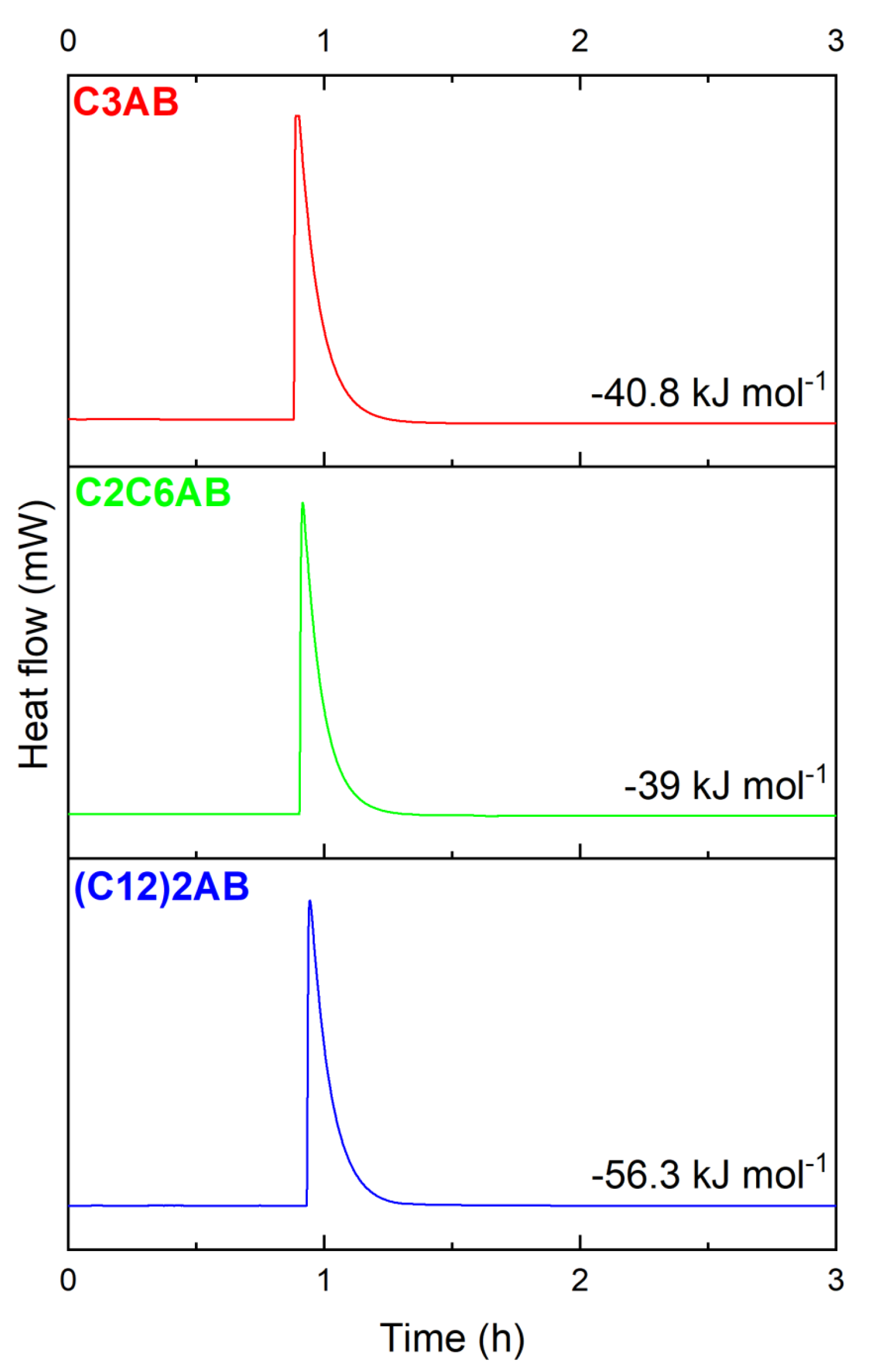
2.1. Syntheses of the ABAs Driven by Calvet Calorimetry
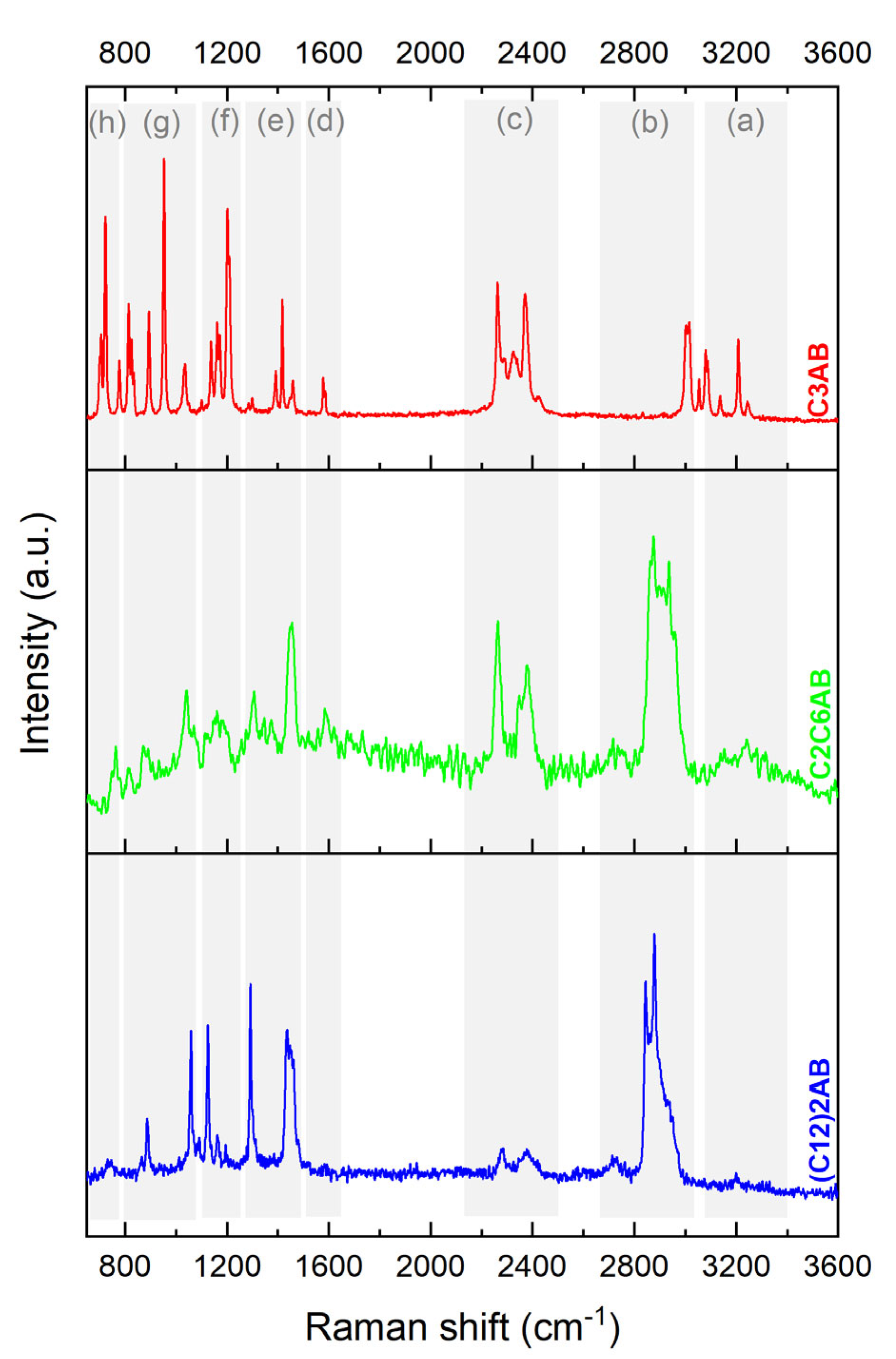
2.2. Molecular Characterization of C3AB
2.3. Molecular Characterization of C2C6AB
2.4. Molecular Characterization of (C12)2AB
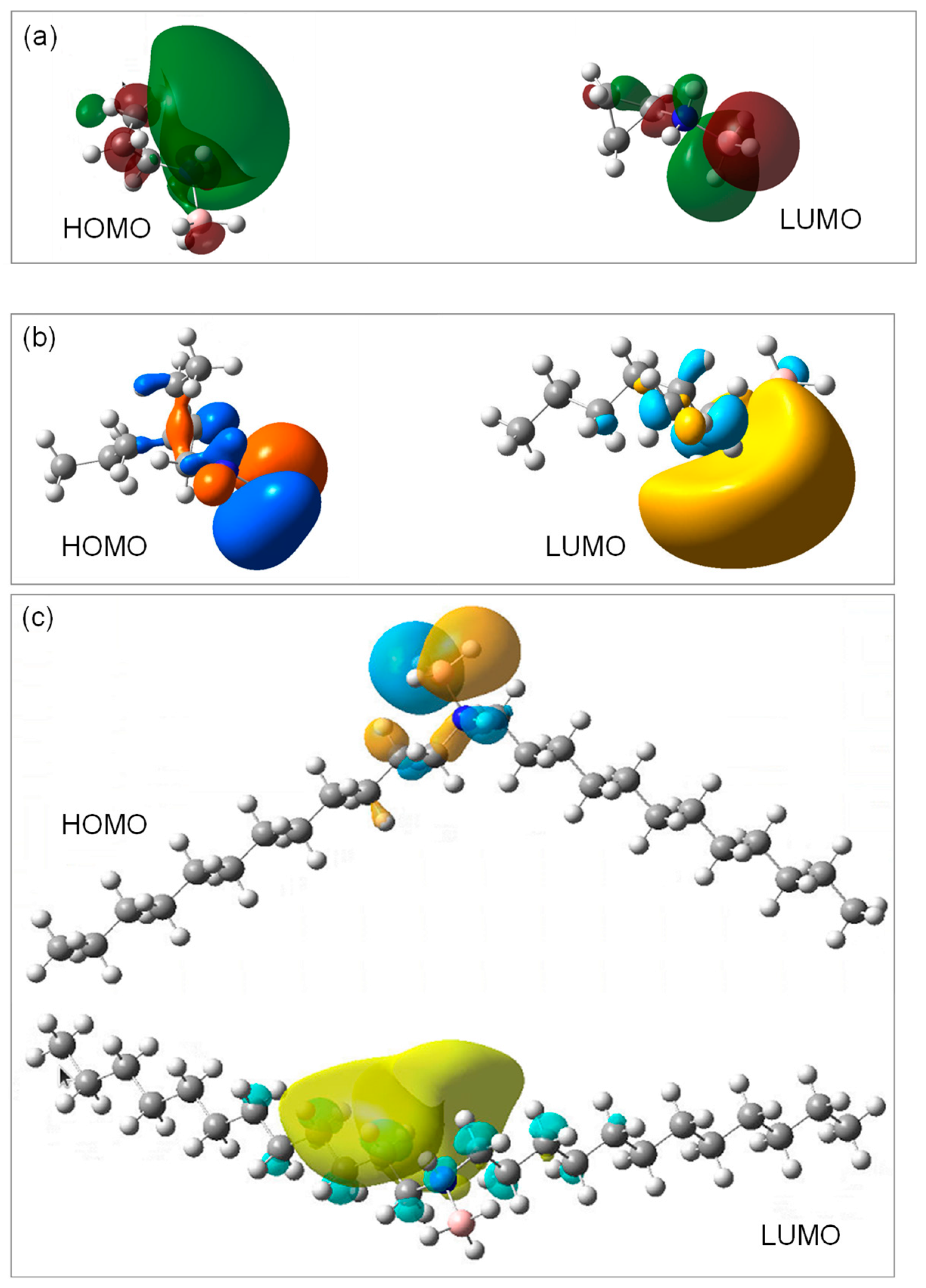
2.5. Further Molecular Analyses by DFT Calculations
2.6. Thermal Properties of the ABAs
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Demirci, U.B. Ammonia borane, a material with exceptional properties for chemical hydrogen storage. Int. J. Hydrogen Energy 2017, 42, 9978–10013. [Google Scholar] [CrossRef]
- Mitoraj, M.P. Bonding in ammonia borane: An analysis based on the natural orbitals for chemical valence and the extended transition state method (ETS-NOCV). J. Phys. Chem. A 2011, 115, 14708–17716. [Google Scholar] [CrossRef] [PubMed]
- Bartell, L.S. On the effects of intramolecular van der Waals forces. J. Chem. Phys. 1960, 32, 827–831. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.P.M.; Sloan, M.E.; Manners, I. Amine- and phosphine-borane adducts: New interest in old molecules. Chem. Rev. 2010, 110, 4023–4078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Morrison, D.; Bao, G.; Yu, H.; Yoon, C.W.; Song, T.; Lee, J.; Ung, A.T.; Huang, Z. An amine-borane system featuring room-temperature dehydrogenation and regeneration. Angew. Chem. Int. Ed. 2021, 60, 11725–11729. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Hamann, H.J.; Mishra, S. Aminoboranes via tandem iodination/dehydroiodination for one-pot borylation. ACS Omega 2022, 7, 14377–14389. [Google Scholar] [CrossRef]
- Liautard, V.; Delgado, M.; Colin, B.; Chabaud, L.; Michaud, G.; Pucheault, M. In situ generation of radical initiators using amine-borane complexes for carbohalogenation of alkenes. Chem. Commun. 2022, 58, 2124–2127. [Google Scholar] [CrossRef]
