


Determining RNA Natural Modifications and Nucleoside Analog-Labeled Sites by a Chemical/Enzyme-Induced Base Mutation Principle

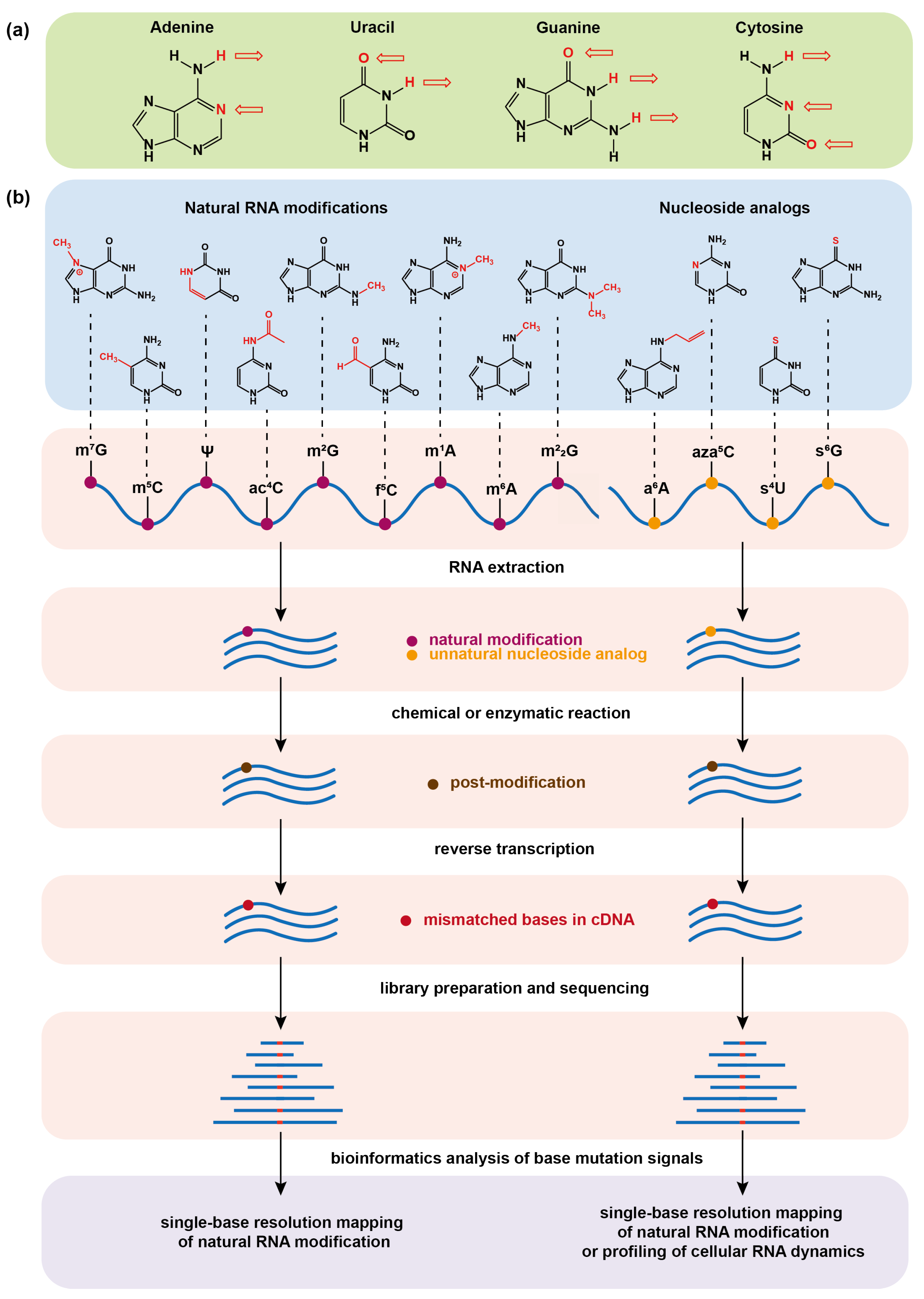
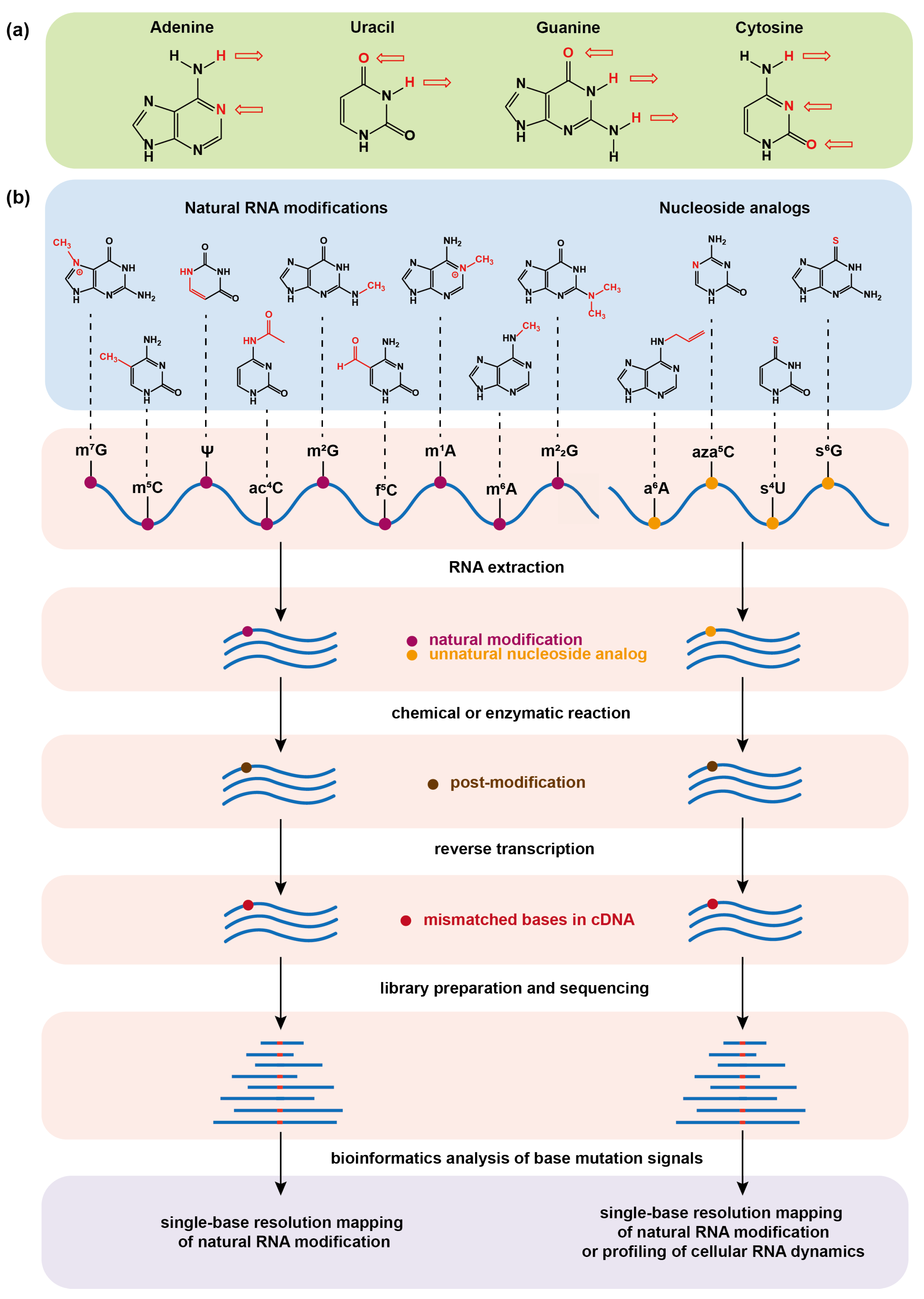{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
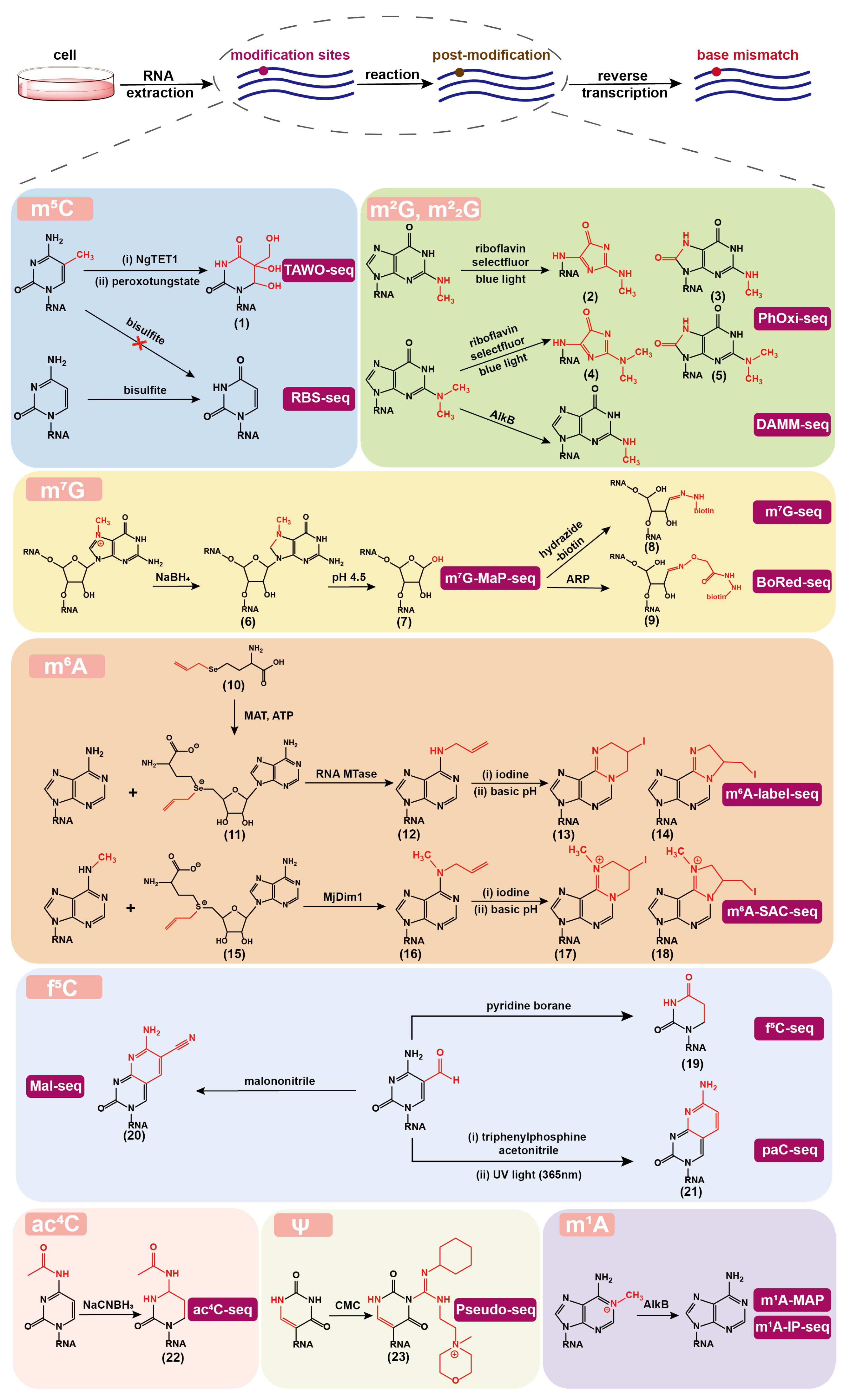
2. Chemical Sequencing Methods for Detecting RNA Natural Modifications
2.1. mC Detection
2.2. mG and mG Detection
2.3. mG Detection
2.4. mA Detection
2.5. fC Detection
2.6. acC Detection
2.7. Detection
2.8. mA Detection
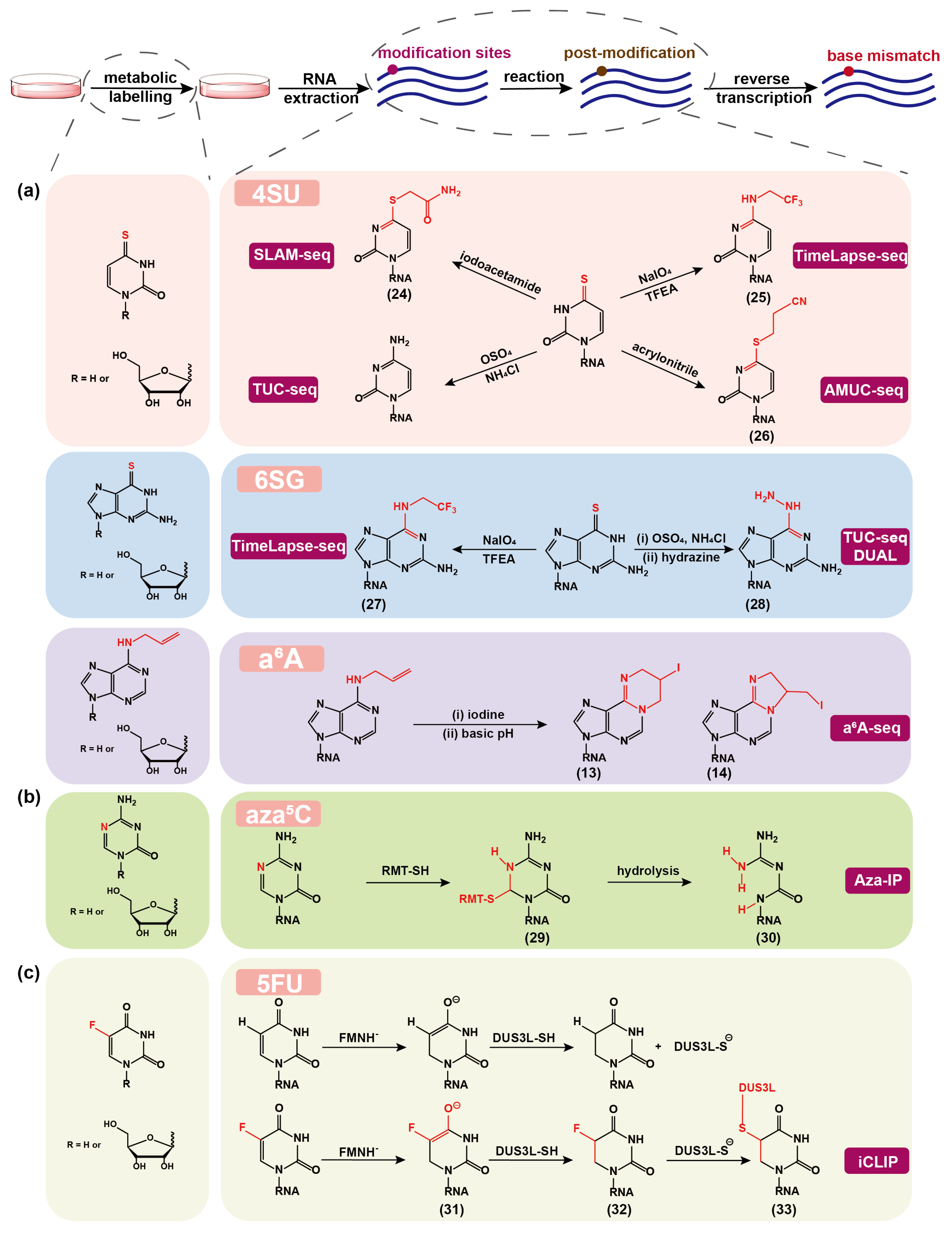
3. Chemical Sequencing Methods for Detecting Artificial Nucleoside Analogs Marked on RNA by Metabolic Labeling
3.1. 4SU for RNA Labeling and Detection
3.2. 6SG for RNA Labeling and Detection
3.3. aA for RNA Labeling and Detection
3.4. azaC for mC Detection
3.5. 5FU for DHU Detection
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Han, X.; Yang, W.-L.; Zhang, M.; Ma, H.-L.; Sun, B.-F.; Li, A.; Xia, J.; Chen, J.; et al. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol. Cell 2019, 75, 1188–1202.e11. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Sun, B.-F.; Chen, Y.-S.; Xu, J.-W.; Lai, W.-Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export — NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.-L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X.; et al. Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol. Cell 2019, 74, 1304–1316. [Google Scholar] [CrossRef]
- Enroth, C.; Poulsen, L.D.; Iversen, S.; Kirpekar, F.; Albrechtsen, A.; Vinther, J. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. 2019, 47, e126. [Google Scholar] [CrossRef]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 2018, 175, 1872–1886. [Google Scholar] [CrossRef]
- Jin, G.; Xu, M.; Zou, M.; Duan, S. The processing, gene regulation, biological functions, and clinical relevance of N4-acetylcytidine on RNA: A systematic review. Mol. Ther.–Nucleic Acids 2020, 20, 13–24. [Google Scholar] [CrossRef]
- Nakano, S.; Suzuki, T.; Kawarada, L.; Iwata, H.; Asano, K.; Suzuki, T. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNAMet. Nat. Chem. Biol. 2016, 12, 546–551. [Google Scholar] [CrossRef]
- Karijolich, J.; Yu, Y.-T. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 2011, 474, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef]
- Hoernes, T.P.; Clementi, N.; Faserl, K.; Glasner, H.; Breuker, K.; Lindner, H.; Huettenhofer, A.; Erlacher, M.D. Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res. 2016, 44, 852–862. [Google Scholar] [CrossRef]
- Elliott, B.A.; Ho, H.-T.; Ranganathan, S.V.; Vangaveti, S.; Ilkayeva, O.; Abou Assi, H.; Choi, A.K.; Agris, P.F.; Holley, C.L. Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat. Commun. 2019, 10, 3401. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef]
