
Ominoxanthone—The First Xanthone Linearly Fused to a γ-Lactone from Cortinarius ominosus Bidaud Basidiomata. CASE- and DFT-Based Structure Elucidation
 , ,
, , 
Abstract
:1. Introduction
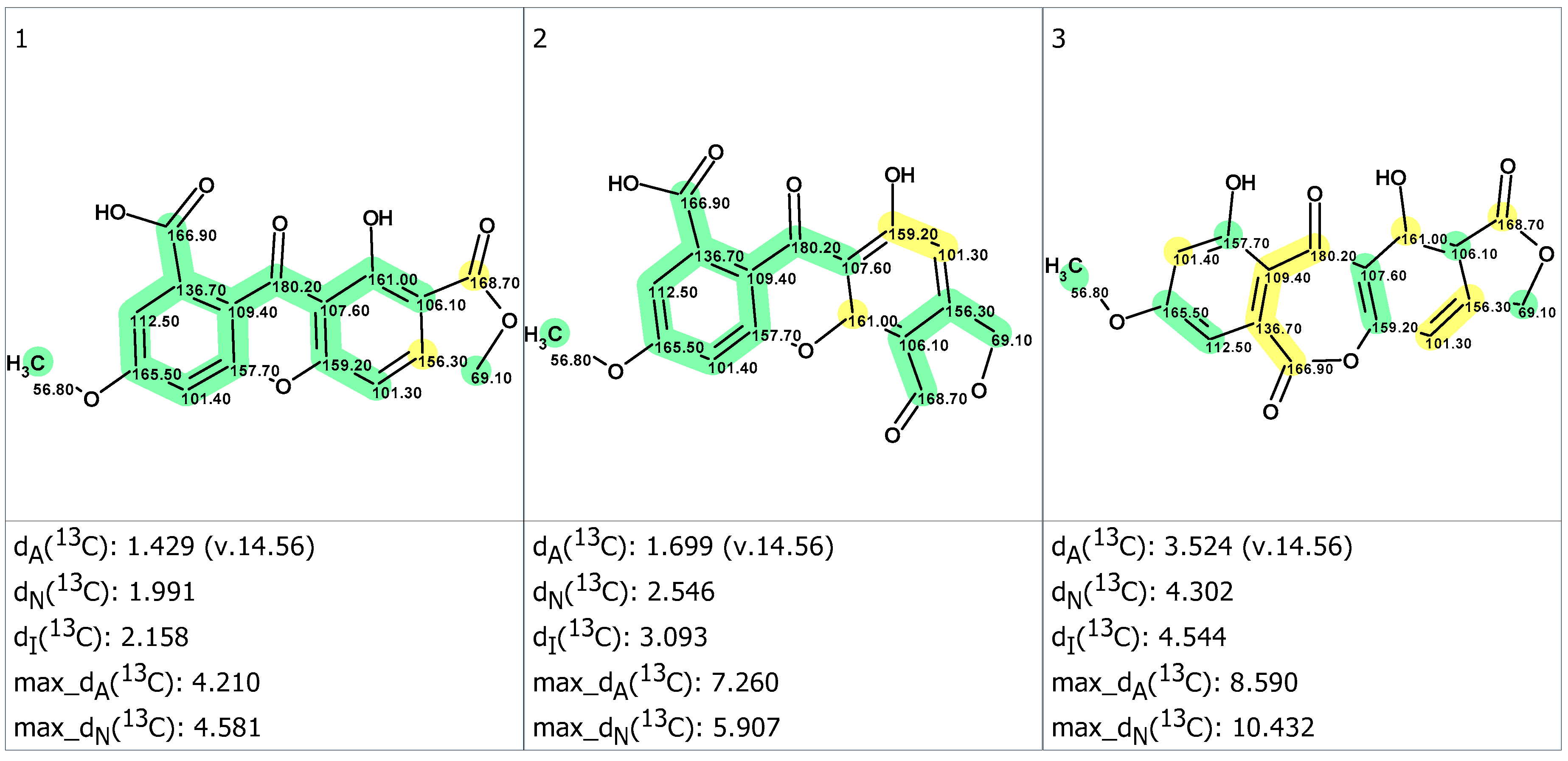
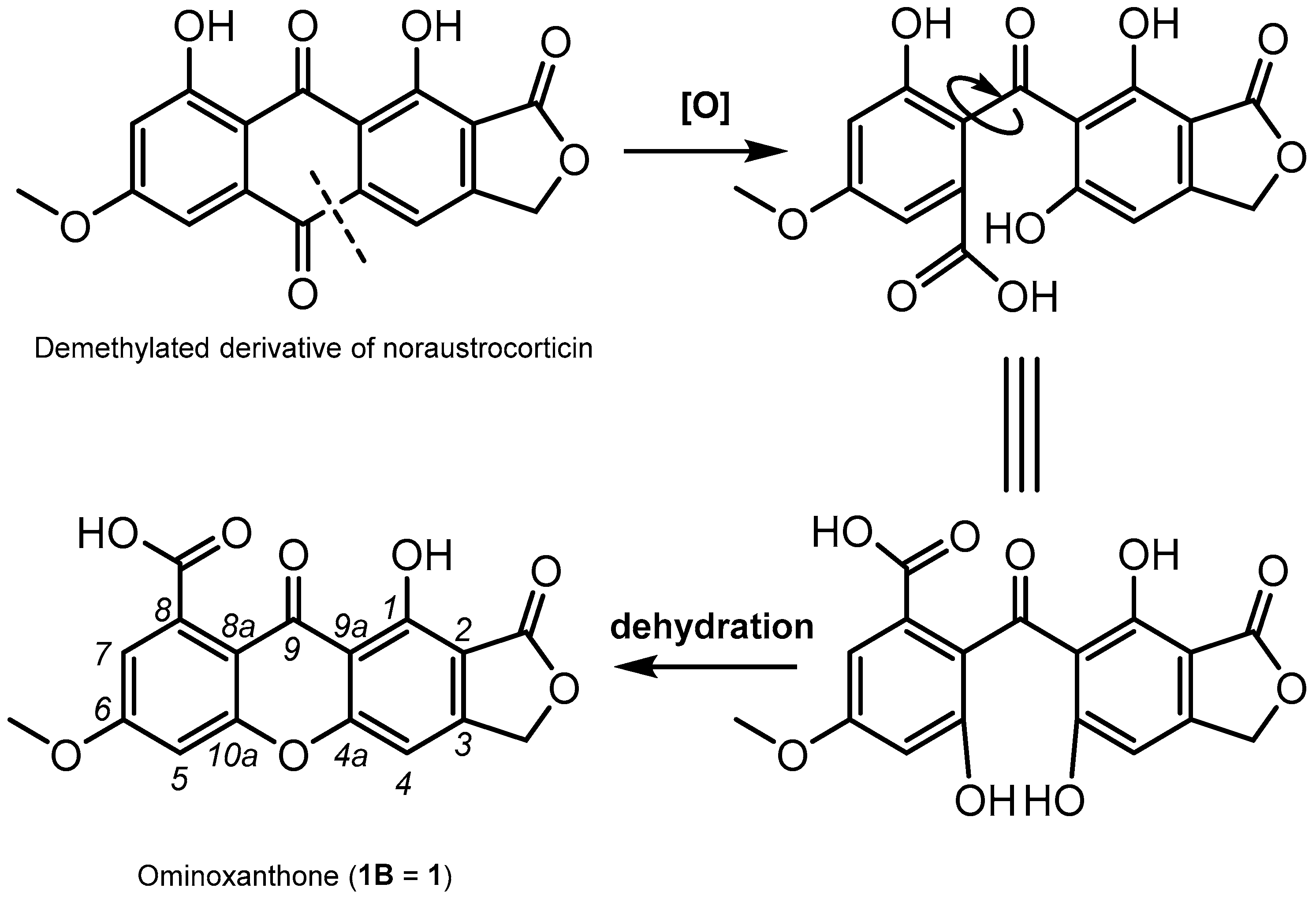
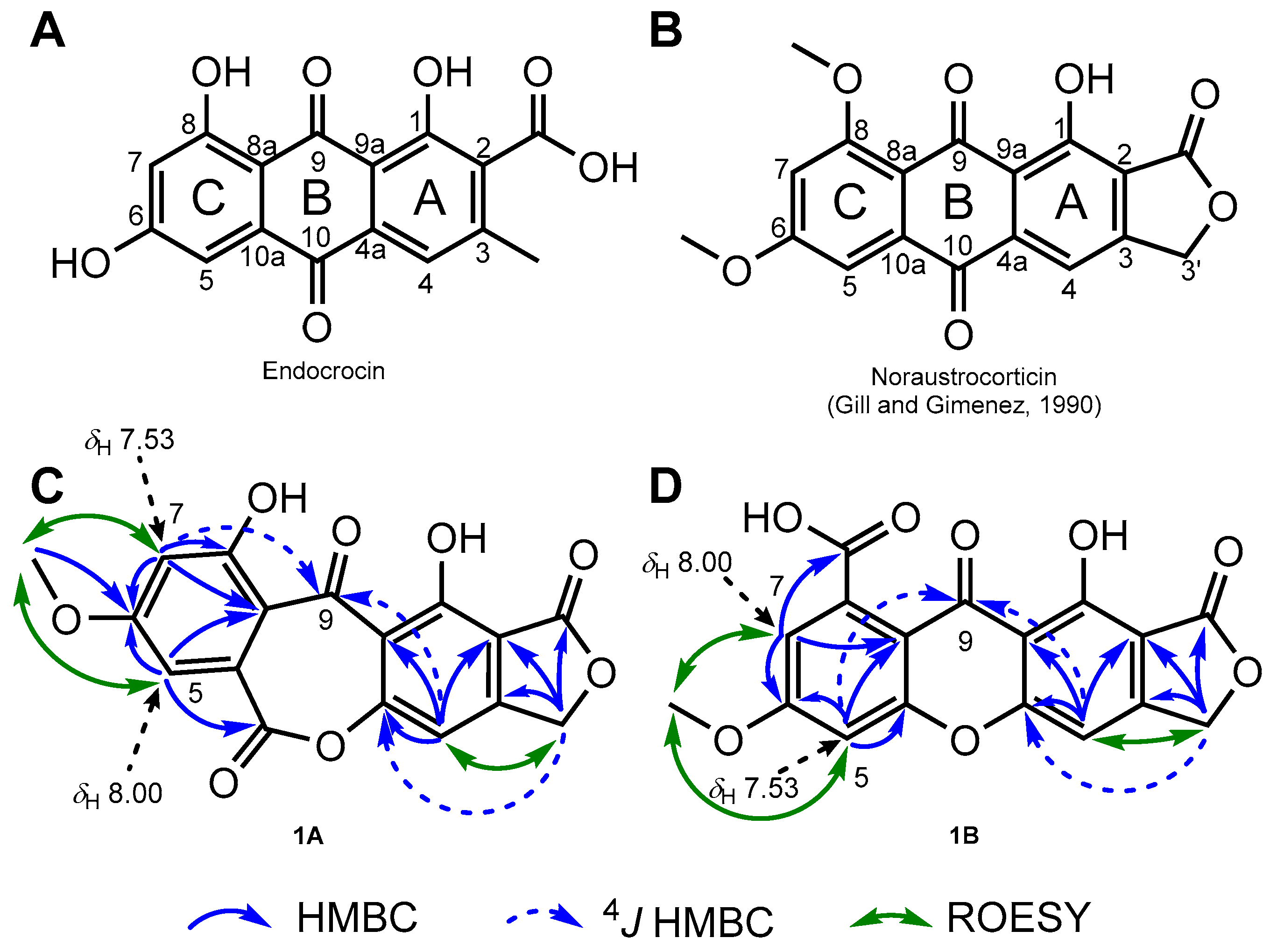
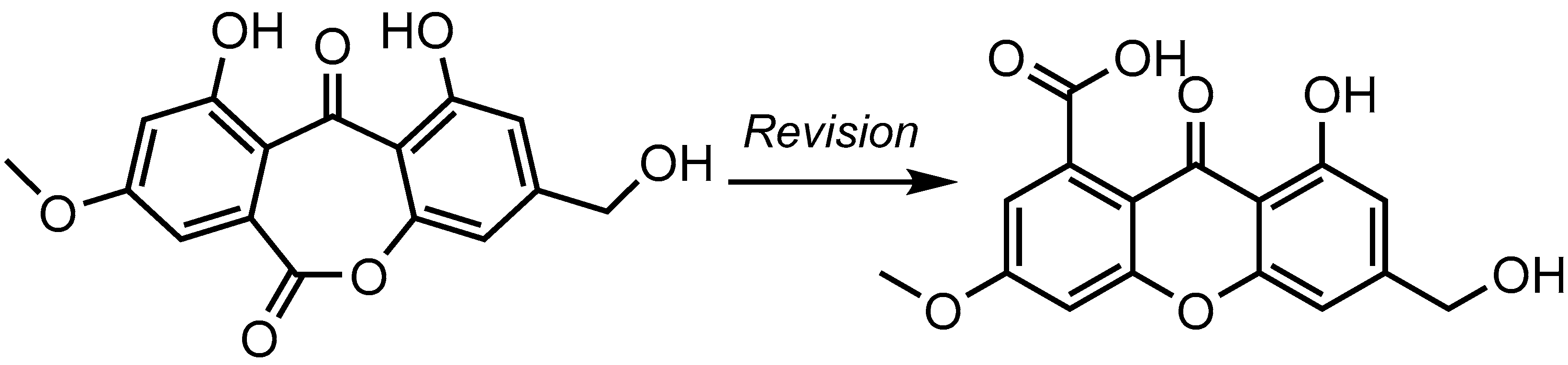
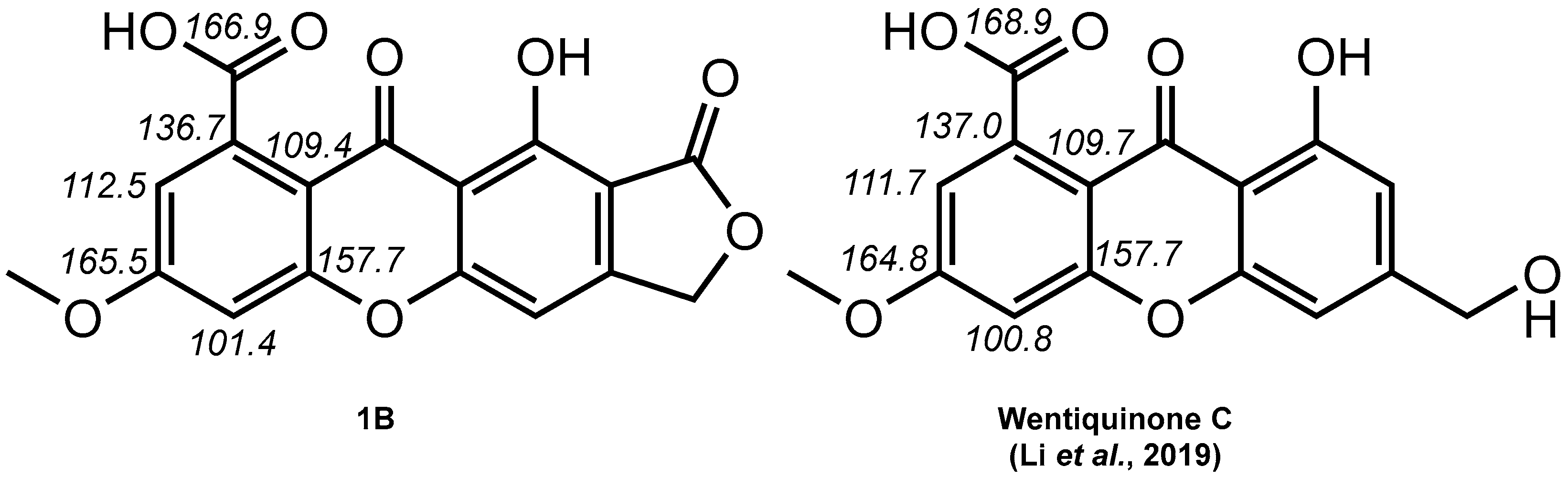
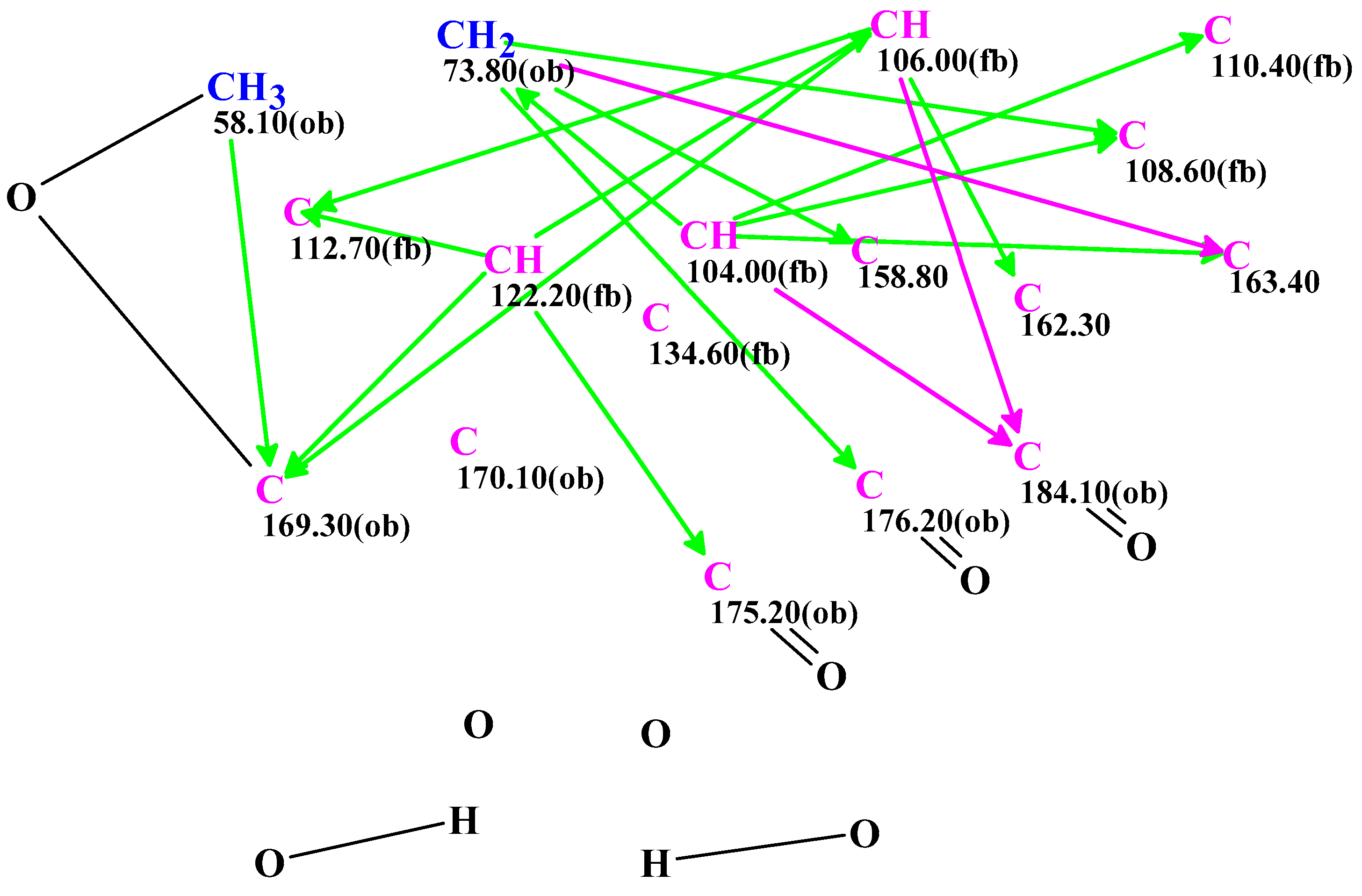
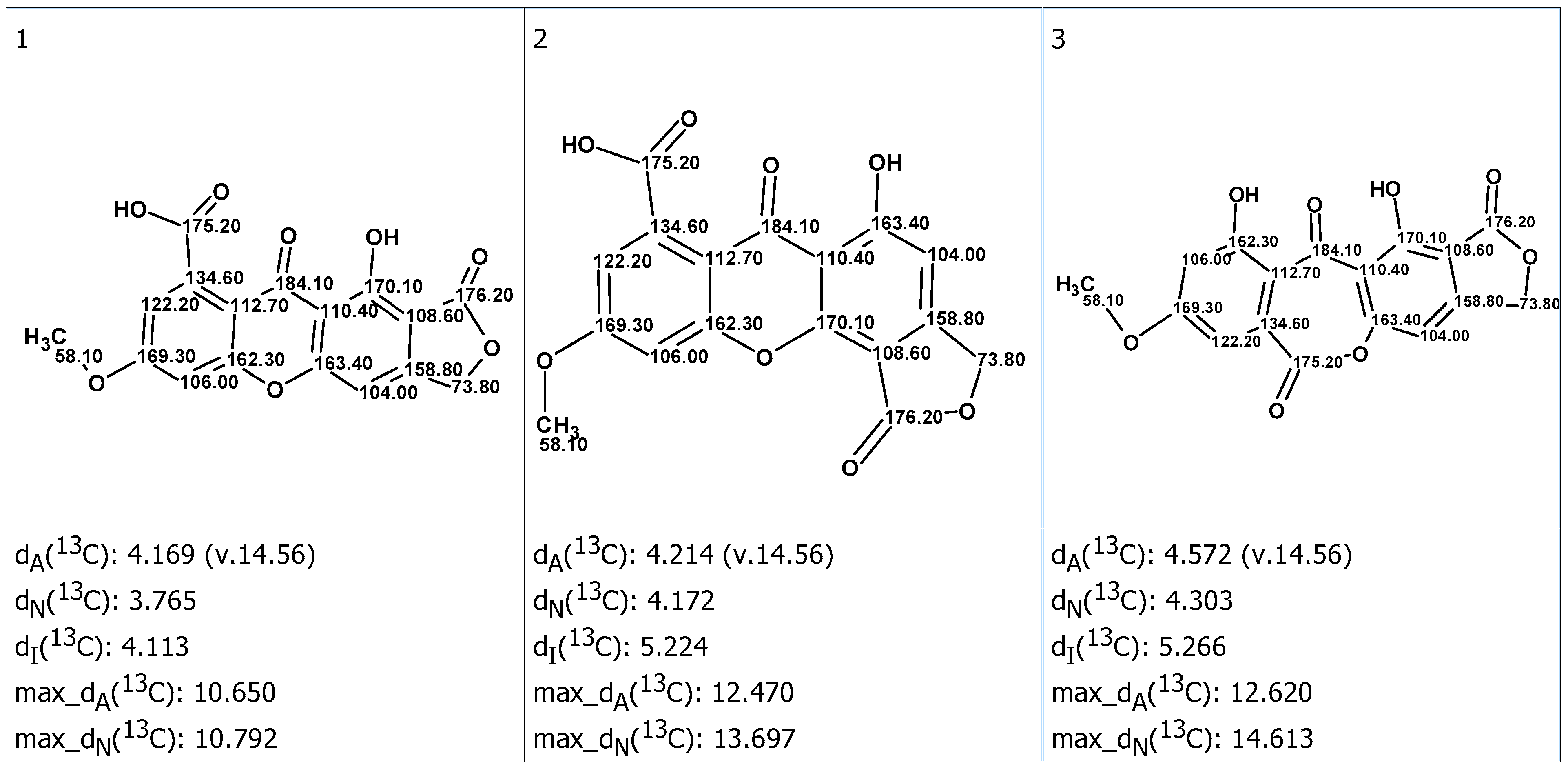
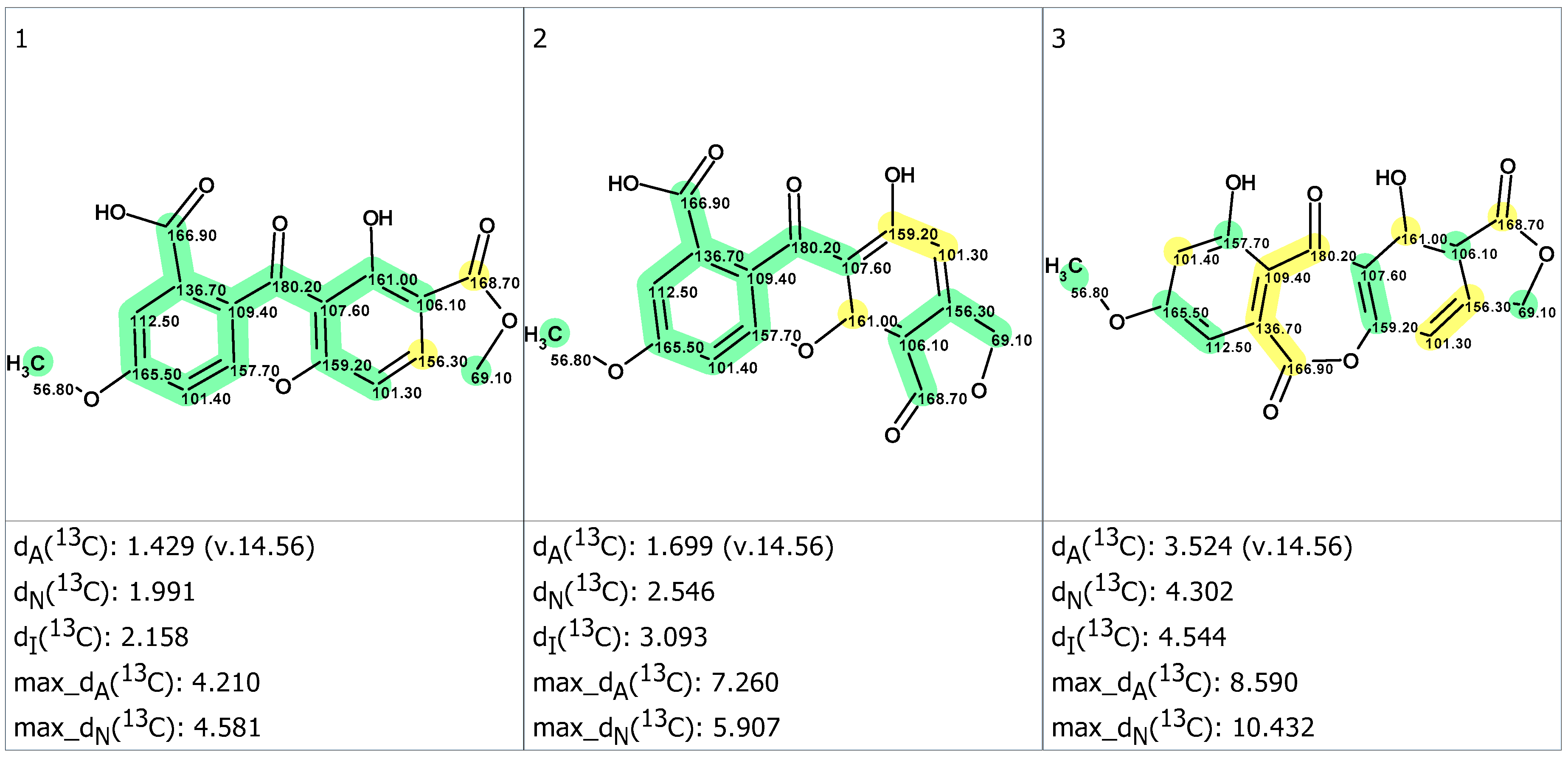
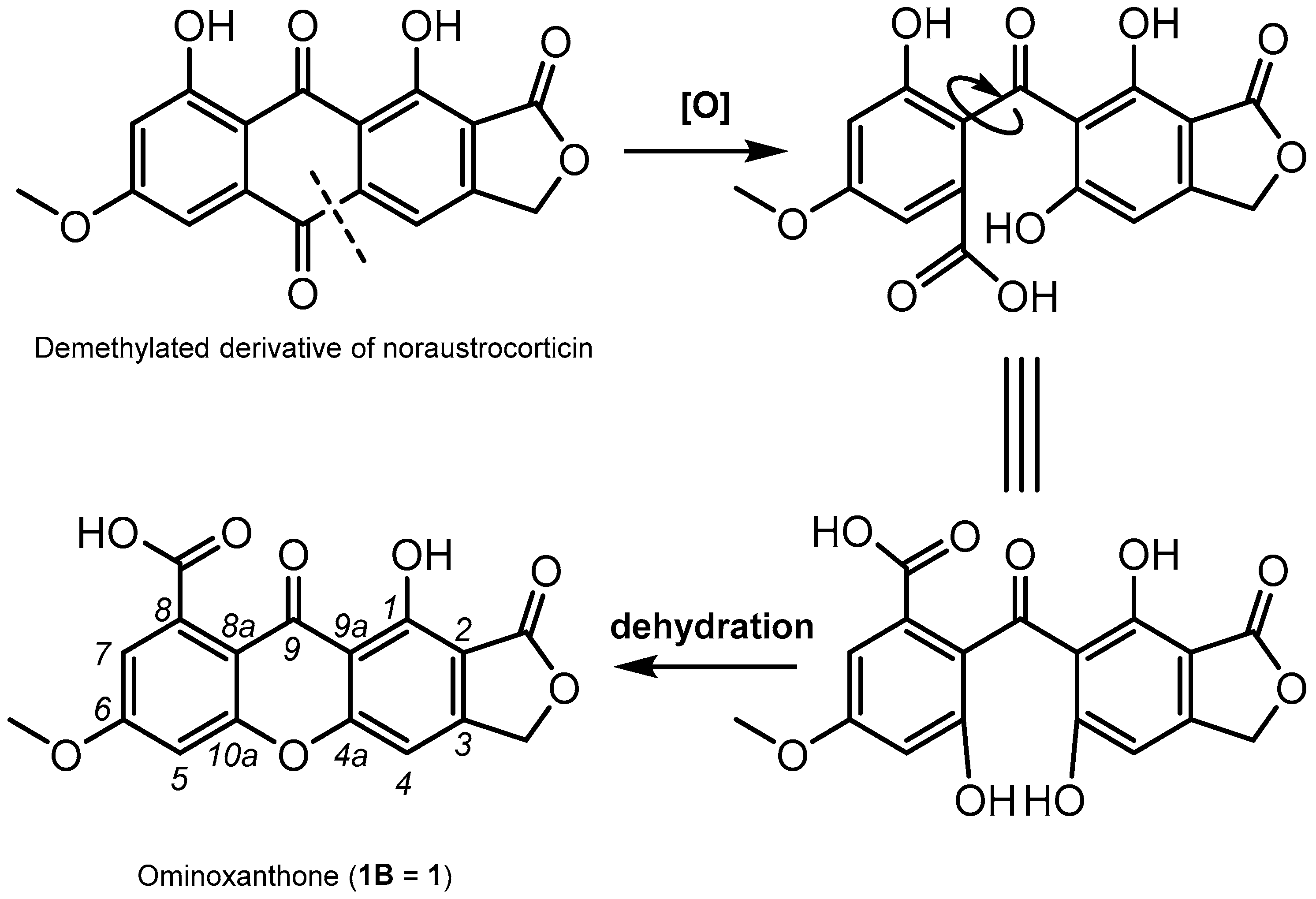
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.4. CASE Analysis
3.5. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Orango-Bourdette, J.O.; Beniddir, M.A.; Otogo N’Nang, E.; Gallard, J.-F.; Ondo, J.P.; Sima Obiang, C.; Rharrabti, S.; Miel, C.; Denis, S.; Obame Engonga, L.C.; et al. Structure Elucidation of a New Lanostane Triterpene from Gabonese Ganoderma orbiforme Fruiting Bodies. Magn. Reson. Chem. 2021, 59, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- DNP. Available online: http://dnp.chemnetbase.com/ (accessed on 30 October 2021).
