Discovery of Novel Chinese Medicine Compounds Targeting 3CL Protease by Virtual Screening and Molecular Dynamics Simulation

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Virtual Screening of TCM
2.2. Molecular Dynamics Simulation of the Top Ligands
2.3. The Time Course of Root-Mean-Square Deviations (RMSD) Analysis
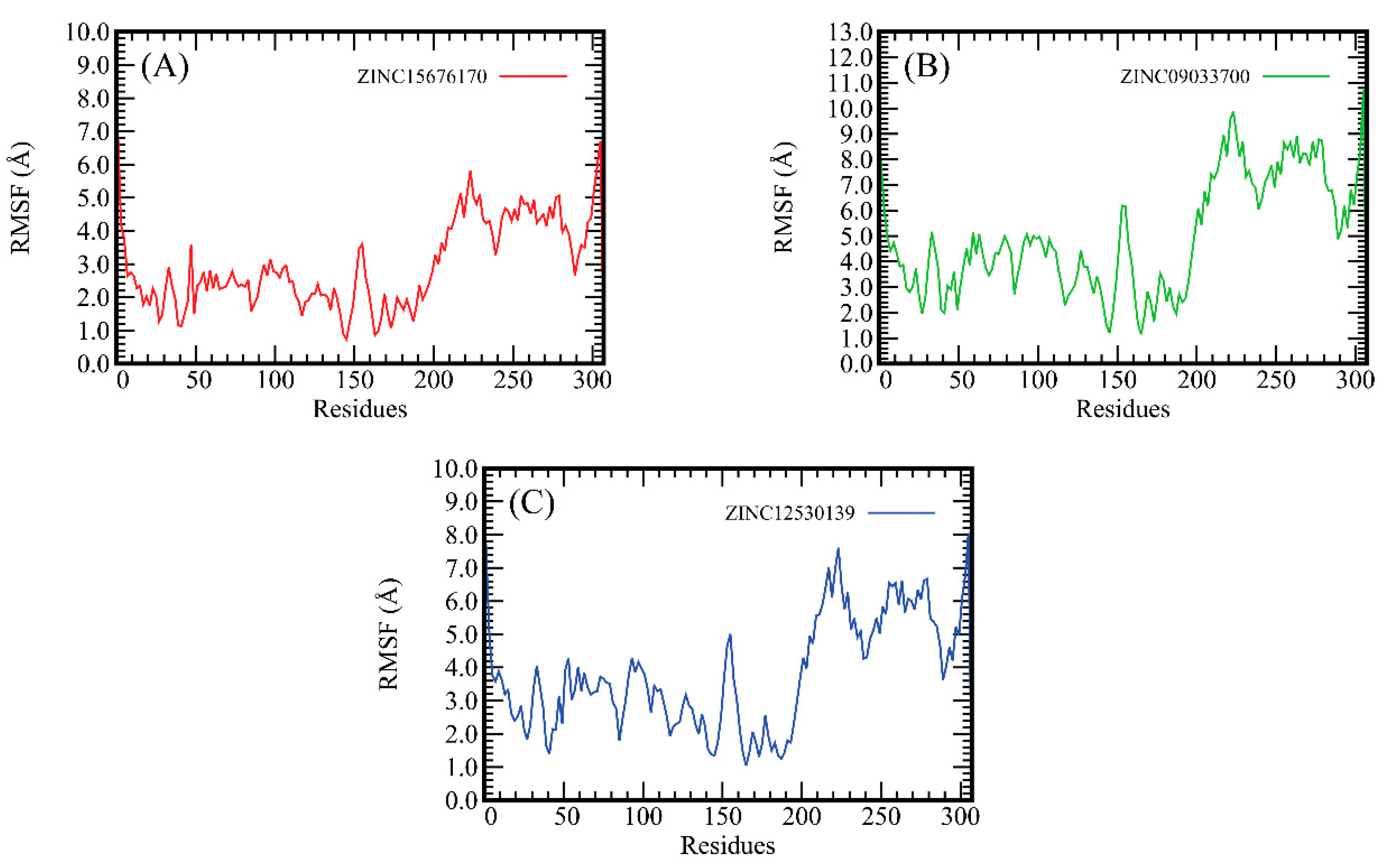
2.4. The Root-Mean-Square Fluctuation (RMSF) Analysis
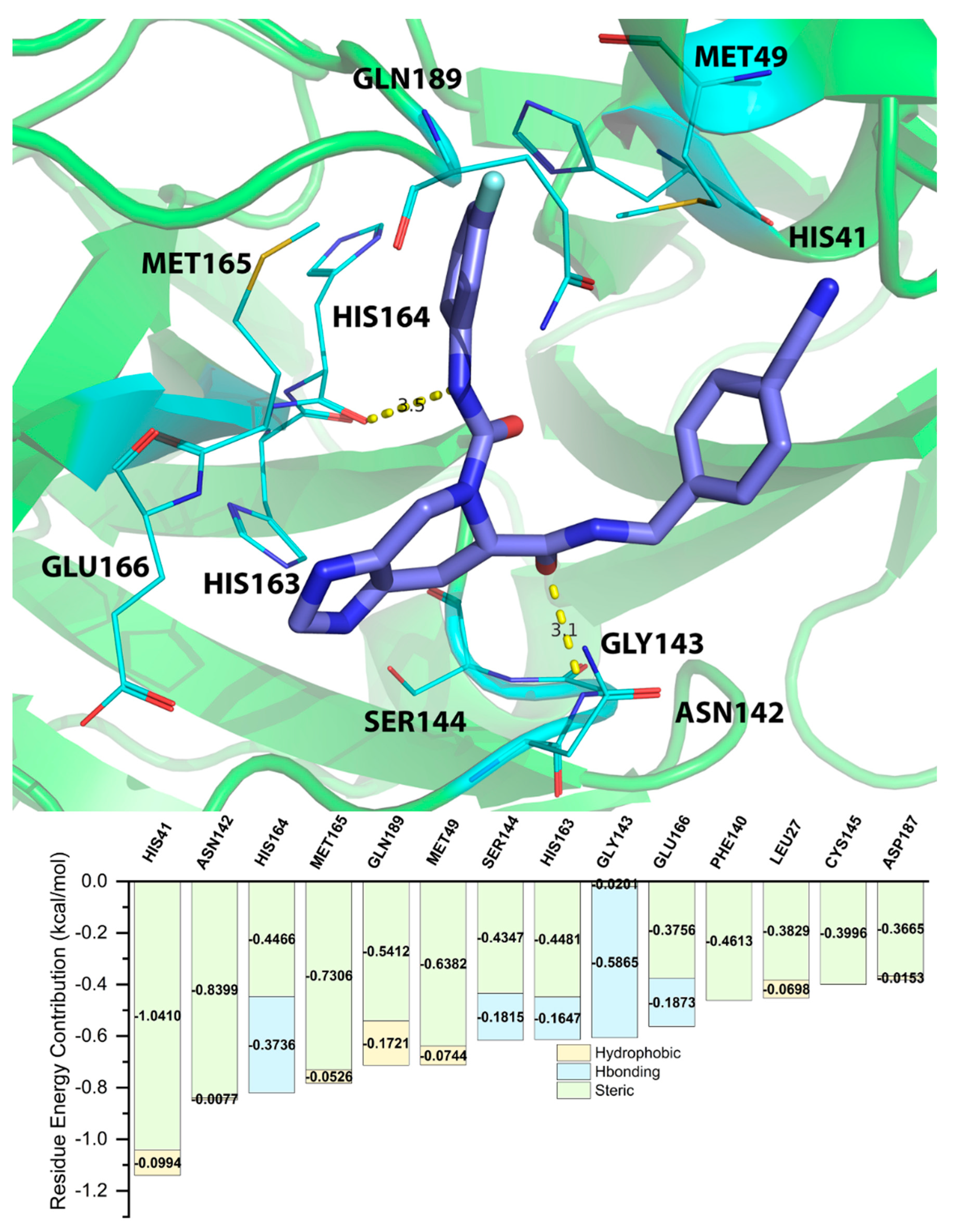
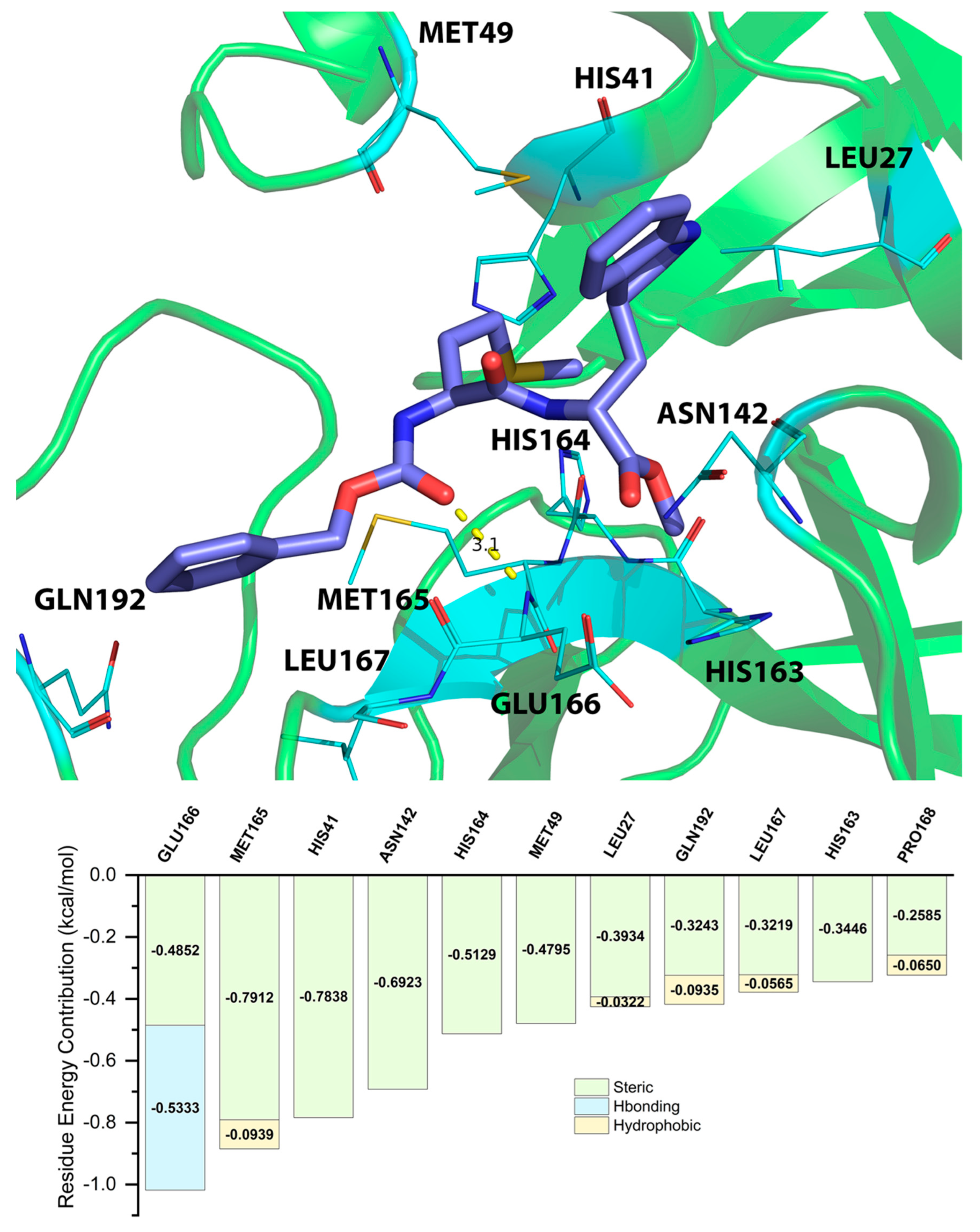
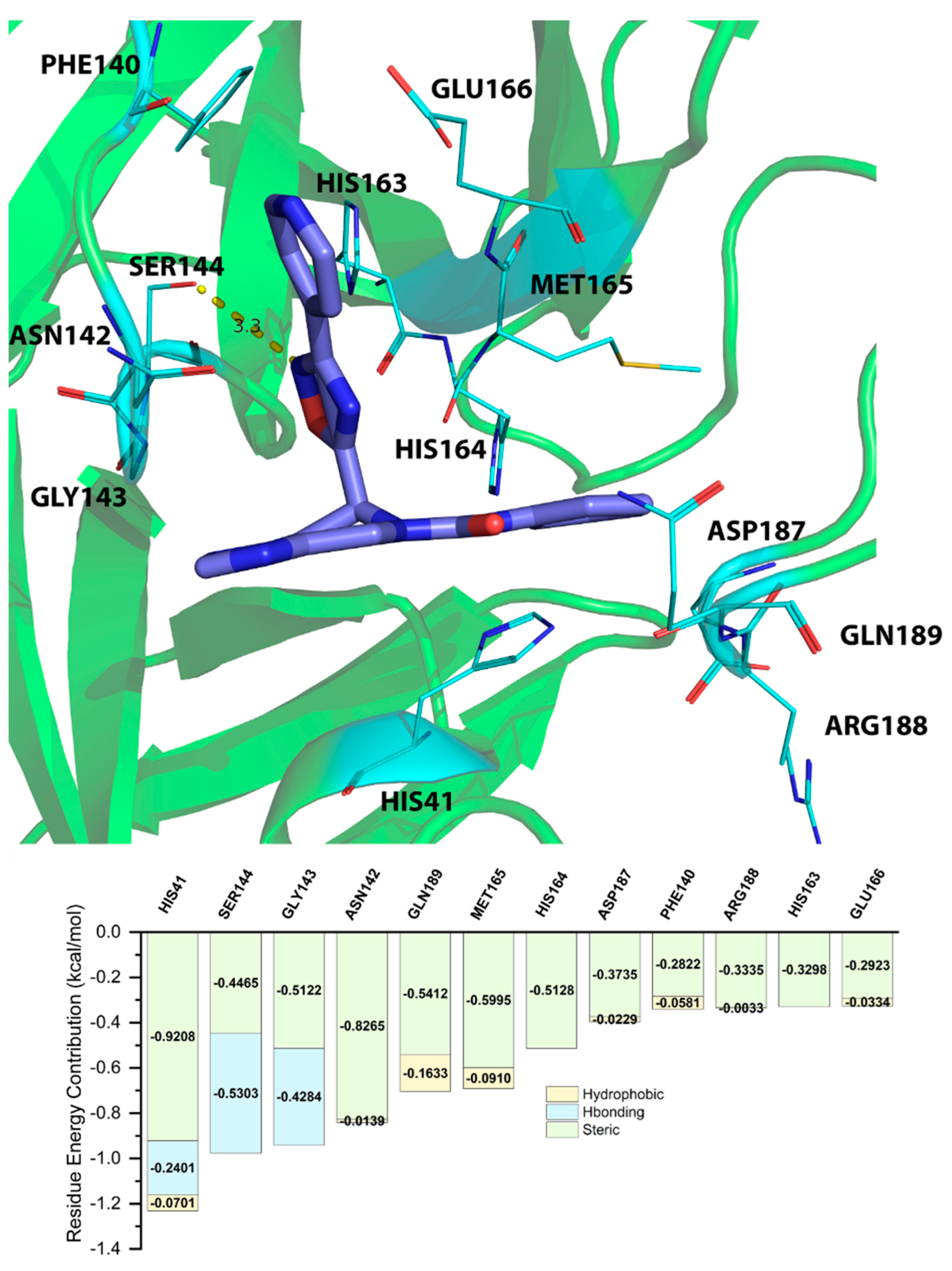
2.5. Energy Decomposition of Three Potential Inhibitors and Their Complex by MM/GBSA Calculation and MCCS
3. Methods
3.1. Protein and Ligand Structure Organization
3.2. TCM Database
3.3. Inhibitor Filtering by Ligand-Based Pharmacophore Model
3.4. Virtual Screening and Selection Method
3.5. Molecular Dynamics (MD) Simulation
3.6. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Calculation
3.7. Molecular Complex Characterizing System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 24 October 2022).
- DiMaio, D.; Enquist, L.W.; Dermody, T.S. A New Coronavirus Emerges, This Time Causing a Pandemic. Annu. Rev. Virol. 2020, 7, iii–v. [Google Scholar] [CrossRef]
- Sarkis, J. Supply chain sustainability: Learning from the COVID-19 pandemic. Int. J. Oper. Prod. Manag. 2021, 41, 63–73. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 13 July 2022).