- Yang, W.-Q.; Xiong, Q.-P.; Ge, J.-Y.; Li, H.; Zhu, W.-Y.; Nie, Y.; Lin, X.; Lv, D.; Li, J.; Lin, H.; et al. THUMPD3–TRMT112 is a m2G methyltransferase working on a broad range of tRNA substrates. Nucleic Acids Res. 2021, 49, 11900–11919. [Google Scholar] [CrossRef]
- Wiener, D.; Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2021, 22, 119–131. [Google Scholar] [CrossRef]
- Harcourt, E.M.; Kietrys, A.M.; Kool, E.T. Chemical and structural effects of base modifications in messenger RNA. Nature 2017, 541, 339–346. [Google Scholar] [CrossRef]
- Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021, 22, 375–392. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Wu, X.; Zhang, J.; Fang, Y.; Pan, Y.; Shu, Y.; Ma, P. The evolving landscape of N6-methyladenosine modification in the tumor microenvironment. Mol. Ther. 2021, 29, 1703–1715. [Google Scholar] [CrossRef]
- Owens, M.C.; Zhang, C.; Liu, K.F. Recent technical advances in the study of nucleic acid modifications. Mol. Cell 2021, 81, 4116–4136. [Google Scholar] [CrossRef]
- Anreiter, I.; Mir, Q.; Simpson, J.T.; Janga, S.C.; Soller, M. New twists in detecting mRNA modification dynamics. Trends Biotechnol. 2021, 39, 72–89. [Google Scholar] [CrossRef]
- Cao, J.; Shu, X.; Feng, X.-H.; Liu, J. Mapping messenger RNA methylations at single base resolution. Curr. Opin. Chem. Biol. 2021, 63, 28–37. [Google Scholar] [CrossRef]
- Moshitch-Moshkovitz, S.; Dominissini, D.; Rechavi, G. The epitranscriptome toolbox. Cell 2022, 185, 764–776. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Shulman, Z.; Stern-Ginossar, N. The RNA modification N6-methyladenosine as a novel regulator of the immune system. Nat. Immunol. 2020, 21, 501–512. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Chen, J. m6A modification in coding and non-coding RNAs: Roles and therapeutic implications in cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Delaunay, S.; Frye, M. RNA modifications regulating cell fate in cancer. Nat. Cell Biol. 2019, 21, 552–559. [Google Scholar] [CrossRef]
- An, Y.; Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 2022, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Moshitch-Moshkovitz, S.; Amariglio, N.; Rechavi, G.; Dominissini, D. The m6A epitranscriptome: Transcriptome plasticity in brain development and function. Nat. Rev. Neurosci. 2020, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kan, R.L.; Chen, J.; Sallam, T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 2022, 38, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- McKenzie, L.K.; El-Khoury, R.; Thorpe, J.D.; Damha, M.J.; Hollenstein, M. Recent progress in non-native nucleic acid modifications. Chem. Soc. Rev. 2021, 50, 5126–5164. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Fazio, M.; Kubota, M.; Nainar, S.; Feng, C.; Li, X.; Atwood, S.X.; Bredy, T.W.; Spitale, R.C. Cell-selective bioorthogonal metabolic labeling of RNA. J. Am. Chem. Soc. 2017, 139, 2148–2151. [Google Scholar] [CrossRef]