- Gill, M. Pigments of Australasian Dermocybe Toadstools. Aust. J. Chem. 1995, 48, 1–26. [Google Scholar] [CrossRef]
- Gill, M. Pigments of Fungi (Macromycetes). Nat. Prod. Rep. 2003, 20, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.; Giménez, A. Pigments of Fungi. Part 11.(+)-Austrocorticin, Austrocorticinic Acid, Austrocorticone, and Related Pigments; the First Naturally Occurring Anthraquinones Derived from a Propionate-Triggered Octaketide. J. Chem. Soc. Perkin Trans. 1 1990, 1159–1167. [Google Scholar] [CrossRef]
- Thomas, R. A Biosynthetic Classification of Fungal and Streptomycete Fused-Ring Aromatic Polyketides. ChemBioChem 2001, 2, 612–627. [Google Scholar] [CrossRef]
- Gill, M.; Giménez, A. Pigments of Fungi. Part 17. (S)-(+)-Dermochrysone, (+)-Dermolactone, Dermoquinone, and Related Pigments; New Nonaketides from the Fungus Dermocybe sanguinea(Sensu Cleland). J. Chem. Soc. Perkin Trans. 1 1990, 2585–2591. [Google Scholar] [CrossRef]
- Steglich, W.; Reininger, W. Pilzpigmente, IX. Anthrachinon-Pigmente Aus Dermocybe cinnabarina (Fr.) Wünsche. Chem. Ber. 1972, 105, 2922–2927. [Google Scholar] [CrossRef]
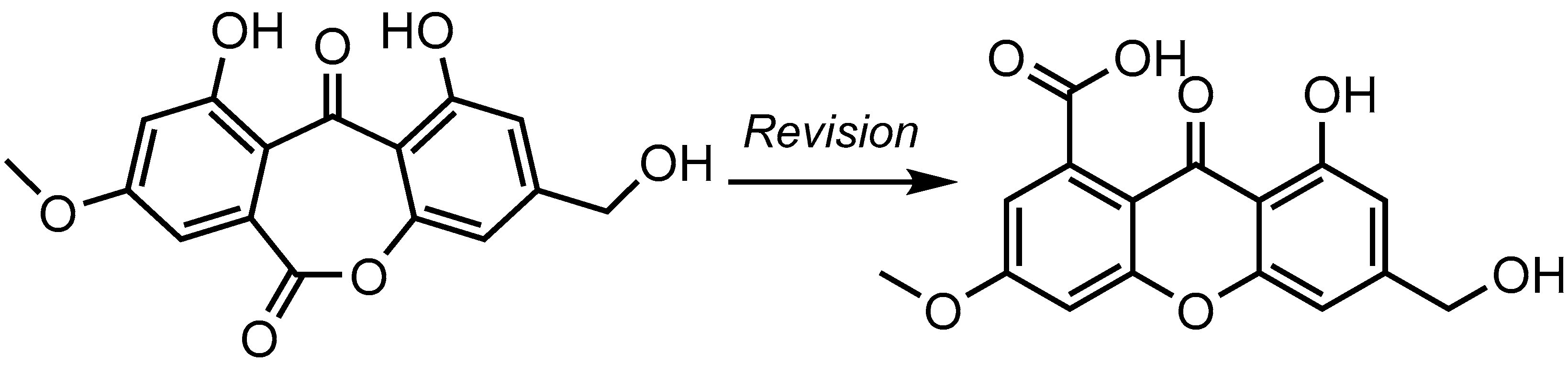
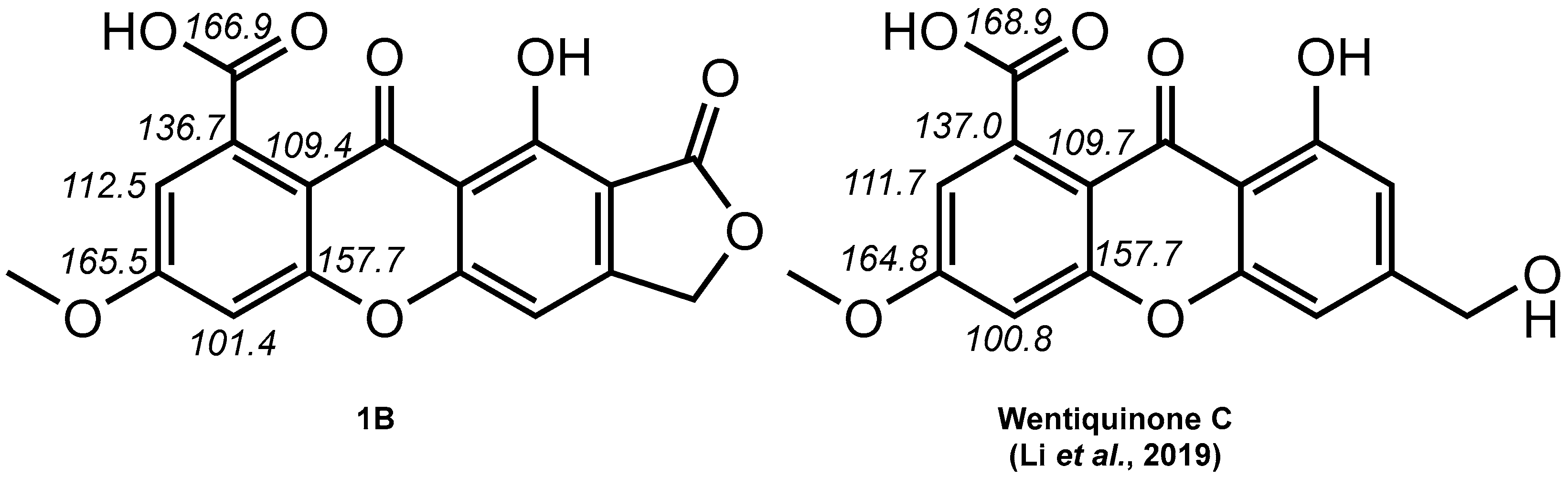