- Nguyen, K.V. Containing the spread of COVID-19 virus facing to its high mutation rate: Approach to intervention using a nonspecific way of blocking its entry into the cells. Nucl. Nucl. Nucleic Acids 2022, 41, 778–814. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.-C. TCM Database@Taiwan: The World’s Largest Traditional Chinese Medicine Database for Drug Screening In Silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Chen, M.; Xue, Y.; Liang, T.; Chen, H.; Zhou, Y.; Nolin, T.D.; Smith, R.B.; Xie, X.Q. MCCS: A novel recognition pattern-based method for fast track discovery of anti-SARS-CoV-2 drugs. Brief. Bioinform. 2021, 22, 946–962. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef]
- Firth, A.E. A putative new SARS-CoV protein, 3c, encoded in an ORF overlapping ORF3a. J. Gen. Virol. 2020, 101, 1085–1089. [Google Scholar] [CrossRef]
- Paul, A.; Sarkar, A.; Saha, S.; Maji, A.; Janah, P.; Kumar Maity, T. Synthetic and computational efforts towards the development of peptidomimetics and small-molecule SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. 2021, 46, 116301. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, B.J.; Zee, R.v.d.; Haan, C.A.M.d.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Saleem, S.; Idrees, M.; Ali, S.S.; Junaid, M.; Chandra Kaushik, A.; Wei, D.