
Organogermanium Analogues of Alkenes, Alkynes, 1,3-Dienes, Allenes, and Vinylidenes


Abstract
:1. Introduction
2. Heavy Analogues of Alkenes
2.1. Homonuclear Derivatives
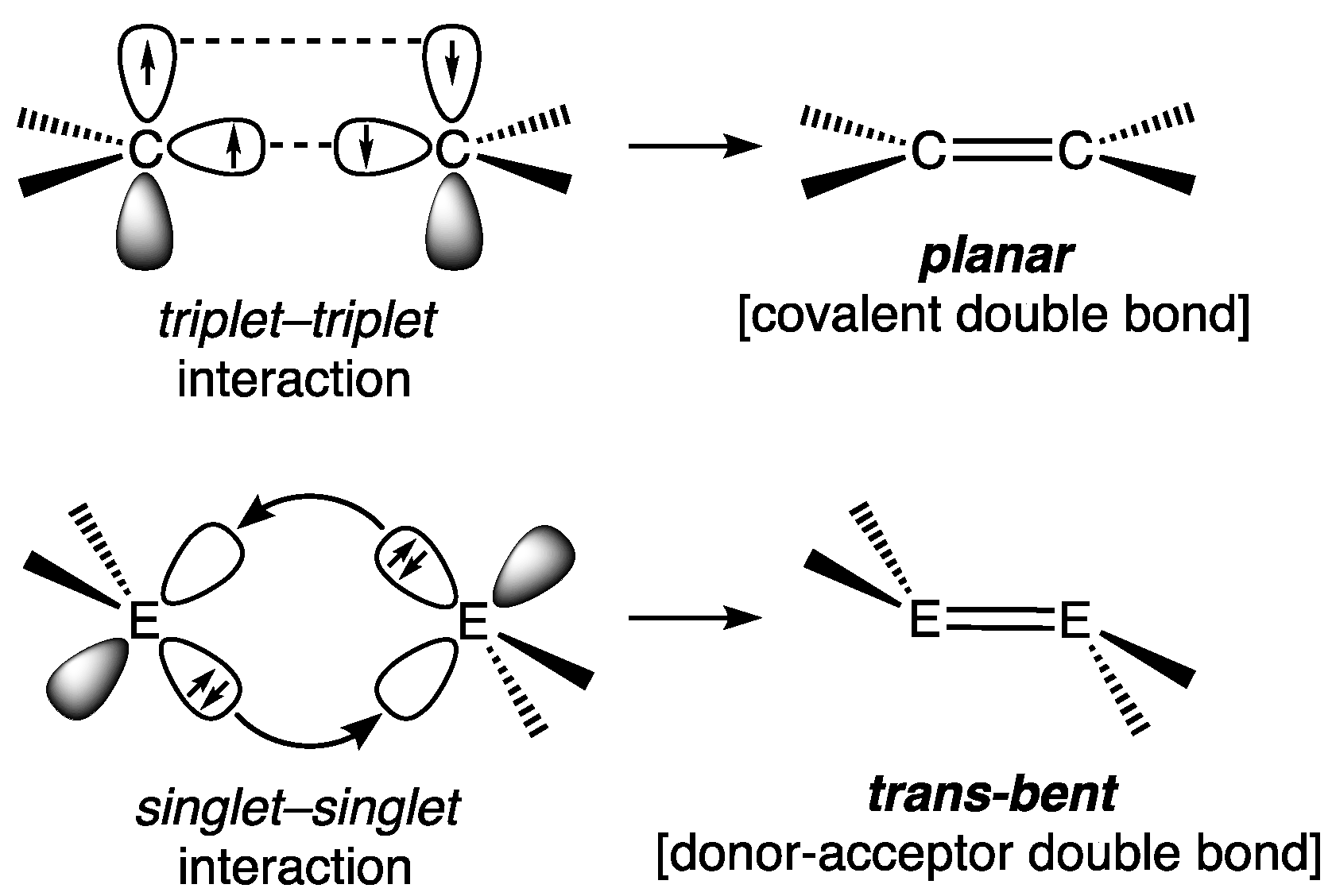
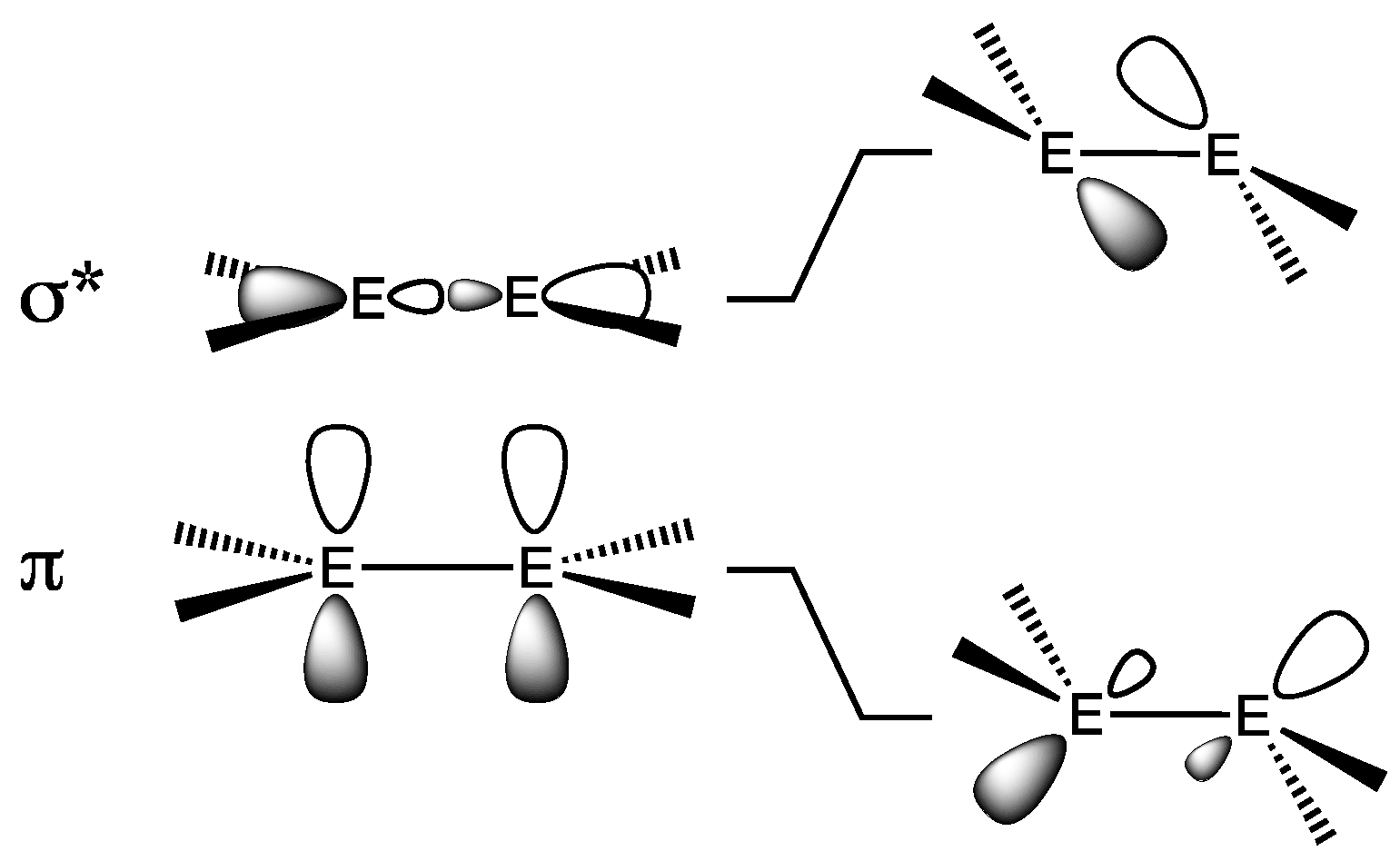
2.1.1. Digermenes >Ge=Ge<
Acyclic Digermenes
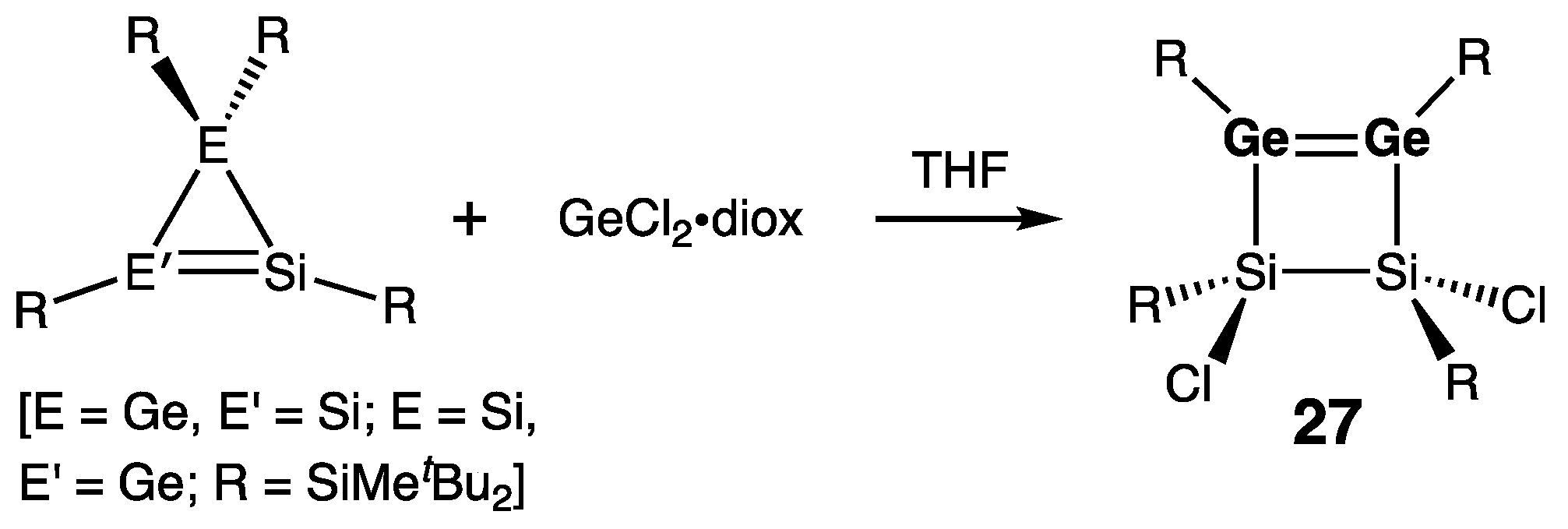
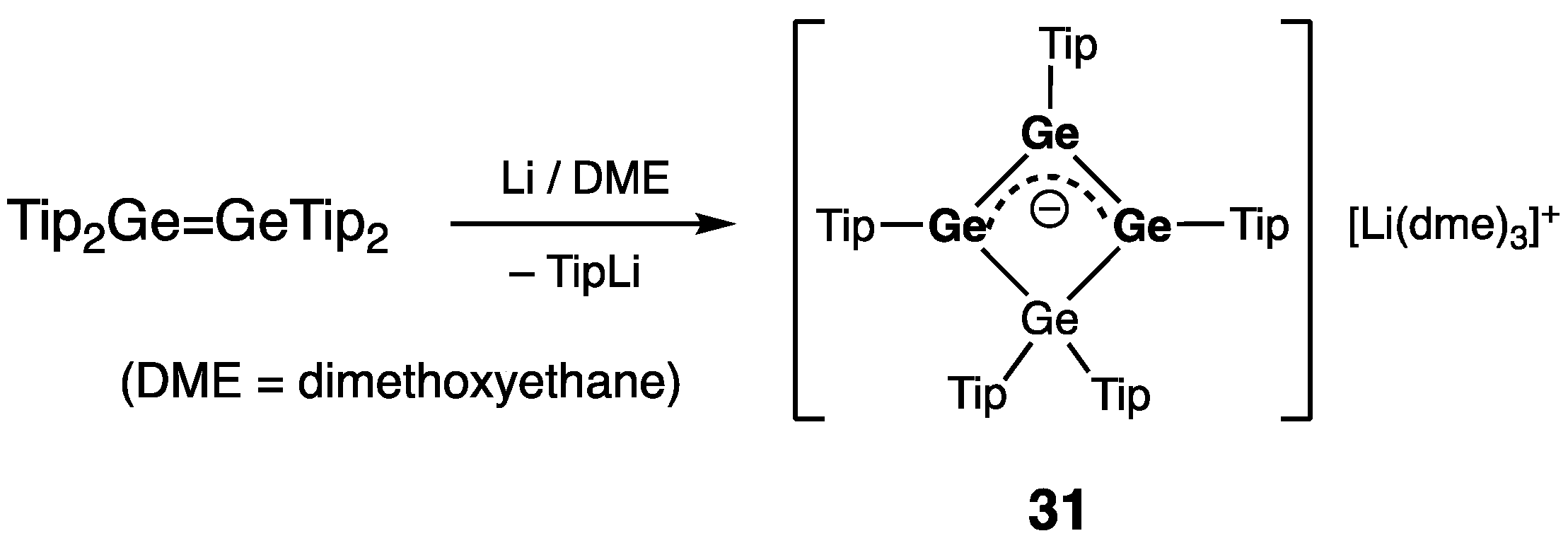
Cyclic Digermenes
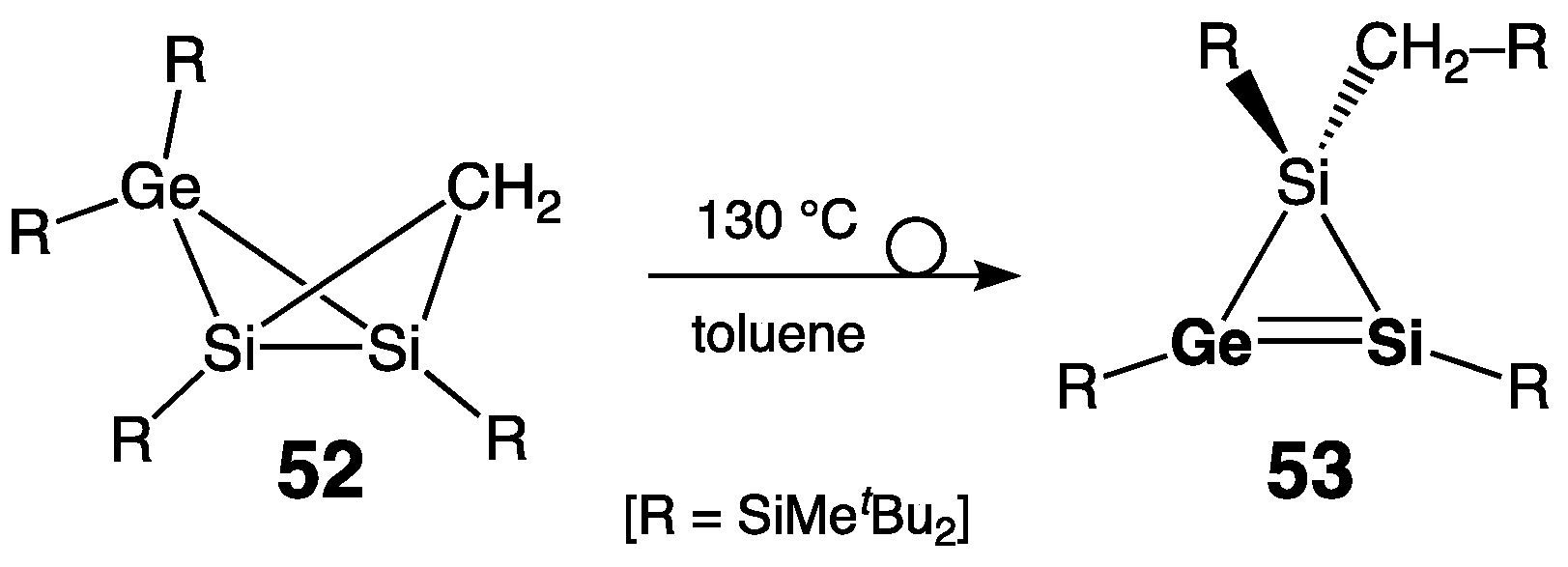
- Four-Membered Ring Compounds
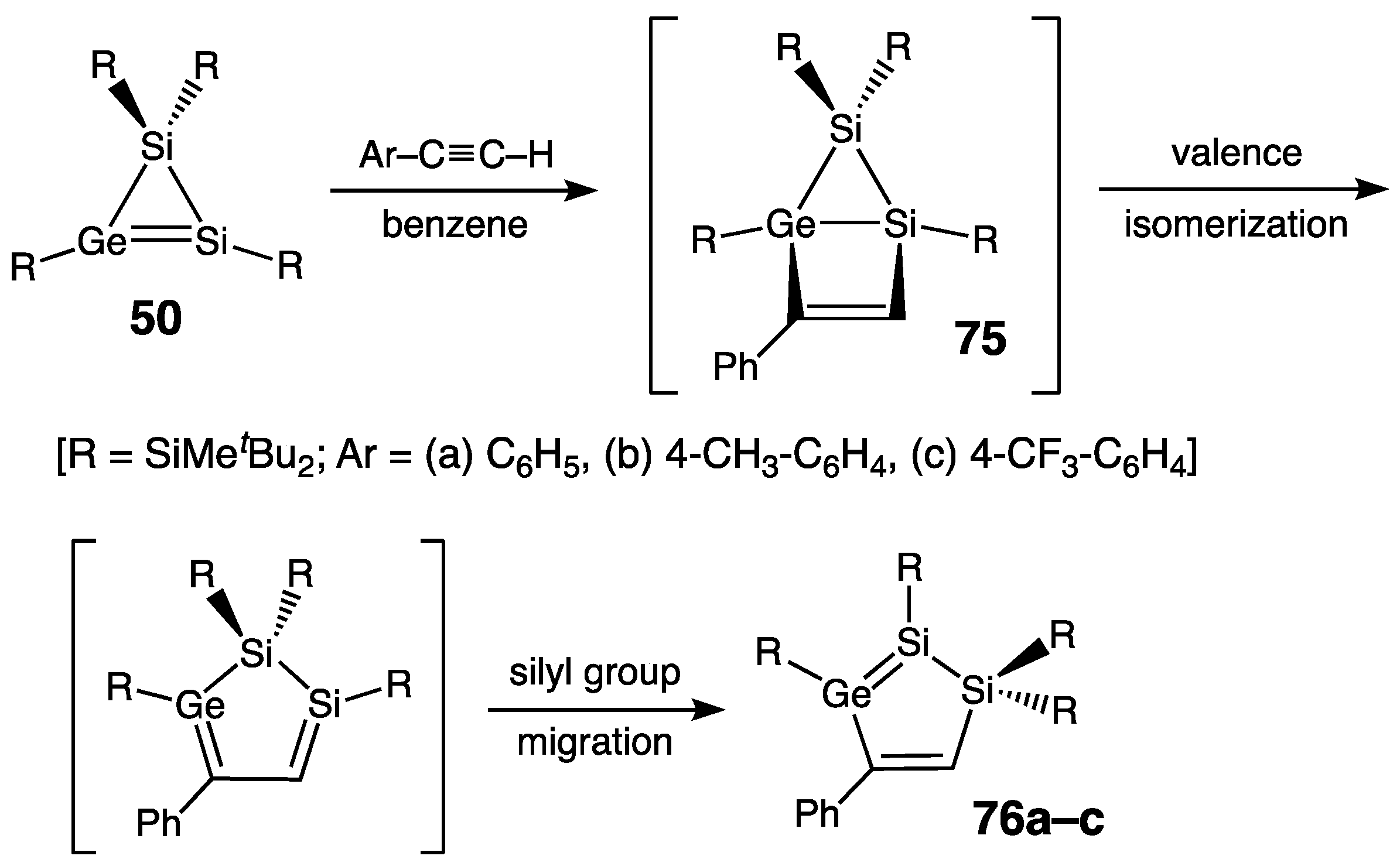
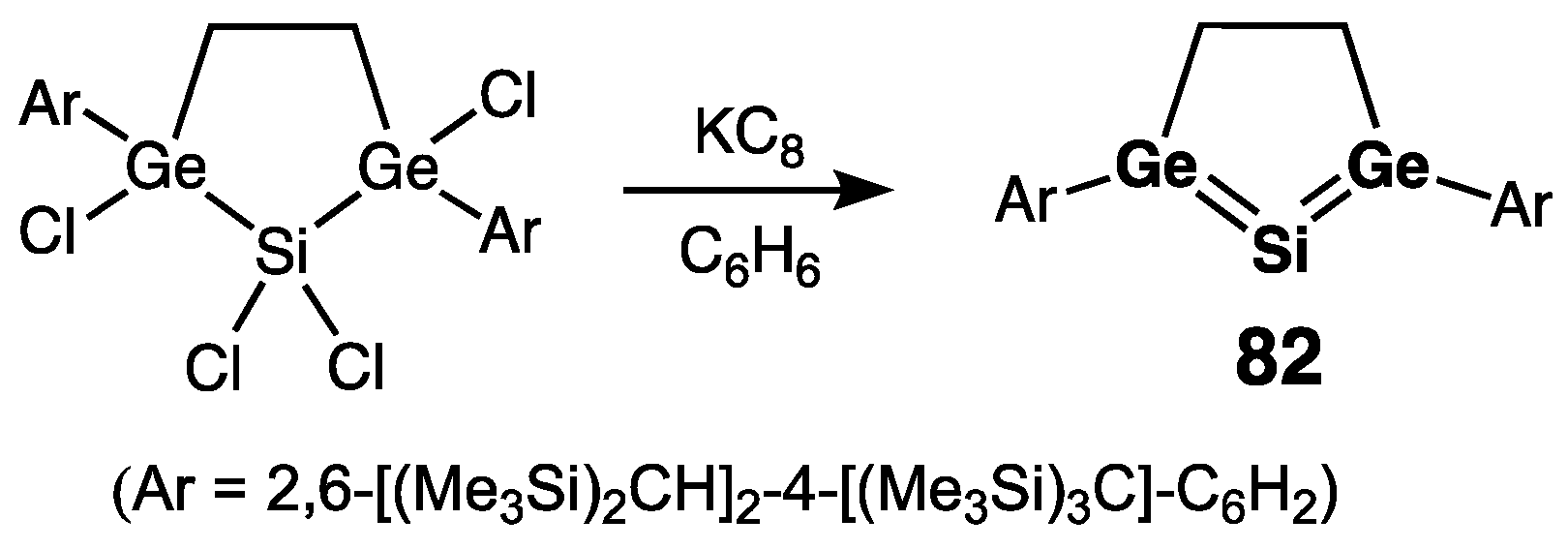
- Five-Membered Ring Compounds
- Six-Membered Ring Compounds
2.2. Heteronuclear Derivatives
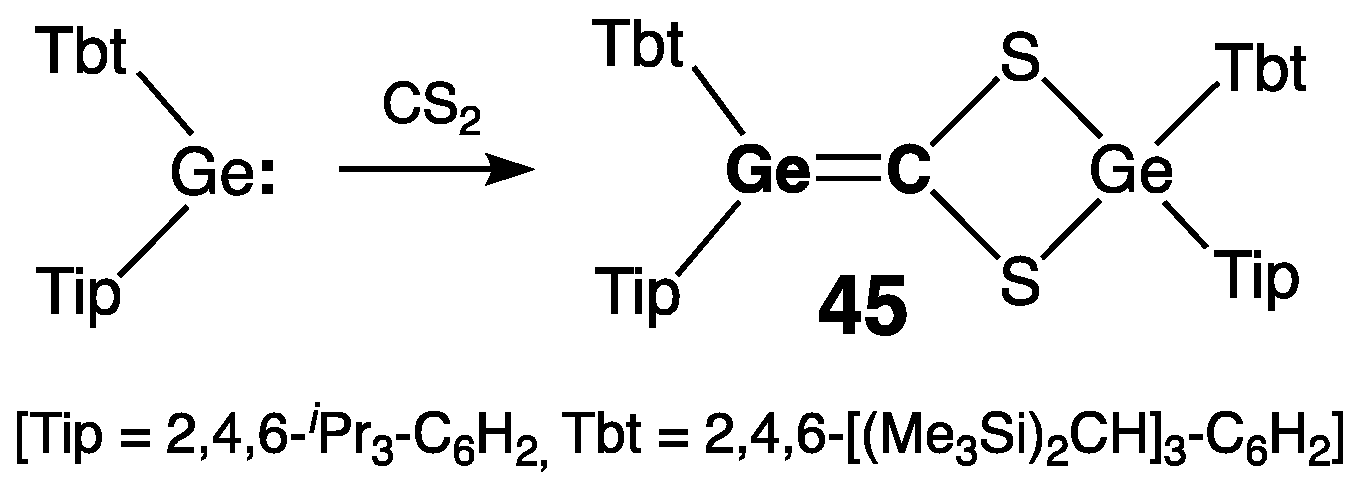
2.2.1. Germenes >Ge=C<
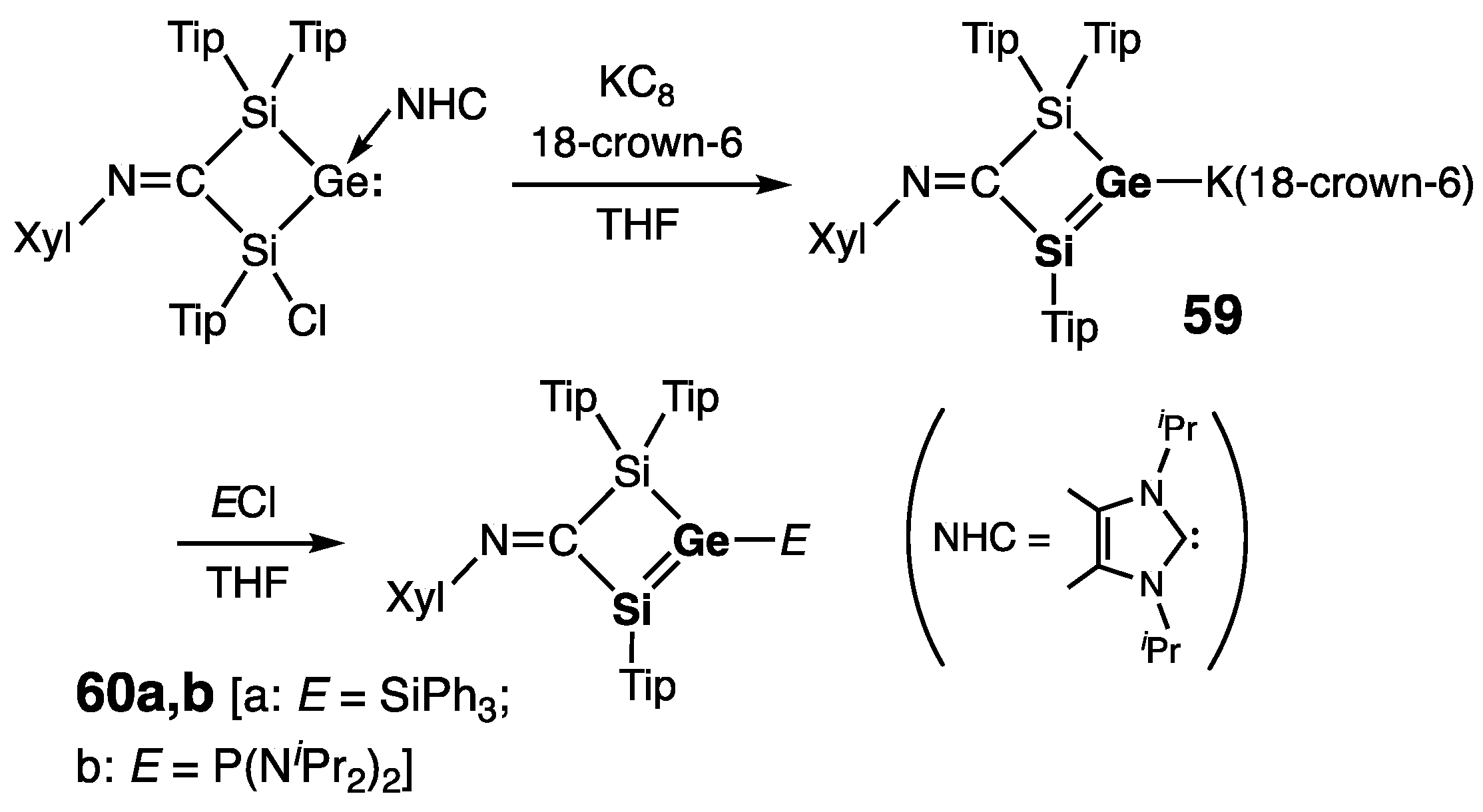
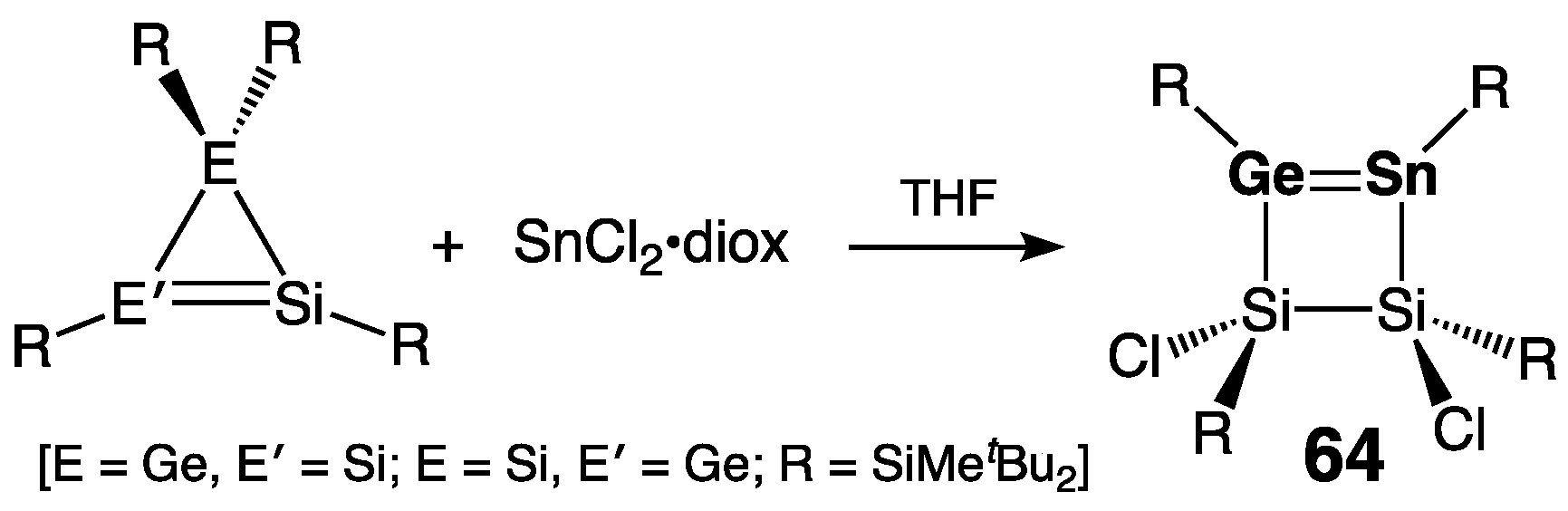
2.2.2. Silagermenes >Si=Ge<
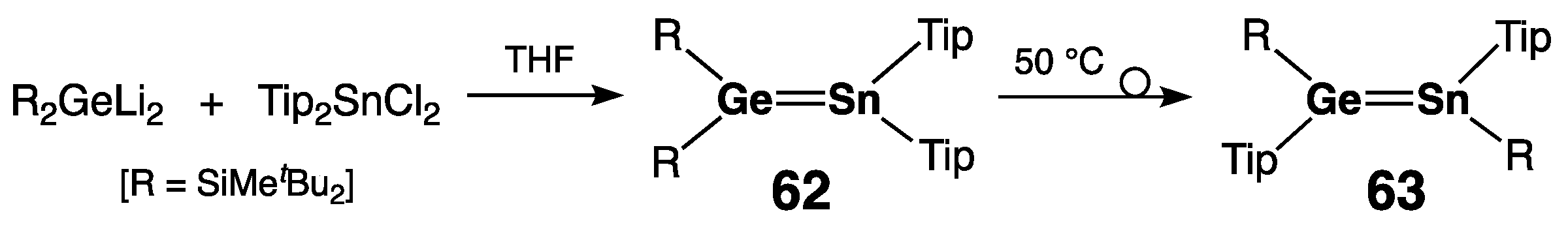
2.2.3. Germastannenes >Ge=Sn<
3. Heavy Analogues of Alkynes
3.1. Homonuclear Derivatives
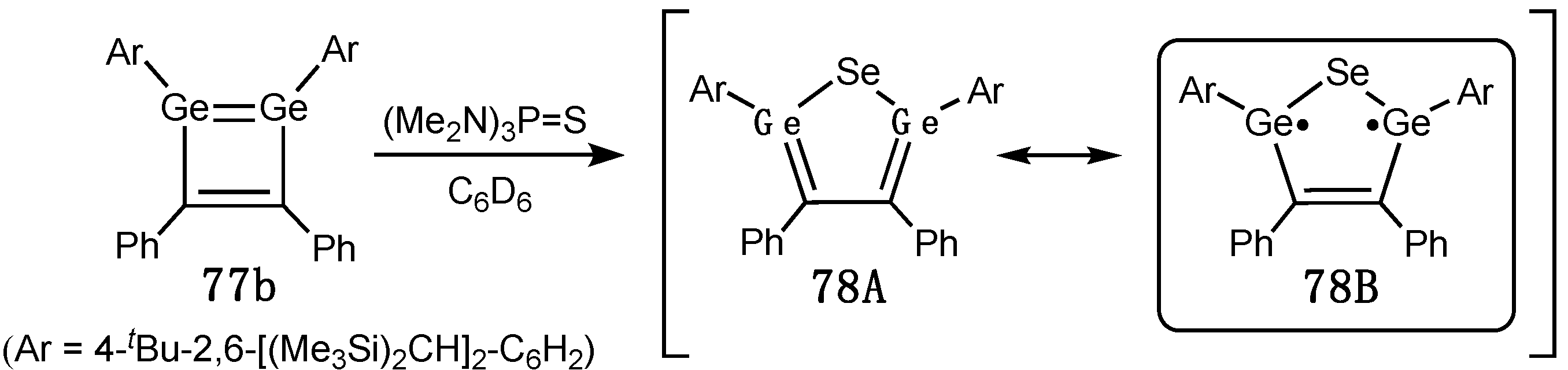
Digermynes –Ge≡Ge–
3.2. Heteronuclear Derivatives
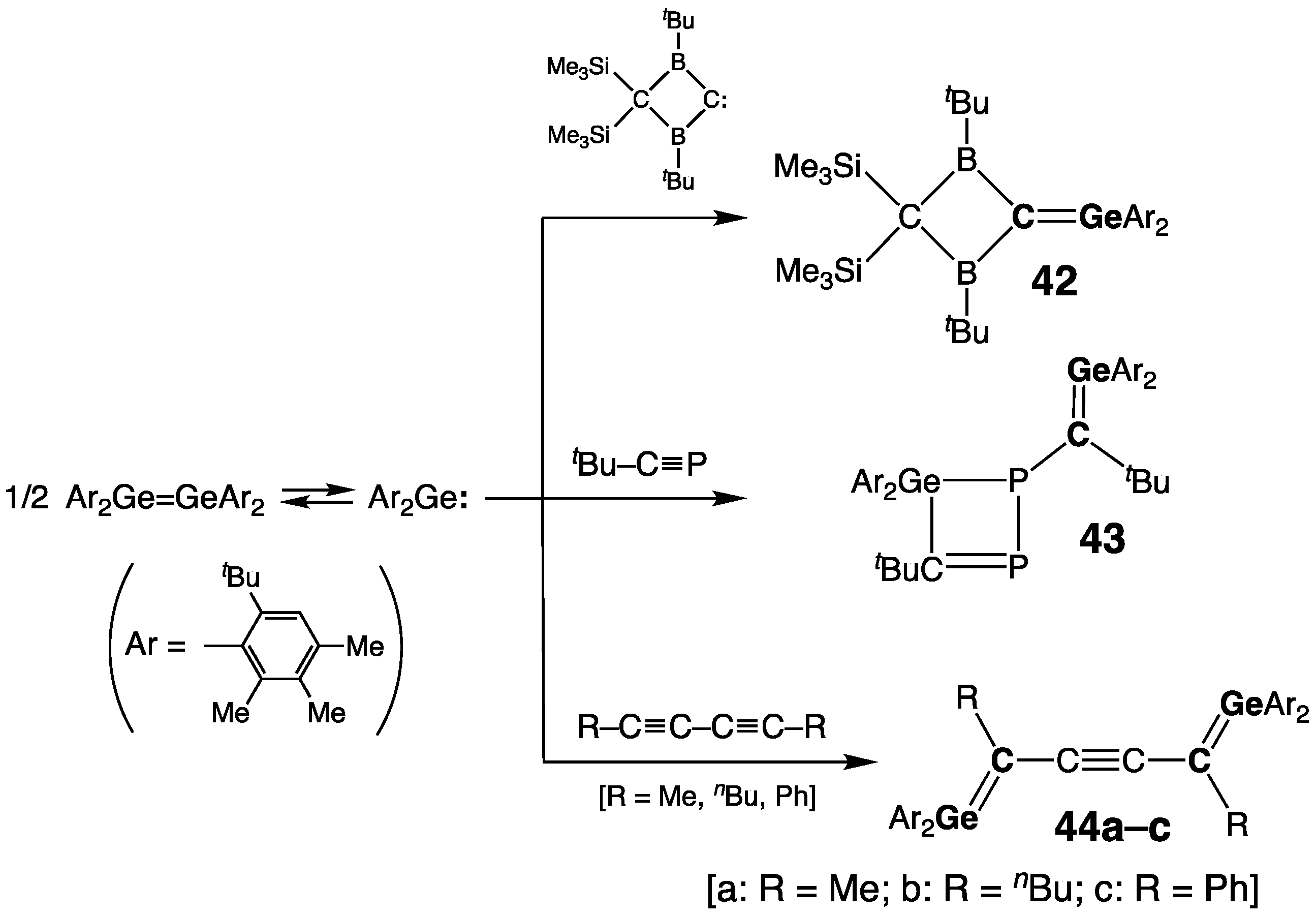
Germynes –Ge≡C–
4. Heavy Analogues of 1,3-Dienes
4.1. Homonuclear Derivatives
Tetra(germa)buta-1,3-dienes >Ge=Ge–Ge=Ge<
4.2. Heteronuclear Derivatives
4.2.1. 1-Sila-2-germabuta-1,3-dienes >Si=Ge–C=C<
4.2.2. 1,2-di(germa)cyclobuta-1,3-diene >Ge=Ge–C=C<
5. Heavy Analogues of Allene
5.1. Tri(germa)allene >Ge=Ge=Ge<, Germadi(sila)allene >Si=Ge=Si<, and Di(germa)silaallene >Ge=Si=Ge<
5.2. Germaallenes >Ge=C=C<
6. Heavy Analogues of Vinylidenes