- Li, X.; Li, X.-M.; Wang, B.-G. Structural Revision of Wentiquinone C and Related Congeners from Anthraquinones to Xanthones Using Chemical Derivatization and NMR Analysis. Mar. Drugs 2018, 17, 8. [Google Scholar] [CrossRef]
- Sun, H.-F.; Li, X.-M.; Meng, L.-H.; Cui, C.-M.; Gao, S.-S.; Li, C.-S.; Wang, B.-G. Two New Secoanthraquinone Derivatives from the Marine-Derived Endophytic Fungus Aspergillus wentii EN-48. Helv. Chim. Acta 2013, 96, 458–462. [Google Scholar] [CrossRef]
- Li, X.; Li, X.-M.; Xu, G.-M.; Li, C.-S.; Wang, B.-G. Antioxidant Metabolites from Marine Alga-Derived Fungus Aspergillus wentii EN-48. Phytochem. Lett. 2014, 7, 120–123. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, Q.L.; Cheng, P.; Liu, C.L.; Lu, Z.Y.; Li, W.; Wang, T.T.; Lu, Y.H.; Tan, R.X.; Ge, H.M. Aromatic Polyketides from a Caterpillar Associated Alternaria sp. Tetrahedron Lett. 2017, 58, 3069–3072. [Google Scholar] [CrossRef]
- Abdissa, N.; Heydenreich, M.; Midiwo, J.O.; Ndakala, A.; Majer, Z.; Neumann, B.; Stammler, H.-G.; Sewald, N.; Yenesew, A. A Xanthone and a Phenylanthraquinone from the Roots of Bulbine frutescens, and the Revision of Six Seco-Anthraquinones into Xanthones. Phytochem. Lett. 2014, 9, 67–73. [Google Scholar] [CrossRef]