-Q. Allosteric ligands for the pharmacologically important Flavivirus target (NS5) from ZINC database based on pharmacophoric points, free energy calculations and dynamics correlation. J. Mol. Graph. Model. 2018, 82, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.S.; Saad, M.M.; Baig, K.; Bahloul, A.; Abdul-Matin, M.; Alaidaroos, A.Y.; Almakhlafi, G.A.; Albarrak, M.M.; Memish, Z.A.; Albarrak, A.M. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: A retrospective cohort study. Lancet Infect. Dis. 2014, 14, 1090–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.; Jain, N.; Nagaich, U. Nanodiamonds with powerful ability for drug delivery and biomedical applications: Recent updates on in vivo study and patents. J. Pharm. Anal. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2021, 39, 3409–3418. [Google Scholar] [CrossRef] [Green Version]
- Park, K. A review of computational drug repurposing. Transl. Clin. Pharmacol. 2019, 27, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Feng, Z.; Wang, S.; Lin, W.; Xie, X.-Q. MCCS, a novel characterization method for protein–ligand complex. Brief. Bioinform. 2020, 22, bbaa239. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 45, D271–D281. [Google Scholar] [CrossRef]
- Salarvand, Z.; Amirnasr, M.; Talebian, M.; Raeissi, K.; Meghdadi, S. Enhanced corrosion resistance of mild steel in 1 M HCl solution by trace amount of 2-phenyl-benzothiazole derivatives: Experimental, quantum chemical calculations and molecular dynamics (MD) simulation studies. Corros. Sci. 2017, 114, 133–145. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. computation. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; Homeyer, N.; Izadi, S.; Janowski, P.J. AMBER 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Hou, T.; Wang, J.; Li, Y.; Wang, W.J. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolym. Orig. Res. Biomol. 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Hu, J.; Feng, Z.; Ma, S.; Zhang, Y.; Tong, Q.; Alqarni, M.H.; Gou, X.; Xie, X.-Q. Difference and influence of inactive and active states of cannabinoid receptor subtype CB2: From conformation to drug discovery. J. Chem. Inf. Model. 2016, 56, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, S.-i.; Higashi-Kuwata, N.; Hayashi, H.; Allu, S.R.; Raghavaiah, J.; Bulut, H.; Das, D.; Anson, B.J.; Lendy, E.K.; Takamatsu, Y.; et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021, 12, 668. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | ZINC ID | Structure | Docking Score (kcal/mol) | Molecular Weight (amu) | LogP Value |
|---|---|---|---|---|---|
| 1 | ZINC15676170 | 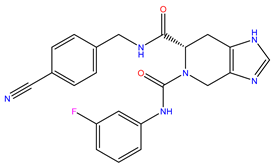 | −9.178 | 418.432 | 3.228 |
| 2 | ZINC15675325 | 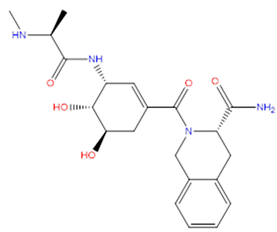 | −9.171 | 417.486 | −2.19 |