6.1. Homonuclear Derivatives
Di(germa)vinylidene >Ge=Ge:
6.2. Heteronuclear Derivatives
6.2.1. Silagermenylidene >Si=Ge:
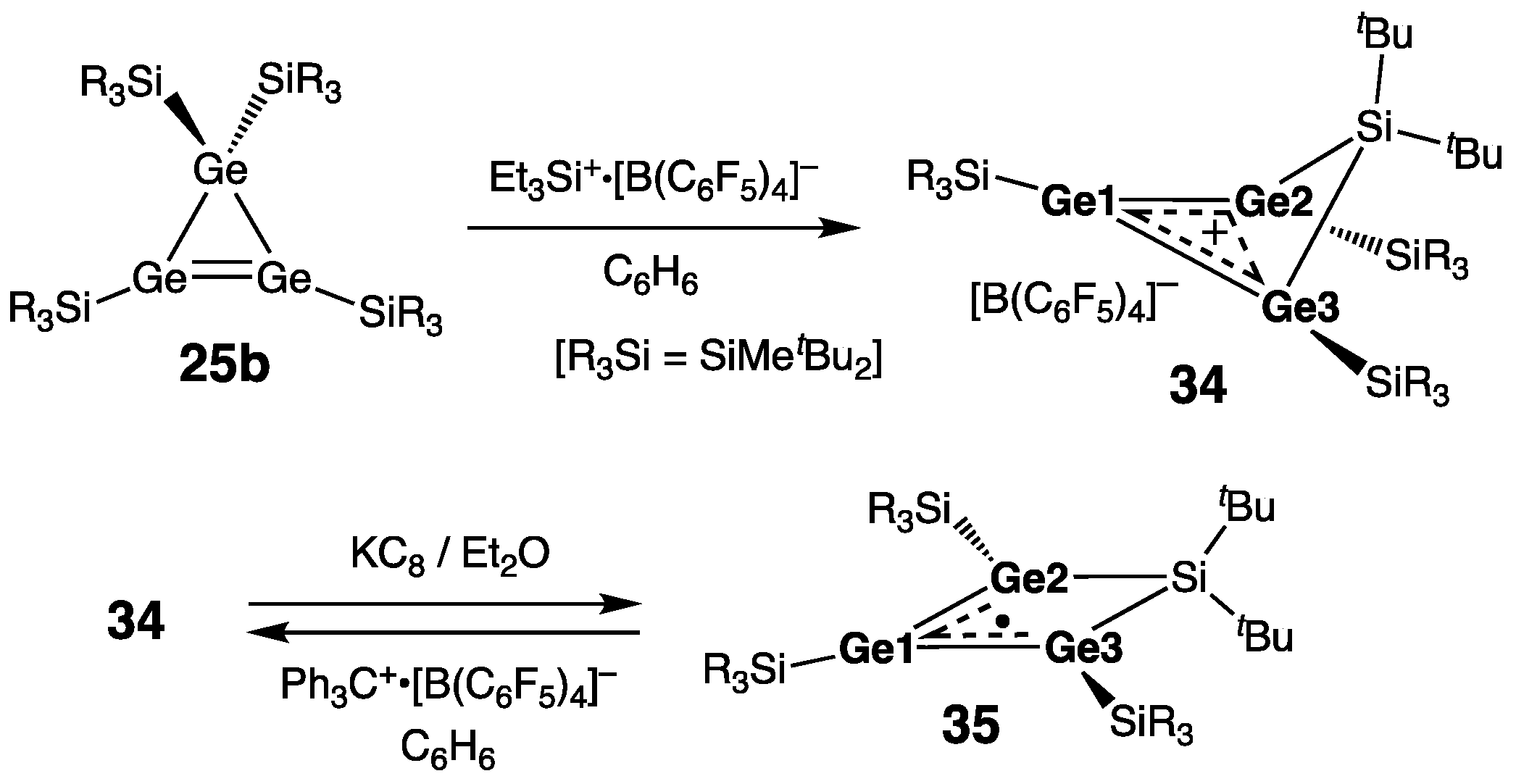
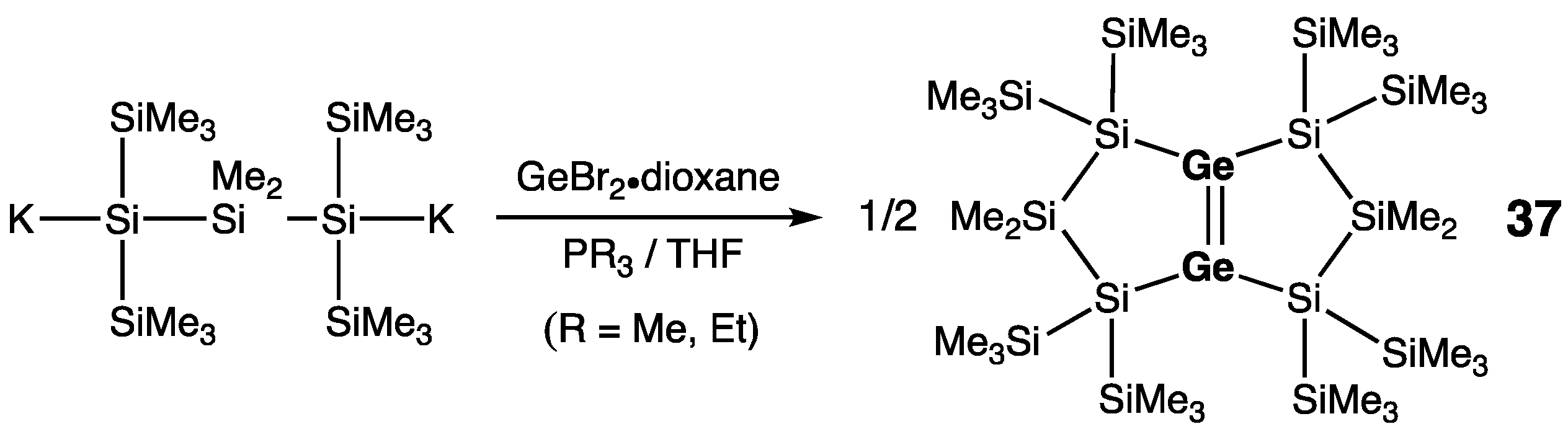
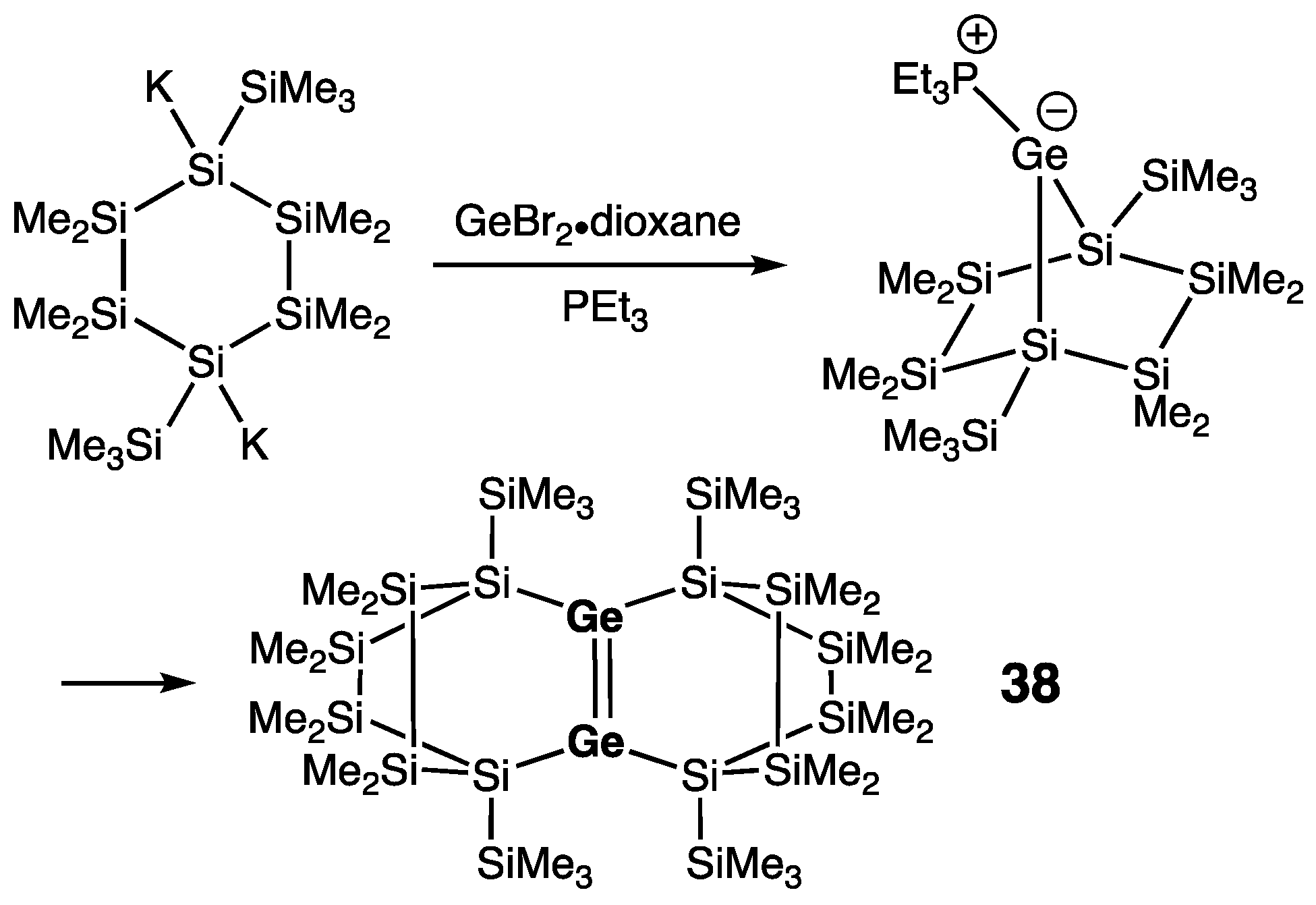
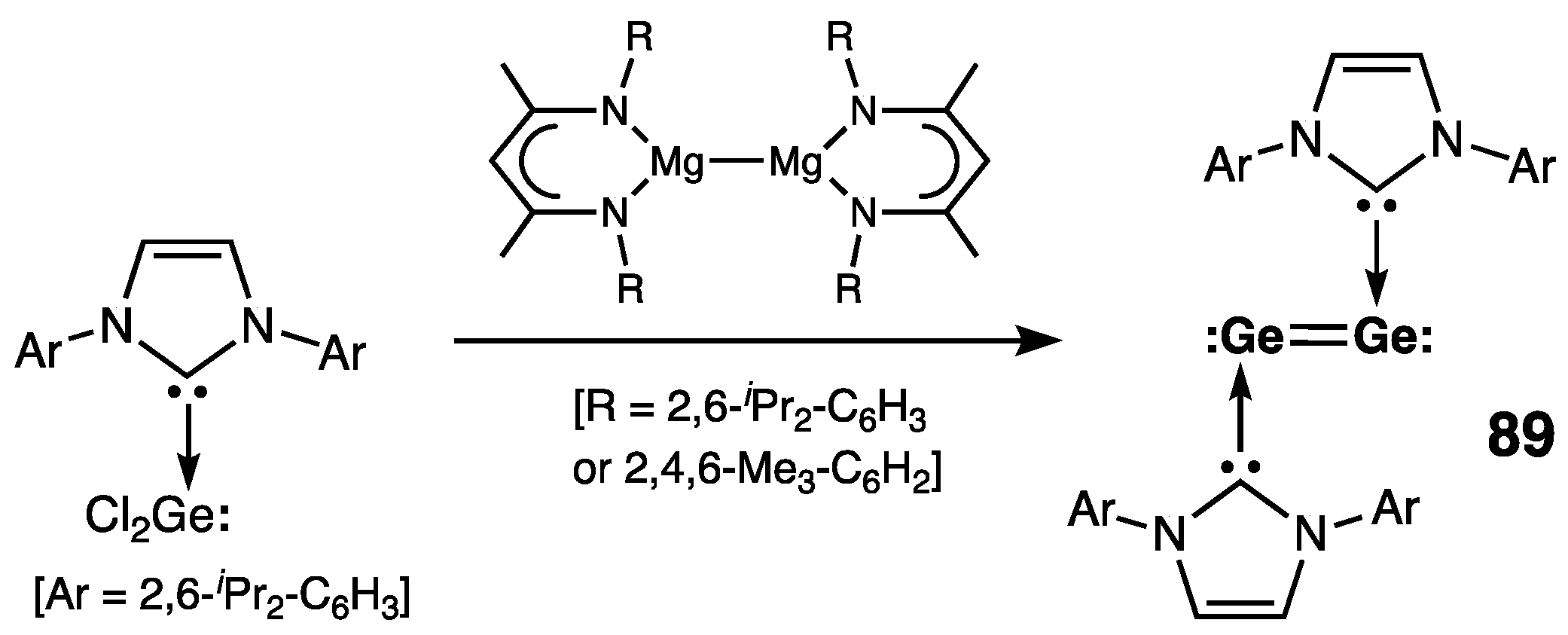
7. Digermanium(0) Complexes :Ge=Ge:
8. Prospects for the Use of Unsaturated Organogermanium Compounds
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADMET | Acyclic Diene METathesis |
| AIM | Atoms in Molecules |
| B3LYP | Becke Three-Parameter Hybrid Functional with the Lee–Yang–Parr Correlation Functional |
| Bbt | 2,6-[(Me3Si)2CH]2-4-[(Me3Si)3C]-C6H2 |
| cc-pVDZ-pp | Correlation-Consistent Polarized Valence Double Zeta with Pseudo Potential Basis Set |
| CV | Cyclic Voltammetry |
| diox | 1,4-dioxane |
| Dis | (Me3Si)2CH |
| Eind | 1,1,3,3,5,5,7,7-octaethyl-s-hydrindacen-4-yl |
| EMind | 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacen-4-yl |
| EPR | Electronic Paramagnetic Resonance |
| Fc | ferrocenyl |
| hffc | hyperfine coupling constant |
| HOMO | Highest Occupied Molecular Orbital |
| IDipp (or IPr) | 1,3-bis(2,6-diisopropylphenyl)-2H-imidazol-2-ylidene |
| LUMO | lowest unoccupied molecular orbital |
| Mes | 2,4,6-Me3-C6H2 |
| MO | Molecular Orbital |
| NHC | N-Heterocyclic Carbene |
| NHSi | N-Heterocyclic Silylene |
| NICS | Nucleus Independent Chemical Shift |
| NMR | Nuclear Magnetic Resonance |
| NPA | Natural Population Analysis |
| Tbb | 2,6-[(Me3Si)2CH]2-4-tBu-C6H2 |
| Tbt | 2,4,6-[(Me3Si)2CH]3-C6H2] |