- Singha, M.; Spitalny, L.; Nguyen, K.; Vandewalle, A.; Spitale, R.C. Chemical methods for measuring RNA expression with metabolic labeling. Wiley Interdiscip. Rev. RNA 2021, 12, e1650. [Google Scholar] [CrossRef]
- Gupta, M.; Levine, S.R.; Spitale, R.C. Probing nascent RNA with metabolic incorporation of modified nucleosides. Acc. Chem. Res. 2022, 55, 2647–2659. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Rabani, M.; Levin, J.Z.; Fan, L.; Adiconis, X.; Raychowdhury, R.; Garber, M.; Gnirke, A.; Nusbaum, C.; Hacohen, N.; Friedman, N.; et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 2011, 29, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Lin, Y.; Wang, W.; Zhao, Y.; Bao, X. Application of nucleoside or nucleotide analogues in RNA dynamics and RNA-binding protein analysis. Wiley Interdiscip. Rev. RNA 2022, 13, e1722. [Google Scholar] [CrossRef]
- Shu, X.; Dai, Q.; Wu, T.; Bothwell, I.R.; Yue, Y.; Zhang, Z.; Cao, J.; Fei, Q.; Luo, M.; He, C.; et al. N6-Allyladenosine: A new small molecule for RNA labeling identified by mutation assay. J. Am. Chem. Soc. 2017, 139, 17213–17216. [Google Scholar] [CrossRef]
- Shu, X.; Huang, C.; Li, T.; Cao, J.; Liu, J. a6A-seq: N6-allyladenosine-based cellular messenger RNA metabolic labelling and sequencing. Fundam. Res. 2023. submitted. [Google Scholar]
- Hartstock, K.; Nilges, B.S.; Ovcharenko, A.; Cornelissen, N.V.; Puellen, N.; Lawrence-Doerner, A.-M.; Leidel, S.A.; Rentmeister, A. Enzymatic or in vivo installation of propargyl groups in combination with click chemistry for the enrichment and detection of methyltransferase target sites in RNA. Angew. Chem. Int. Ed. 2018, 57, 6342–6346. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Cao, J.; Cheng, M.; Xiang, S.; Gao, M.; Li, T.; Ying, X.; Wang, F.; Yue, Y.; Lu, Z.; et al. A metabolic labeling method detects m6A transcriptome-wide at single base resolution. Nat. Chem. Biol. 2020, 16, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, J.; Liu, B.; Xu, J.; Cai, B.; Yang, H.; Straube, J.; Yu, X.; Ma, T. Biological roles of RNA m5C modification and its implications in cancer immunotherapy. Biomark. Res. 2022, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; Mcdonald, L.; Millar, D.; Collis, C.; Watt, F.; Grigg, G.; Molloy, P.; Paul, C. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Ma, H.-L.; Yang, Y.; Lai, W.-Y.; Sun, B.-F.; Yang, Y.-G. 5-methylcytosine analysis by RNA-BisSeq. In Epitranscriptomics: Methods and Protocols; Methods in Molecular Biology; Wajapeyee, N., Gupta, R., Eds.; Springer: New York, NY, USA, 2019; pp. 237–248. ISBN 978-1-4939-8808-2. [Google Scholar]