| 3 | ZINC12529667 | 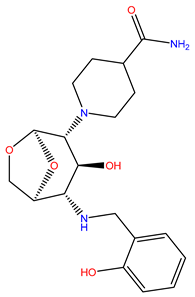 | −9.124 | 378.449 | −1.228 |
| 4 | ZINC13550544 | 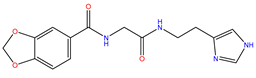 | −9.031 | 317.325 | −0.354 |
| 5 | ZINC03838803 |  | −8.947 | 286.352 | −2.335 |
| 6 | ZINC12664661 | 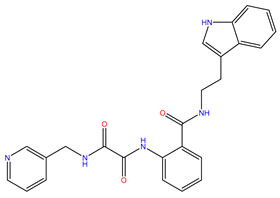 | −8.845 | 442.499 | 2.476 |
| 7 | ZINC09033700 |  | −8.799 | 483.589 | 3.683 |
| 8 | ZINC12530139 | 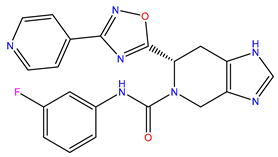 | −8.774 | 407.409 | 2.525 |
| 9 | ZINC00198624 | 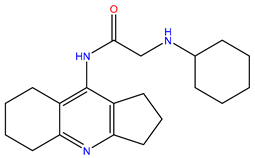 | −8.704 | 329.488 | 1.703 |
| 10 | ZINC08299537 |  | −8.602 | 442.54 | −1.292 |
| Rank | ZINC ID | MD Binding Free Energy (kcal/mol) |
|---|---|---|
| 1 | ZINC09033700 | −6.8953 |
| 2 | ZINC15676170 | −4.3351 |
| 3 | ZINC12530139 | −3.0082 |
| 4 | ZINC15675325 | −1.8083 |
| 5 | ZINC12529667 | −1.3637 |
| 6 | ZINC00198624 | −0.1468 |
| 7 | ZINC12664661 | 0.0053 |
| 8 | ZINC03838803 | 0.4986 |
| 9 | ZINC08299537 | 0.6072 |
| 10 | ZINC13550544 | 8.7553 |
| Residue | EVDW | EEEL | EINT | GPOL | ESAS | EMM | EEELPOL | EVDWEELPOL | GGBSA | GMM/GBSA |
|---|---|---|---|---|---|---|---|---|---|---|
| CYS145 | −1.8152 | −1.767 | 0 | 0.0997 | −1.2208 | −3.5822 | −1.6673 | −3.4824 | −1.121 | −4.7032 |
| ASN142 | −2.2802 | −0.8786 | 0 | 0.0751 | −1.3824 | −3.1588 | −0.8035 | −3.0838 | −1.3073 | −4.4662 |