| THF | tetrahydrofuran |
| Tip | 2,4,6-iPr3-C6H2 |
| UV | ultraviolet |
| WBI | Wiberg Bond Index |
References
- Power, P.P. π-Bonding and the lone pair effect in multiple bonds between heavier main group elements. Chem. Rev. 1999, 99, 3463–3503. [Google Scholar] [CrossRef] [PubMed]
- Tokitoh, N.; Okazaki, R. Multiply bonded germanium, tin and lead compounds. In The Chemistry of Organic Germanium, Tin and Lead Compounds; Rappoport, Z., Ed.; Wiley: Chichester, UK, 2002; Volume 2, Part 1, Chapter 13. [Google Scholar]
- Power, P.P. Silicon, germanium, tin and lead analogues of acetylenes. Chem. Commun. 2003, 2091–2101. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.C.; Power, P.P. π-Bonding and the lone pair effect in multiple bonds involving heavier main group elements: Developments in the new millennium. Chem. Rev. 2010, 110, 3877–3923. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.Y.; Sekiguchi, A. Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds; Wiley: Chichester, UK, 2010; Chapter 5 (Heavy analogs of alkenes, 1,3-dienes, allenes and alkynes: Multiply bonded derivatives of Si, Ge, Sn and Pb). [Google Scholar]
- Lee, V.Y.; Sekiguchi, A. Multiply bonded compounds of the heavy group 14 elements. In Comprehensive Inorganic Chemistry II.; Reedijk, J., Poeppelmeier, K.R., Eds.; Elsevier: Oxford, UK, 2013; Volume 1 (Chivers, T., Volume Ed.), Chapter 1.11. [Google Scholar]
- Lee, V.Y. Multiple bonds to germanium. In Organogermanium Compounds (Theory, Experiment, and Applications); Lee, V.Y., Ed.; Wiley: Hoboken, NJ, USA, 2023; Chapter 10, in press. [Google Scholar]