- Gurram, S.; Srivastava, G.; Badve, V.; Nandre, V.; Gundu, S.; Doshi, P. Pyridine borane as alternative reducing agent to sodium cyanoborohydride for PEGylation of L-asparaginase. Appl. Biochem. Biotechnol. 2022, 194, 827–847. [Google Scholar] [CrossRef]
- Guo, X.; Unglaube, F.; Kragl, U.; Mejia, E. B(C6F5)3-Catalyzed transfer hydrogenation of esters and organic carbonates towards alcohols with ammonia borane. Chem. Commun. 2022, 58, 6144–6147. [Google Scholar] [CrossRef]
- Jackson, K.T.; Reich, T.E.; El-Kaderi, H.M. Targeted synthesis of a porous borazine-linked covalent organic framework. Chem. Commun. 2012, 48, 8823–8825. [Google Scholar] [CrossRef]
- Leardini, F.; Flores, E.; Galvis, A.R.; Ferrer, I.J.; Ares, J.R.; Sánchez, C.; Molina, P.; van der Meulen, H.P.; Navarro, C.G.; López Polin, G.; et al. Chemical vapor deposition growth of boron–carbon–nitrogen layers from methylamine borane thermolysis products. Nanotechnology 2018, 29, 025603. [Google Scholar] [CrossRef]
- Hutchins, R.O.; Learn, K.; Nazer, B.; Pytlewski, D.; Pelter, A. Amine boranes as selective reducing and hydroborating agents. A review. Org. Prep. Proced. Int. 1984, 16, 335–372. [Google Scholar] [CrossRef]
- Burnham, B.S. Synthesis and pharmacological activities of amine-boranes. Curr. Med. Chem. 2005, 12, 1995–2010. [Google Scholar] [CrossRef]
- Kalidindi, S.B.; Sanyal, U.; Jagirdar, B.R. Chemical synthesis of metal nanoparticles using amine-boranes. Chem. Sus. Chem. 2011, 4, 317–324. [Google Scholar] [CrossRef]
- Rossin, A.; Peruzzini, M. Ammonia-borane and amine-borane dehydrogenation mediated by complex metal hydrides. Chem. Rev. 2016, 116, 8848–8872. [Google Scholar] [CrossRef]
- Colebatch, A.L.; Weller, A.S. Amine-borane dehydropolymerization: Challenges and opportunities. Chem. Eur. J. 2019, 25, 1379–1390. [Google Scholar] [CrossRef]
- Han, D.; Anke, F.; Trose, T.; Beweries, T. Recent advances in transition metal catalysed dehydropolymerisation of amine boranes and phosphine boranes. Coord. Chem. Rev. 2019, 308, 260–286. [Google Scholar] [CrossRef]