- Yang, B.-J.; Chen, G.-D.; Li, Y.-J.; Hu, D.; Guo, L.-D.; Xiong, P.; Gao, H. A New Xanthone Glycoside from the Endolichenic Fungus Sporormiella irregularis. Molecules 2016, 21, 764. [Google Scholar] [CrossRef] [PubMed]
- White, K.N.; Amagata, T.; Oliver, A.G.; Tenney, K.; Wenzel, P.J.; Crews, P. Structure Revision of Spiroleucettadine, a Sponge Alkaloid with a Bicyclic Core Meager in H-Atoms. J. Org. Chem. 2008, 73, 8719–8722. [Google Scholar] [CrossRef]
- Sidebottom, P.J. Crews’ Rule—Still Useful but Often Misquoted. Magn. Reson. Chem. 2021, 59, 752–753. [Google Scholar] [CrossRef]
- Elyashberg, M.; Argyropoulos, D. Computer Assisted Structure Elucidation (CASE): Current and Future Perspectives. Magn. Reson. Chem. 2021, 59, 669–690. [Google Scholar] [CrossRef]
- ACD. Structure Elucidator; V.14.56; Advanced Chemistry Development Inc.: Toronto, ON, Canada, 2020. [Google Scholar]
- Elyashberg, M.E.; Williams, A.; Blinov, K. Contemporary Computer-Assisted Approaches to Molecular Structure Elucidation; Royal Society of Chemistry: Cambridge, UK, 2011. [Google Scholar]