- Goldberg, D.E.; Harris, D.H.; Lappert, M.F.; Thomas, K.M. A new synthesis of divalent group 4B alkyls M[CH(SiMe3)2]2 (M = Ge or Sn), and the crystal and molecular structure of the tin compound. J. Chem. Soc., Chem. Commun. 1976, 261–262. [Google Scholar] [CrossRef]
- Wedler, H.B.; Wendelboe, P.; Power, P.P. Second-order Jahn–Teller (SOJT) structural distortions in multiply bonded higher main group compounds. Organometallics 2018, 37, 2929–2936. [Google Scholar] [CrossRef]
- Richards, A.F.; Brynda, M.; Power, P.P. Effects of the alkali metal counter ions on the germanium–germanium double bond length in a heavier group 14 element ethenide salt. Chem. Commun. 2004, 1592–1593. [Google Scholar] [CrossRef]
- Davidson, P.J.; Harris, D.H.; Lappert, M.F. Subvalent group 4B metal alkyl and amides. Part I. The synthesis and physical properties of kinetically stable bis[bis(trimethylsilyl)methyl]-germanium(II), -tin(II), and -lead(II). J. Chem. Soc., Dalton Trans. 1976, 2268–2274. [Google Scholar] [CrossRef]
- Goldberg, D.E.; Hitchcock, P.B.; Lappert, M.F.; Thomas, K.M.; Thorne, A.J.; Fjeldberg, T.; Haaland, A.; Schilling, B.E.R. Subvalent group 4B metal alkyl and amides. Part 9. Germanium and tin alkene analogues, the dimetallenes M2R4 [M = Ge or Sn, R = CH(SiMe3)2]: X-ray structures, molecular orbital calculations for M2H4, and trends in the series M2R′4 [M = C, Si, Ge, or Sn; R′ = R, Ph, C6H2Me3-2,4,6, or C6H3Et2-2,6]. J. Chem. Soc. Dalton Trans. 1986, 2387–2394. [Google Scholar] [CrossRef]