- Faverio, C.; Boselli, M.F.; Medici, F.; Benaglia, M. Ammonia borane as a reducing agent in organic synthesis. Org. Biomol. Chem. 2020, 18, 7789–7813. [Google Scholar] [CrossRef]
- Reddy, D.O. A short chronological review on the syntheses of amine-boranes. Chem. Rev. Lett. 2020, 3, 184–191. [Google Scholar]
- Lau, S.; Gasperini, D.; Webster, R.L. Amine-boranes as transfer hydrogenation and hydrogenation reagents: A mechanistic perspective. Angew. Chem. Int. Ed. 2021, 60, 14272–14294. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, J.C.; Shore, S.G. The roles of dihydrogen bonds in amine borane chemistry. Acc. Chem. Res. 2013, 46, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Klooster, W.T.; Koetzle, T.F.; Siegbahn, P.E.M.; Richardson, T.B.; Crabtree, R.H. Study of the N-H···H-B dihydrogen bond including the crystal structure of BH3NH3 by neutron diffraction. J. Am. Chem. Soc. 1999, 121, 6337–6343. [Google Scholar] [CrossRef]
- Morrison, C.A.; Siddick, M.M. Dihydrogen bonds in solid BH3NH3. Ang. Chem. Int. Ed. 2004, 43, 4780–4782. [Google Scholar] [CrossRef]
- Al-Kukhun, A.; Hwang, H.T.; Varma, A. Mechanistic studies of ammonia borane dehydrogenation. Int. J. Hydrogen Energy 2013, 38, 169–179. [Google Scholar] [CrossRef]
- Tao, J.; Lv, N.; Wen, L.; Qi, Y.; Lv, X. Hydrogen-release mechanisms in LiNH2BH3·NH3BH3: A theoretical study. J. Mol. Struct. 2012, 1081, 437–442. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, J.; Hamilton, E.J.M.; Chen, X. The continuing story of the diammoniate of diborane. J. Organomet. Chem. 2015, 798, 24–29. [Google Scholar] [CrossRef]
- Shore, S.G.; Parry, R.W. Chemical evidence for the structure of the “diammoniate of diborane”. II. The preparation of ammonia-borane. J. Am. Chem. Soc. 1958, 80, 8–12. [Google Scholar] [CrossRef]