- Elyashberg, M.E.; Williams, A.J.; Martin, G.E. Computer-Assisted Structure Verification and Elucidation Tools in NMR-Based Structure Elucidation. Prog. Nucl. Magn. Reson. Spectrosc. 2008, 53, 1–104. [Google Scholar] [CrossRef]
- Buevich, A.V.; Elyashberg, M.E. Synergistic Combination of CASE Algorithms and DFT Chemical Shift Predictions: A Powerful Approach for Structure Elucidation, Verification, and Revision. J. Nat. Prod. 2016, 79, 3105–3116. [Google Scholar] [CrossRef]
- Buevich, A.V.; Elyashberg, M.E. Towards Unbiased and More Versatile NMR-Based Structure Elucidation: A Powerful Combination of CASE Algorithms and DFT Calculations. Magn. Reson. Chem. 2018, 56, 493–504. [Google Scholar] [CrossRef]
- Ezell, E.L.; Smith, L.L. 1H-and 13C-NMR Spectra of Camptothecin and Derivatives. J. Nat. Prod. 1991, 54, 1645–1650. [Google Scholar] [CrossRef]
- Räisänen, R. Fungal Colorants in Applications—Focus on Cortinarius Species. Color. Technol. 2019, 135, 22–31. [Google Scholar] [CrossRef]
- Le Pogam, P.; Boustie, J. Xanthones of Lichen Source: A 2016 Update. Molecules 2016, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Kopanski, L.; Klaar, M.; Steglich, W. Pilzpigmente, 40. Leprocybin, Der Fluoreszenzstoff von Cortinarius cotoneus Und Verwandten Leprocyben (Agaricales). Liebigs Ann. Chem. 1982, 1982, 1280–1296. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- CHESHIRE. CHEmical SHift REpository with Coupling Constants Added Too. Available online: http://cheshirenmr.info (accessed on 5 January 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
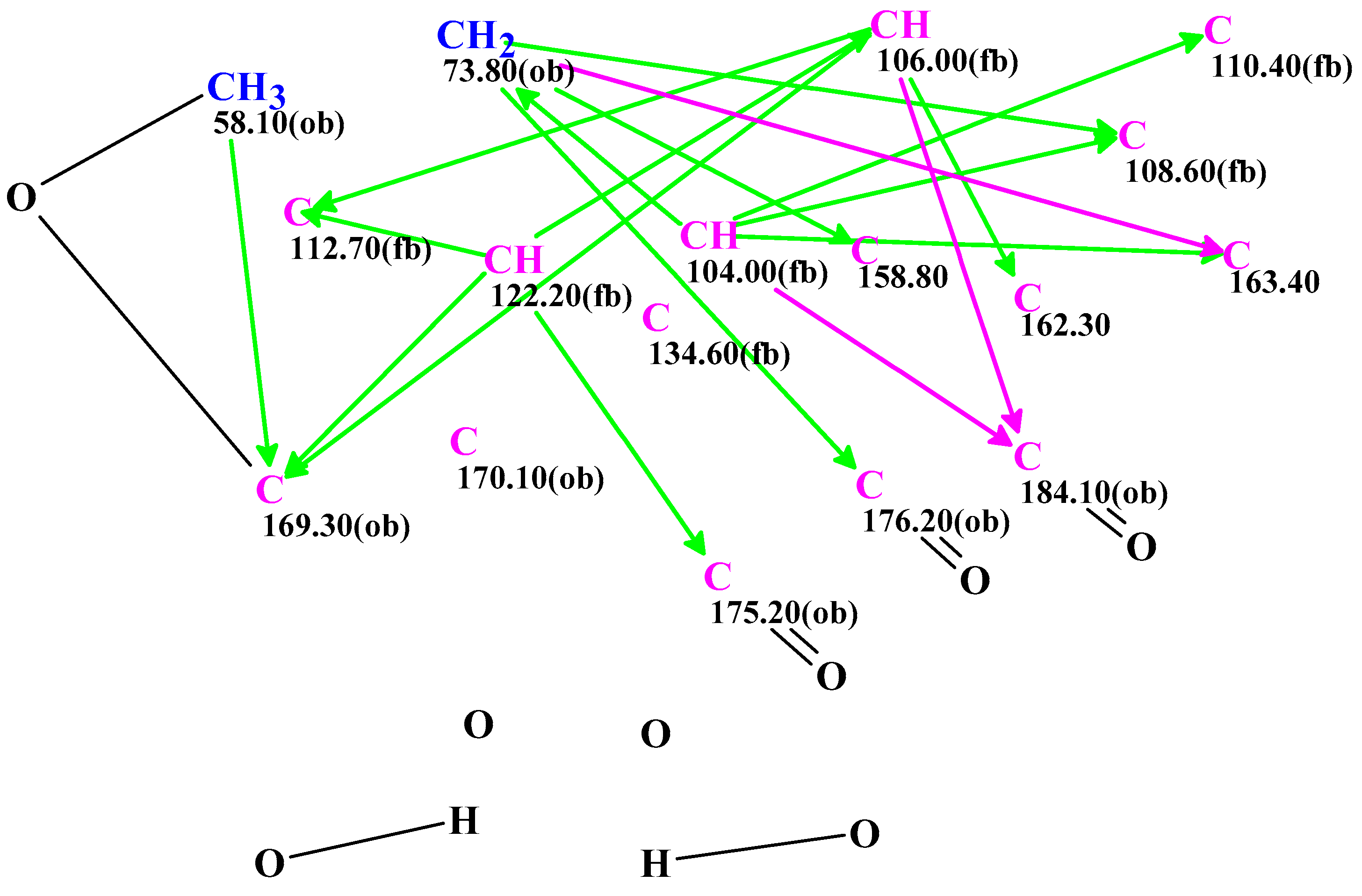
| 1 (TFA-d) | 1 (DMSO-d6) a | |||
|---|---|---|---|---|
| position | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC |
| 1 | 170.1 | 161.0 | ||
| 2 | 108.6 | 106.1 | ||
| 3 | 158.8 | 156.3 | ||
| 4 | 7.47 (1H, s) | 104.0 | 7.25 (1H, br s) | 101.3 |
| 4a | 163.4 | 159.2 | ||
| 5 | 7.53 (1H, d, 2.2) | 106.0 | 7.30 (1H, br s) | 101.4 |
| 6 | 169.3 | 165.5 | ||
| 7 | 8.00 (1H, d, 2.2) | 122.2 | 7.07 (1H, br s) | 112.5 |
| 8 | 134.6 | 136.7 | ||
| 8a | 112.7 | 109.4 | ||
| 9 | 184.1 | 180.2 | ||
| 9a | 110.4 | 107.6 | ||
| 10a | 162.3 | 157.7 | ||
| 2-COO | 176.2 | 168.7 | ||
| 3-CH2O | 5.82 (2H, s) | 73.8 | 5.42 (2H, s) | 69.1 |
| 6-OCH3 | 4.34 (3H, s) | 58.1 | 3.98 (3H, s) | 56.8 |
| 8-COOH | 175.2 | 166.9 | ||
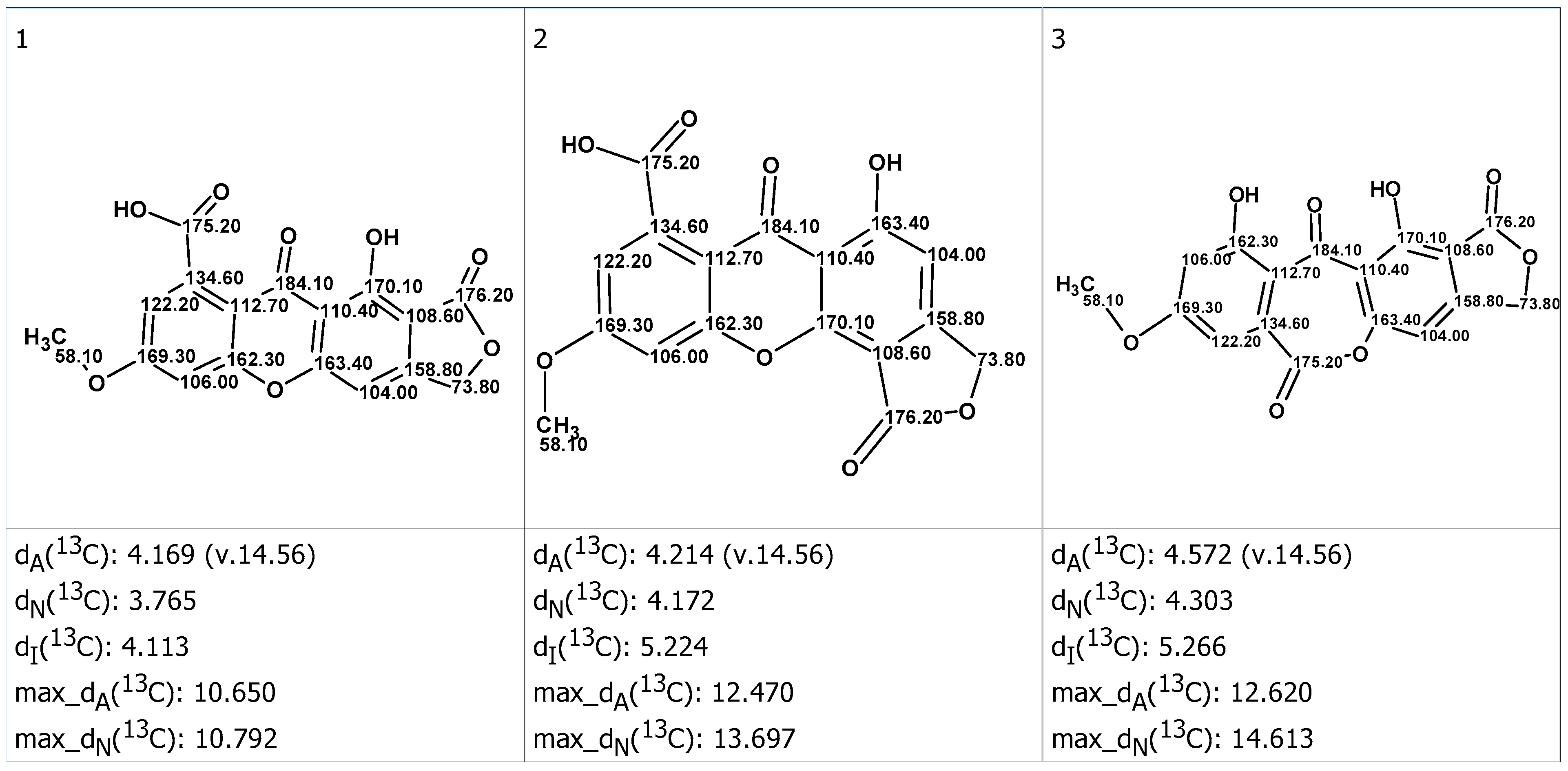