- Snow, J.T.; Murakami, S.; Masamune, S.; Williams, D.J. Synthesis and characterization of tetrakis(2,6-diethylphenyl)digermene. Tetrahedron Lett. 1984, 25, 4191–4194. [Google Scholar] [CrossRef]
- Hurni, K.L.; Rupar, P.A.; Payne, N.C.; Baines, K.M. On the synthesis, structure, and reactivity of tetramesityldigermene. Organometallics 2007, 26, 5569–5575. [Google Scholar] [CrossRef]
- Stender, M.; Pu, L.; Power, P.P. Stabilized terphenyl-substituted digermene derivatives of simple organic groups and their halide precursors: Preference for symmetrically bonded structures. Organometallics 2001, 20, 1820–1824. [Google Scholar] [CrossRef]
- Lei, H.; Fettinger, J.C.; Power, P.P. Synthesis and structures of low-valent alkynyl tin and germanium complexes supported by terphenyl ligands: Heavier group 14 element enediyne analogues. Organometallics 2010, 29, 5585–5590. [Google Scholar] [CrossRef]
- Pampuch, B.; Saak, W.; Weidenbruch, M. New compounds with Ge/Ge and Ge/C multiple bonds. J. Organomet. Chem. 2006, 691, 3540–3544. [Google Scholar] [CrossRef]
- Sasamori, T.; Sugiyama, Y.; Takeda, N.; Tokitoh, N. Structure and properties of an overcrowded 1,2-dibromodigermene. Organometallics 2005, 24, 3309–3314. [Google Scholar] [CrossRef]
- Sasamori, T.; Sugahara, T.; Agou, T.; Guo, J.-D.; Nagase, S.; Streubel, R.; Tokitoh, N. Synthesis and characterization of a 1,2-digermabenzene. Organometallics 2015, 34, 2106–2109. [Google Scholar] [CrossRef]