- Khoddami, V.; Yerra, A.; Mosbruger, T.L.; Fleming, A.M.; Burrows, C.J.; Cairns, B.R. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 6784–6789. [Google Scholar] [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Song, J.; Yi, C. Reading chemical modifications in the transcriptome. J. Mol. Biol. 2020, 432, 1824–1839. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Bi, Y.; Siejka-Zielinska, P.; Zhou, Y.-L.; Zhang, X.-X.; Song, C.-X. Bisulfite-free and base-resolution analysis of 5-methylcytidine and 5-hydroxymethylcytidine in RNA with peroxotungstate. Chem. Commun. 2019, 55, 2328–2331. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Guerrero, C.R.; Zhong, N.; Amato, N.J.; Liu, Y.; Liu, S.; Cai, Q.; Ji, D.; Jin, S.-G.; Niedernhofer, L.J.; et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 2014, 136, 11582–11585. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.C.K.; Mahdavi-Amiri, Y.; Korfmann, C.; Hili, R. PhOxi-Seq: Single-nucleotide resolution sequencing of N2-methylation at guanosine in RNA by photoredox catalysis. J. Am. Chem. Soc. 2022, 144, 5723–5727. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Xiong, Q.-P.; Perez, S.P.; Liu, C.; Wei, J.; Le, C.; Zhang, L.; Harada, B.T.; Dai, Q.; Feng, X.; et al. ALKBH7-mediated demethylation regulates mitochondrial polycistronic RNA processing. Nat. Cell Biol. 2021, 23, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Zheng, G.; Schwartz, M.H.; Clark, W.C.; Pan, T. Selective enzymatic demethylation of N2,N2-dimethylguanosine in RNA and its application in high-throughput tRNA sequencing. Angew. Chem. Int. Ed. 2017, 56, 5017–5020. [Google Scholar] [CrossRef]
- Wang, Y.; Katanski, C.D.; Watkins, C.; Pan, J.N.; Dai, Q.; Jiang, Z.; Pan, T. A high-throughput screening method for evolving a demethylase enzyme with improved and new functionalities. Nucleic Acids Res. 2021, 49, e30. [Google Scholar] [CrossRef]
- Cowling, V.H. Regulation of mRNA cap methylation. Biochem. J. 2010, 425, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Guy, M.P.; Phizicky, E.M. Two-subunit enzymes involved in eukaryotic post-transcriptional tRNA modification. RNA Biol. 2014, 11, 1608–1618. [Google Scholar] [CrossRef]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.L.J.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yao, Y.; Wu, P.; Zi, X.; Sun, N.; He, J. The potential role of N7-methylguanosine (m7G) in cancer. J. Hematol. Oncol. 2022, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 promotes let-7 microRNA processing via m7G methylation. Mol. Cell 2019, 74, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.K.; Koutmou, K.S. Chemical modifications to mRNA nucleobases impact translation elongation and termination. Biophys. Chem. 2022, 285, 106780. [Google Scholar] [CrossRef]
- Ma, L.; He, L.; Kang, S.; Gu, B.; Gao, S.; Zuo, Z. Advances in detecting N6-methyladenosine modification in circRNAs. Methods 2022, 205, 234–246. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Dong, S.; Yu, Q.; Jia, G. Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol. 2020, 16, 896–903. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.G. SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chem. Biol. 2016, 11, 583–597. [Google Scholar] [CrossRef] [Green Version]