- Merten, C.; Berger, C.J.; McDonald, R.; Xu, Y. Evidence of dihydrogen bonding of a chiral amine-borane complex in solution by VCD spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 9940–9943. [Google Scholar] [CrossRef]
- Theorodatou, A.; Turani-I-Belloto, K.; Petit, E.; Dourdain, S.; Alauzun, J.G.; Demirci, U.B. Synthesis of n-dodecylamine borane C12H25NH2BH3, its stability against hydrolysis, and its characterization in THF. J. Mol. Struct. 2022, 1248, 131484. [Google Scholar] [CrossRef]
- Staubitz, A.; Sloan, M.E.; Robertson, A.P.M.; Friedrich, A.; Schneider, S.; Gates, P.J.; Schmedt auf der Grüne, J.; Manners, I. Catalytic dehydrocoupling/dehydrogenation of N-methylamine-borane and ammonia-borane: Synthesis and characterization of high molecular weight polyaminoboranes. J. Am. Chem. Soc. 2010, 132, 13332–13345. [Google Scholar] [CrossRef]
- Turani-I-Belloto, K.; Valero-Pedraza, M.J.; Chiriac, R.; Toche, F.; Granier, D.; Cot, D.; Petit, E.; Yot, P.G.; Alauzun, J.G.; Demirci, U.B. A series of primary alkylamine borane adducts CxH2x+1NH2BH3: Synthesis and properties. ChemistrySelect 2021, 6, 9853–9860. [Google Scholar] [CrossRef]
- Turani-I-Belloto, K.; Valero-Pedraza, M.J.; Petit, E.; Chiriac, R.; Toche, F.; Granier, D.; Yot, P.G.; Alauzun, J.G.; Demirci, U.B. Solid-state structures of primary long-chain alkylamine borane adducts—Synthesis, properties and computational analysis. ChemistrySelect 2022, 7, e202203533. [Google Scholar] [CrossRef]
- Brown, R.J.C.; Brown, R.F.C. Melting point and molecular symmetry. J. Chem. Educ. 2000, 77, 724–731. [Google Scholar] [CrossRef]
- Smith, J.; Seshadri, K.S.; White, D. Infrared spectra of matric isolated BH3NH3, BD3ND3, and BH3ND3. J. Mol. Spectr. 1973, 45, 327–337. [Google Scholar] [CrossRef]
- Wolff, H.; Gamer, G. Hydrogen bonding and complex formation of dimethylamine. Infrared investigations on the NH stretching vibration bands. J. Phys. Chem. 1972, 76, 871–876. [Google Scholar] [CrossRef]