- Al-Rafia, S.M.I.; Momeni, M.R.; Ferguson, M.J.; McDonald, R.; Brown, A.; Rivard, E. Stable complexes of parent digermene: An inorganic analogue of ethylene. Organometallics 2013, 32, 6658–6665. [Google Scholar] [CrossRef]
- Kira, M.; Iwamoto, T.; Maruyama, T.; Kabuto, C.; Sakurai, H. Tetrakis(trialkylsilyl)digermenes. Salient effects of trialkylsilyl substituents on planarity around the Ge=Ge Bond and remarkable thermochromism. Organometallics 1996, 15, 3767–3769. [Google Scholar] [CrossRef]
- Iwamoto, T.; Okita, J.; Yoshida, N.; Kira, M. Structure and reactions of an isolable Ge=Si doubly bonded compound, tetra(t-butyldimethylsilyl)germasilene. Silicon 2010, 2, 209–216. [Google Scholar] [CrossRef]
- Tokitoh, N.; Kishikawa, K.; Okazaki, R.; Sasamori, T.; Nakata, N.; Takeda, N. Synthesis and characterization of an extremely hindered tetraaryl-substituted digermene and its unique properties in the solid state and in solution. Polyhedron 2002, 21, 563–577. [Google Scholar] [CrossRef]
- Sasamori, T.; Miyamoto, H.; Sakai, H.; Furukawa, Y.; Tokitoh, N. 1,2-Bis(ferrocenyl)digermene: A d–π electron system containing a Ge=Ge unit. Organometallics 2012, 31, 3904–3910. [Google Scholar] [CrossRef]
- Spikes, G.H.; Fettinger, J.C.; Power, P.P. Facile activation of dihydrogen by an unsaturated heavier main group compound. J. Am. Chem. Soc. 2005, 127, 12232–12233. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.F.; Phillips, A.D.; Olmstead, M.M.; Power, P.P. Isomeric forms of divalent heavier group 14 element hydrides: Characterization of Ar′(H)GeGe(H)Ar′ and Ar′(H)2GeGeAr′•PMe3 (Ar′ = C6H3-2,6-Dipp2; Dipp = C6H3-2,6-Pri2). J. Am. Chem. Soc. 2003, 125, 3204–3205. [Google Scholar] [CrossRef] [PubMed]