- Rudenko, A.Y.; Mariasina, S.S.; Sergiev, P.; Polshakov, V. Analogs of S-adenosyl-L-methionine in studies of methyltransferases. Mol. Biol. 2022, 56, 229–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Islam, K.; Liu, Y.; Zheng, W.; Tang, H.; Lailler, N.; Blum, G.; Deng, H.; Luo, M. Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J. Am. Chem. Soc. 2013, 135, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Popadic, D.; Mhaindarkar, D.; Dang Thai, M.H.N.; Hailes, H.C.; Mordhorst, S.; Andexer, J.N. A bicyclic S-adenosylmethionine regeneration system applicable with different nucleosides or nucleotides as cofactor building blocks. RSC Chem. Biol. 2021, 2, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, S.; Peng, Y.; Ge, R.; Su, R.; Senevirathne, C.; Harada, B.T.; Dai, Q.; Wei, J.; Zhang, L.; et al. m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol. 2022, 40, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Siejka-Zielinska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Bockler, B.; Song, C.-X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Z.; Zhang, X.; Weng, X.; Deng, J.; Yang, W.; Wu, F.; Han, S.; Xia, C.; Zhou, Y.; et al. Single-base resolution mapping reveals distinct 5-formylcytidine in saccharomyces cerevisiae mRNAs. ACS Chem. Biol. 2022, 17, 77–84. [Google Scholar] [CrossRef]
- Link, C.N.; Thalalla Gamage, S.; Gallimore, D.; Kopajtich, R.; Evans, C.; Nance, S.; Fox, S.D.; Andresson, T.; Chari, R.; Ivanic, J.; et al. Protonation-dependent sequencing of 5-formylcytidine in RNA. Biochemistry 2022, 61, 535–544. [Google Scholar] [CrossRef]
- Van Haute, L.; Minczuk, M. Detection of 5-formylcytosine in mitochondrial transcriptome. In Mitochondrial Gene Expression: Methods and Protocols; Methods in Molecular Biology; Minczuk, M., Rorbach, J., Eds.; Springer: New York, NY, USA, 2021; pp. 59–68. ISBN 978-1-07-160834-0. [Google Scholar]
- Zhu, C.; Gao, Y.; Guo, H.; Xia, B.; Song, J.; Wu, X.; Zeng, H.; Kee, K.; Tang, F.; Yi, C. Single-cell 5-formylcytosine landscapes of mammalian early embryos and ESCs at single-base resolution. Cell Stem Cell 2017, 20, 720–731. [Google Scholar] [CrossRef]
- Li, A.; Sun, X.; Arguello, A.E.; Kleiner, R.E. Chemical method to sequence 5-formylcytosine on RNA. ACS Chem. Biol. 2022, 17, 503–508. [Google Scholar] [CrossRef]
- Jin, X.-Y.; Huang, Z.-R.; Xie, L.-J.; Liu, L.; Han, D.-L.; Cheng, L. Photo-facilitated detection and sequencing of 5-formylcytidine RNA. Angew. Chem. Int. Ed. 2022, 61, e202210652. [Google Scholar] [CrossRef] [PubMed]
- Arango, D.; Sturgill, D.; Yang, R.; Kanai, T.; Bauer, P.; Roy, J.; Wang, Z.; Hosogane, M.; Schiffers, S.; Oberdoerffer, S. Direct epitranscriptomic regulation of mammalian translation initiation through N4-acetylcytidine. Mol. Cell 2022, 82, 2797–2814.e11. [Google Scholar] [CrossRef]