| MET165 | −2.4885 | 0.1252 | 0 | −0.396 | −1.5208 | −2.3633 | −0.2709 | −2.7594 | −1.9169 | −4.2802 |
| HIS164 | −2.066 | −1.1163 | 0 | 0.3195 | −0.9614 | −3.1823 | −0.7968 | −2.8628 | −0.6419 | −3.8242 |
| GLY143 | −0.7966 | −1.5772 | 0 | −0.053 | −0.6921 | −2.3738 | −1.63 | −2.4266 | −0.7448 | −3.1187 |
| HIS163 | −0.9209 | −1.5059 | 0 | −0.055 | −0.576 | −2.4268 | −1.5613 | −2.4822 | −0.6314 | −3.0582 |
| MET49 | −1.3443 | −0.2809 | 0 | 0.1433 | −1.1549 | −1.6252 | −0.1376 | −1.4819 | −1.0116 | −2.6368 |
| GLU166 | −0.8813 | −2.6098 | 0 | 1.815 | −0.877 | −3.4911 | −0.7948 | −1.6761 | 0.938 | −2.553 |
| PHE140 | −0.8967 | −0.9905 | 0 | 0.4139 | −0.4562 | −1.8872 | −0.5766 | −1.4733 | −0.0423 | −1.9295 |
| HIS41 | −1.1417 | 0.2094 | 0 | −0.212 | −0.7811 | −0.9322 | −0.0026 | −1.1442 | −0.993 | −1.9253 |
| Residue | EVDW | EEEL | EINT | GPOL | ESAS | EMM | EEELPOL | EVDWEELPOL | GGBSA | GMM/GBSA |
|---|---|---|---|---|---|---|---|---|---|---|
| MET165 | −3.6331 | −2.335 | 0 | 0.827 | −2.2815 | −5.9681 | −1.508 | −5.1411 | −1.4545 | −7.4226 |
| ASN142 | −2.2616 | −3.8489 | 0 | 1.0485 | −1.8162 | −6.1106 | −2.8004 | −5.062 | −0.7676 | −6.8782 |
| HIS164 | −2.005 | −3.4553 | 0 | 0.29 | −1.2107 | −5.4603 | −3.1653 | −5.1703 | −0.9207 | −6.381 |
| MET49 | −2.363 | −1.6125 | 0 | 0.8082 | −1.6008 | −3.9755 | −0.8043 | −3.1673 | −0.7926 | −4.7681 |