- Summerscales, O.T.; Caputo, C.A.; Knapp, C.E.; Fettinger, J.C.; Power, P.P. The role of group 14 element hydrides in the activation of C–H bonds in cyclic olefins. J. Am. Chem. Soc. 2012, 134, 14595–14603. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Hermann, M.; Li, J.; Frenking, G.; Jones, C. Activation of H2 by a multiply bonded amido-digermyne: Evidence for the formation of a hydrido-germylene. Angew. Chem. Int. Ed. 2013, 52, 10199–10203. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Li, J.; Hermann, M.; Davey, A.; Frenking, G.; Jones, C. Reactivity of amido-digermynes, LGeGeL (L = bulky amide), toward olefins and related molecules: Facile reduction, C–H activation, and reversible cycloaddition of unsaturated substrates. Organometallics 2015, 34, 3175–3185. [Google Scholar] [CrossRef]
- Kelly, J.A.; Juckel, M.; Hadlington, T.J.; Fernández, I.; Frenking, G.; Jones, C. Synthesis and reactivity studies of amido-substituted germanium(I)/tin(I) dimers and clusters. Chem. Eur. J. 2019, 25, 2773–2785. [Google Scholar] [CrossRef]
- Lee, V.Y.; McNiece, K.; Ito, Y.; Sekiguchi, A. A blue digermene (t-Bu2MeSi)2Ge=Ge(SiMet-Bu2)2. Chem. Commun. 2011, 47, 3272–3274. [Google Scholar] [CrossRef]
- Lee, V.Y.; McNiece, K.; Ito, Y.; Sekiguchi, A.; Geinik, N.; Becker, J.Y. Tetrakis(di-tert-butylmethylsilyl)digermene: Synthesis, structure, electrochemical properties, and reactivity. Heteroatom Chem. 2014, 25, 313–319. [Google Scholar] [CrossRef]