- Thomas, J.M.; Briney, C.A.; Nance, K.D.; Lopez, J.E.; Thorpe, A.L.; Fox, S.D.; Bortolin-Cavaille, M.-L.; Sas-Chen, A.; Arango, D.; Oberdoerffer, S.; et al. A chemical signature for cytidine acetylation in RNA. J. Am. Chem. Soc. 2018, 140, 12667–12670. [Google Scholar] [CrossRef] [PubMed]
- Sas-Chen, A.; Thomas, J.M.; Matzov, D.; Taoka, M.; Nance, K.D.; Nir, R.; Bryson, K.M.; Shachar, R.; Liman, G.L.S.; Burkhart, B.W.; et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature 2020, 583, 638–643. [Google Scholar] [CrossRef]
- Thalalla Gamage, S.; Sas-Chen, A.; Schwartz, S.; Meier, J.L. Quantitative nucleotide resolution profiling of RNA cytidine acetylation by ac4C-seq. Nat. Protoc. 2021, 16, 2286–2307. [Google Scholar] [CrossRef]
- Davis, F.; Allen, F. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 1957, 227, 907–915. [Google Scholar] [CrossRef]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; Leon-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 2014, 9, e110799. [Google Scholar] [CrossRef]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef]
- Bakin, A.; Ofengand, J. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: Analysis by the application of a new sequencing technique. Biochemistry 1993, 32, 9754–9762. [Google Scholar] [CrossRef]
- Zhou, K.I.; Clark, W.C.; Pan, D.W.; Eckwahl, M.J.; Dai, Q.; Pan, T. Pseudouridines have context-dependent mutation and stop rates in high-throughput sequencing. RNA Biol. 2018, 15, 892–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macon, J.; Wolfenden, R. 1-methyladenosine—Dimroth rearrangement and reversible reduction. Biochemistry 1968, 7, 3453–3458. [Google Scholar] [CrossRef]
- Schwartz, S. m1A within cytoplasmic mRNAs at single nucleotide resolution: A reconciled transcriptome-wide map. RNA 2018, 24, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 2017, 68, 993–1005. [Google Scholar] [CrossRef]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef]
- Erhard, F.; Saliba, A.-E.; Lusser, A.; Toussaint, C.; Hennig, T.; Prusty, B.K.; Kirschenbaum, D.; Abadie, K.; Miska, E.A.; Friedel, C.C.; et al. Time-resolved single-cell RNA-seq using metabolic RNA labelling. Nat. Rev. Methods Primers 2022, 2, 77. [Google Scholar] [CrossRef]
- Young, J.D.; Yao, S.Y.M.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Hinze, F.; Drewe-Boss, P.; Milek, M.; Ohler, U.; Landthaler, M.; Gotthardt, M. Expanding the map of protein-RNA interaction sites via cell fusion followed by PAR-CLIP. RNA Biol. 2018, 15, 359–368. [Google Scholar] [CrossRef]