- Vijay, A.; Sathyanarayana, D.N. Theoretical investigation of equilibrium structure, harmonic force field and vibrational spectra of borane diammine: Effects of basis set and electron correlation. J. Mol. Struct. 1996, 375, 127–141. [Google Scholar]
- Krueger, P.J.; Jan, J. Infrared spectra and the molecular conformations of some aliphatic amines. Can. J. Chem. 1970, 48, 3229–3235. [Google Scholar] [CrossRef]
- Durig, J.R.; Lindsay, N.E.; Hizer, T.J.; Odom, J.D. Infrared and raman spectra, conformational stability and normal coordinate analysis of ethyldimethylamine-borane. J. Mol. Struct. 1988, 189, 257–277. [Google Scholar] [CrossRef]
- Eaton, G.R. NMR of boron compounds. J. Chem Educ. 1969, 46, 547–556. [Google Scholar] [CrossRef]
- Flores-Parra, A.; Guadarrama-Pérez, C.; Galvez Ruiz, J.C.; Sanchez Ruiz, S.A.; Suarez-Moreno, G.V.; Contreras, R. Mono- and di-alkyl-[1,3,5]-dithiazinanes and their N–borane adducts revisited. Structural and theoretical study. J. Mol. Struct. 2013, 1047, 149–159. [Google Scholar] [CrossRef]
- Kobayashi, T.; Gupta, S.; Caporini, M.A.; Pecharsky, V.K.; Pruski, M. Mechanism of solid-state thermolysis of ammonia borane: A 15N NMR study using fast magic-angle spinning and dynamic nuclear polarization. J. Phys. Chem. C 2014, 118, 19548–19555. [Google Scholar] [CrossRef]
- Sinton, S.W. Complexation chemistry of sodium borate with poly(vinyl alcohol) and small diols. A 11B NMR study. Macromolecules 1987, 20, 2430–2441. [Google Scholar] [CrossRef]
- Roy, B.; Pal, U.; Bishnoi, A.; O’Dell, L.A.; Sharma, P. Exploring the homopolar dehydrocoupling of ammonia borane by solid-state multinuclear NMR spectroscopy. Chem. Commun. 2021, 57, 1887–1890. [Google Scholar] [CrossRef] [PubMed]
- Hermanek, S. Boron-11 NMR spectra of boranes, main-group heteroboranes, and substituted derivatives. Factors influencing chemical shifts of skeletal atoms. Chem. Rev. 1992, 92, 325–362. [Google Scholar] [CrossRef]