| GLU166 | −1.7504 | 2.5706 | 0 | −3.6206 | −1.093 | 0.8203 | −1.0499 | −2.8003 | −4.7136 | −3.8933 |
| CYS145 | −1.5458 | −0.2229 | 0 | −0.1515 | −1.2428 | −1.7687 | −0.3743 | −1.9201 | −1.3943 | −3.1629 |
| HIS41 | −1.3246 | −0.288 | 0 | −0.015 | −1.01 | −1.6126 | −0.303 | −1.6276 | −1.025 | −2.6376 |
| LEU167 | −1.2952 | 0.2722 | 0 | −0.3641 | −0.8727 | −1.023 | −0.0919 | −1.3871 | −1.2368 | −2.2598 |
| ASP187 | −1.1167 | −1.877 | 0 | 1.5806 | −0.7113 | −2.9937 | −0.2963 | −1.4131 | 0.8693 | −2.1244 |
| PRO168 | −0.9398 | −0.0493 | 0 | −0.0512 | −0.9239 | −0.9892 | −0.1006 | −1.0404 | −0.9751 | −1.9643 |
| Residue | EVDW | EEEL | EINT | GPOL | ESAS | EMM | EEELPOL | EVDWEELPOL | GGBSA | GMM/GBSA |
|---|---|---|---|---|---|---|---|---|---|---|
| ASN142 | −2.8483 | −0.7788 | 0 | 0.1051 | −1.774 | −3.627 | −0.6737 | −3.522 | −1.669 | −5.296 |
| GLN189 | −1.2062 | −2.8307 | 0 | 0.3067 | −1.3396 | −4.0368 | −2.524 | −3.7301 | −1.0329 | −5.0697 |
| HIS164 | −1.8092 | −2.6969 | 0 | 0.5538 | −0.9579 | −4.5061 | −2.1431 | −3.9523 | −0.4041 | −4.9101 |
| MET165 | −2.4619 | −0.3918 | 0 | 0.0996 | −1.6995 | −2.8537 | −0.2922 | −2.7541 | −1.5999 | −4.4536 |
| GLY143 | −1.6115 | −1.0566 | 0 | 0.2761 | −0.9806 | −2.6681 | −0.7805 | −2.392 | −0.7045 | −3.3726 |
| CYS145 | −1.2837 | −0.0759 | 0 | −0.2514 | −0.873 | −1.3596 | −0.3273 | −1.611 | −1.1244 | −2.484 |