- Doelken, L.; Ruzsics, Z.; Raedle, B.; Friedel, C.C.; Zimmer, R.; Mages, J.; Hoffmann, R.; Dickinson, P.; Forster, T.; Ghazal, P.; et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 2008, 14, 1959–1972. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, V.A.; Reichholf, B.; Neumann, T.; Rescheneder, P.; Bhat, P.; Burkard, T.R.; Wlotzka, W.; von Haeseler, A.; Zuber, J.; Ameres, S.L. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 2017, 14, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Baptista, M.A.P.; Krammer, T.; Hennig, T.; Lange, M.; Arampatzi, P.; Juerges, C.S.; Theis, F.J.; Saliba, A.-E.; Doelken, L. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 2019, 571, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, G.-J.; Jung, L.A.; Larsson, A.J.M.; Lidschreiber, M.; Forsman, O.A.; Lidschreiber, K.; Cramer, P.; Sandberg, R. NASC-seq monitors RNA synthesis in single cells. Nat. Commun. 2019, 10, 3138. [Google Scholar] [CrossRef]
- Schofield, J.A.; Duffy, E.E.; Kiefer, L.; Sullivan, M.C.; Simon, M.D. TimeLapse-seq: Adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat. Methods 2018, 15, 221–225. [Google Scholar] [CrossRef]
- Qiu, Q.; Hu, P.; Qiu, X.; Govek, K.W.; Camara, P.G.; Wu, H. Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq. Nat. Methods 2020, 17, 991–1001. [Google Scholar] [CrossRef]
- Riml, C.; Amort, T.; Rieder, D.; Gasser, C.; Lusser, A.; Micura, R. Osmium-mediated transformation of 4-thiouridine to cytidine as key to study RNA dynamics by sequencing. Angew. Chem. Int. Ed. 2017, 56, 13479–13483. [Google Scholar] [CrossRef]
- Lusser, A.; Gasser, C.; Trixl, L.; Piatti, P.; Delazer, I.; Rieder, D.; Bashin, J.; Riml, C.; Amort, T.; Micura, R. Thiouridine-to-cytidine conversion sequencing (TUC-Seq) to measure mRNA transcription and degradation rates. In Eukaryotic RNA Exosome: Methods and Protocols; LaCava, J., Vanacova, S., Eds.; Humana Press Inc.: Innsbruck, Austria, 2020; Volume 2062, pp. 191–211. ISBN 978-1-4939-9821-0. [Google Scholar]
- Chen, Y.; Wu, F.; Chen, Z.; He, Z.; Wei, Q.; Zeng, W.; Chen, K.; Xiao, F.; Yuan, Y.; Weng, X.; et al. Acrylonitrile-mediated nascent RNA sequencing for transcriptome-wide profiling of cellular RNA dynamics. Adv. Sci. 2020, 7, 1900997. [Google Scholar] [CrossRef]
- Boileau, E.; Altmueller, J.; Naarmann-de Vries, I.S.; Dieterich, C. A comparison of metabolic labeling and statistical methods to infer genome-wide dynamics of RNA turnover. Briefings Bioinf. 2021, 22, bbab219. [Google Scholar] [CrossRef]
- Miller, C.; Schwalb, B.; Maier, K.; Schulz, D.; Duemcke, S.; Zacher, B.; Mayer, A.; Sydow, J.; Marcinowski, L.; Doelken, L.; et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 2011, 7, 458. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, W.; Li, X.; Zhang, J.; Chen, S.; Zhang, J.-L.; Yang, L.; Chen, L.-L. The biogenesis of nascent circular RNAs. Cell Rep. 2016, 15, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Kim, B.; Kim, V.N. Emerging roles of RNA modification: m6A and U-tail. Cell 2014, 158, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, L.; Schofield, J.A.; Simon, M.D. Expanding the nucleoside recoding toolkit: Revealing RNA population dynamics with 6-thioguanosine. J. Am. Chem. Soc. 2018, 140, 14567–14570. [Google Scholar] [CrossRef] [PubMed]