| Carbons | Exp. (DMSO) | 1 | 2 | 3 |
|---|---|---|---|---|
| 1 | 161.0 | 161.46 | 151.87 | 161.23 |
| 2 | 106.1 | 105.27 | 102.44 | 108.32 |
| 3 | 156.3 | 156.89 | 157.33 | 157.22 |
| 4 | 101.3 | 99.43 | 103.36 | 103.88 |
| 4a | 159.2 | 158.10 | 167.07 | 154.57 |
| 5 | 101.4 | 99.28 | 99.24 | 110.08 |
| 6 | 165.5 | 164.28 | 164.21 | 163.65 |
| 7 | 112.5 | 115.09 | 114.93 | 109.95 |
| 8 | 136.7 | 137.15 | 136.96 | 128.13 |
| 8a | 109.4 | 109.32 | 109.57 | 114.74 |
| 9 | 180.2 | 177.70 | 176.91 | 191.89 |
| 9a | 107.6 | 107.02 | 107.32 | 115.48 |
| 10a | 157.7 | 156.47 | 155.91 | 163.10 |
| 2-COO | 168.7 | 166.94 | 166.62 | 167.46 |
| 3-CH2O | 69.1 | 68.39 | 68.68 | 68.48 |
| 6-OCH3 | 56.8 | 53.95 | 54.17 | 53.75 |
| 8-COOH | 166.9 | 169.15 | 169.18 | 162.13 |
| RMSD, ppm | 1.66 | 3.59 | 5.48 | |
| Max_dev, ppm | 2.83 | 9.12 | 11.64 | |
| R2 | 0.99921 | 0.99558 | 0.98980 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trac, A.; Issaad, C.; Beniddir, M.A.; Bellanger, J.-M.; Gallard, J.-F.; Buevich, A.V.; Elyashberg, M.E.; Le Pogam, P. Ominoxanthone—The First Xanthone Linearly Fused to a γ-Lactone from Cortinarius ominosus Bidaud Basidiomata. CASE- and DFT-Based Structure Elucidation. Molecules 2023, 28, 1557. https://doi.org/10.3390/molecules28041557