| GLU166 | −1.9215 | 2.6406 | 0 | −1.892 | −1.2823 | 0.7191 | 0.7486 | −1.173 | −3.1743 | −2.4553 |
| SER144 | −1.0346 | −0.6017 | 0 | 0.1506 | −0.5429 | −1.6363 | −0.4511 | −1.4857 | −0.3923 | −2.0286 |
| LEU141 | −1.0447 | −0.5713 | 0 | 0.0816 | −0.4866 | −1.616 | −0.4897 | −1.5344 | −0.405 | −2.021 |
| HIS41 | −0.7741 | −0.919 | 0 | 0.4292 | −0.6046 | −1.6931 | −0.4898 | −1.2639 | −0.1755 | −1.8685 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, J.; Hao, Y.; Shi, Q.; Hou, G.; Wang, Y.; Wang, Y.; Xiao, W.; Othman, J.; Qi, J.; Wang, Y.; et al. Discovery of Novel Chinese Medicine Compounds Targeting 3CL Protease by Virtual Screening and Molecular Dynamics Simulation. Molecules 2023, 28, 937. https://doi.org/10.3390/molecules28030937
Cheng J, Hao Y, Shi Q, Hou G, Wang Y, Wang Y, Xiao W, Othman J, Qi J, Wang Y, et al. Discovery of Novel Chinese Medicine Compounds Targeting 3CL Protease by Virtual Screening and Molecular Dynamics Simulation. Molecules. 2023; 28(3):937. https://doi.org/10.3390/molecules28030937
Chicago/Turabian StyleCheng, Jin, Yixuan Hao, Qin Shi, Guanyu Hou, Yanan Wang, Yong Wang, Wen Xiao, Joseph Othman, Junnan Qi, Yuanqiang Wang, and et al. 2023. "Discovery of Novel Chinese Medicine Compounds Targeting 3CL Protease by Virtual Screening and Molecular Dynamics Simulation" Molecules 28, no. 3: 937. https://doi.org/10.3390/molecules28030937