- Courvan, M.C.S.; Niederer, R.O.; Vock, I.W.; Kiefer, L.; Gilbert, W.; Simon, M.D. Internally controlled RNA sequencing comparisons using nucleoside recoding chemistry. Nucleic Acids Res. 2022, 50. [Google Scholar] [CrossRef]
- Gasser, C.; Delazer, I.; Neuner, E.; Pascher, K.; Brillet, K.; Klotz, S.; Trixl, L.; Himmelstoss, M.; Ennifar, E.; Rieder, D.; et al. Thioguanosine conversion enables mRNA-lifetime evaluation by RNA sequencing using double metabolic labeling (TUC-seq DUAL). Angew. Chem. Int. Ed. 2020, 59, 6881–6886. [Google Scholar] [CrossRef]
- Gladysz, M.; Andralojc, W.; Czapik, T.; Gdaniec, Z.; Kierzek, R. Thermodynamic and structural contributions of the 6-thioguanosine residue to helical properties of RNA. Sci. Rep. 2019, 9, 4385. [Google Scholar] [CrossRef]
- Khoddami, V.; Cairns, B.R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013, 31, 458–464. [Google Scholar] [CrossRef]
- Koenig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef]
- Finet, O.; Yague-Sanz, C.; Krüger, L.K.; Tran, P.; Migeot, V.; Louski, M.; Nevers, A.; Rougemaille, M.; Sun, J.; Ernst, F.G.M.; et al. Transcription-wide mapping of dihydrouridine reveals that mRNA dihydrouridylation is required for meiotic chromosome segregation. Mol. Cell 2022, 82, 404–419.e9. [Google Scholar] [CrossRef]
- Dai, W.; Li, A.; Yu, N.J.; Nguyen, T.; Leach, R.W.; Wuhr, M.; Kleiner, R.E. Activity-based RNA-modifying enzyme probing reveals DUS3L-mediated dihydrouridylation. Nat. Chem. Biol. 2021, 17, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Bregeon, D.; Pecqueur, L.; Toubdji, S.; Sudol, C.; Lombard, M.; Fontecave, M.; de Crecy-Lagard, V.; Motorin, Y.; Helm, M.; Hamdane, D. Dihydrouridine in the transcriptome: New life for this ancient RNA chemical modification. ACS Chem. Biol. 2022, 17, 1638–1657. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, Z.; Li, T.; Liu, J. Determining RNA Natural Modifications and Nucleoside Analog-Labeled Sites by a Chemical/Enzyme-Induced Base Mutation Principle. Molecules 2023, 28, 1517. https://doi.org/10.3390/molecules28041517
Bao Z, Li T, Liu J. Determining RNA Natural Modifications and Nucleoside Analog-Labeled Sites by a Chemical/Enzyme-Induced Base Mutation Principle. Molecules. 2023; 28(4):1517. https://doi.org/10.3390/molecules28041517
Chicago/Turabian StyleBao, Ziming, Tengwei Li, and Jianzhao Liu. 2023. "Determining RNA Natural Modifications and Nucleoside Analog-Labeled Sites by a Chemical/Enzyme-Induced Base Mutation Principle" Molecules 28, no. 4: 1517. https://doi.org/10.3390/molecules28041517
APA StyleBao, Z., Li, T., & Liu, J. (2023). Determining RNA Natural Modifications and Nucleoside Analog-Labeled Sites by a Chemical/Enzyme-Induced Base Mutation Principle. Molecules, 28(4), 1517. https://doi.org/10.3390/molecules28041517