Trac A, Issaad C, Beniddir MA, Bellanger J-M, Gallard J-F, Buevich AV, Elyashberg ME, Le Pogam P. Ominoxanthone—The First Xanthone Linearly Fused to a γ-Lactone from Cortinarius ominosus Bidaud Basidiomata. CASE- and DFT-Based Structure Elucidation. Molecules. 2023; 28(4):1557. https://doi.org/10.3390/molecules28041557
Chicago/Turabian StyleTrac, Alice, Célia Issaad, Mehdi A. Beniddir, Jean-Michel Bellanger, Jean-François Gallard, Alexei V. Buevich, Mikhail E. Elyashberg, and Pierre Le Pogam. 2023. "Ominoxanthone—The First Xanthone Linearly Fused to a γ-Lactone from Cortinarius ominosus Bidaud Basidiomata. CASE- and DFT-Based Structure Elucidation" Molecules 28, no. 4: 1557. https://doi.org/10.3390/molecules28041557
APA StyleTrac, A., Issaad, C., Beniddir, M. A., Bellanger, J.-M., Gallard, J.-F., Buevich, A. V., Elyashberg, M. E., & Le Pogam, P. (2023). Ominoxanthone—The First Xanthone Linearly Fused to a γ-Lactone from Cortinarius ominosus Bidaud Basidiomata. CASE- and DFT-Based Structure Elucidation. Molecules, 28(4), 1557. https://doi.org/10.3390/molecules28041557