- Carboni, B.; Monnier, L. Recent developments in the chemistry of amine- and phosphine-boranes. Tetrahedron 1999, 55, 1197–1248. [Google Scholar] [CrossRef]
- Kumar, R.; Karkamkar, A.; Bowden, M.; Autrey, T. Solid-state hydrogen rich boron–nitrogen compounds for energy storage. Chem. Soc. Rev. 2019, 48, 5350–5380. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C3AB | C2C6AB | (C12)2AB | |
|---|---|---|---|
| B | −0.028 | 0.049 | 0.243 |
| N | −0.449 | −0.402 | −0.278 |
| H of BH3 | −0.092 to −0.086 | −0.114 to −0.091 | −0.131 to −0.109 |
| H of NH2 or NH | 0.278 and 0.297 | 0.293 and 0.306 | 0.278 |
| alpha C | 0.156 | −0.632 | −0.496 and −0.743 |
| beta C | −0.283 | 0.861 | 0.298 and −0.017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turani-I-Belloto, K.; Chiriac, R.; Toche, F.; Petit, E.; Yot, P.G.; Alauzun, J.G.; Demirci, U.B. Synthesis: Molecular Structure, Thermal-Calorimetric and Computational Analyses, of Three New Amine Borane Adducts. Molecules 2023, 28, 1469. https://doi.org/10.3390/molecules28031469
Turani-I-Belloto K, Chiriac R, Toche F, Petit E, Yot PG, Alauzun JG, Demirci UB. Synthesis: Molecular Structure, Thermal-Calorimetric and Computational Analyses, of Three New Amine Borane Adducts. Molecules. 2023; 28(3):1469. https://doi.org/10.3390/molecules28031469
Chicago/Turabian StyleTurani-I-Belloto, Kevin, Rodica Chiriac, François Toche, Eddy Petit, Pascal G. Yot, Johan G. Alauzun, and Umit B. Demirci. 2023. "Synthesis: Molecular Structure, Thermal-Calorimetric and Computational Analyses, of Three New Amine Borane Adducts" Molecules 28, no. 3: 1469. https://doi.org/10.3390/molecules28031469
APA StyleTurani-I-Belloto, K., Chiriac, R., Toche, F., Petit, E., Yot, P. G., Alauzun, J. G., & Demirci, U. B. (2023). Synthesis: Molecular Structure, Thermal-Calorimetric and Computational Analyses, of Three New Amine Borane Adducts. Molecules, 28(3), 1469. https://doi.org/10.3390/molecules28031469