- Aysin, R.R.; Bukalov, S.S.; Leites, L.A.; Lee, V.Y.; Sekiguchi, A. Electronic structure and conformational isomerism of the digermene (tBu2MeSi)2Ge=Ge(SiMet-Bu2)2 as studied by temperature-dependent Raman and UV–vis spectra and quantum-chemistry calculations. J. Organomet. Chem. 2019, 892, 18–23. [Google Scholar] [CrossRef]
- Jana, A.; Huch, V.; Rzepa, H.S.; Scheschkewitz, D. A Multiply functionalized base-coordinated GeII compound and its reversible dimerization to the digermene. Angew. Chem. Int. Ed. 2015, 54, 289–292. [Google Scholar] [CrossRef]
- Nieder, D.; Klemmer, L.; Kaiser, Y.; Huch, V.; Scheschkewitz, D. Isolation and reactivity of a digerma analogue of vinyllithiums: A lithium digermenide. Organometallics 2018, 37, 632–635. [Google Scholar] [CrossRef]
- Klemmer, L.; Kaiser, Y.; Huch, V.; Zimmer, M.; Scheschkewitz, D. Persistent digermenes with acyl and α-chlorosilyl functionalities. Chem. Eur. J. 2019, 25, 12187–12195. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, N.; Sugahara, T.; Numata, Y.; Kawaai, H.; Yamatani, K.; Nishimura, S.; Goda, S.; Suzuki, Y.; Tanikawa, T.; Nakai, H.; et al. 1,2-Dihalodigermenes bearing bulky Eind groups: Synthesis, characterization, and conversion to halogermylenoids. Dalton Trans. 2018, 47, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Mangan, R.J.; Rit, A.; Sindlinger, C.P.; Tirfoin, R.; Campos, J.; Hicks, J.; Christensen, K.E.; Niu, H.; Aldridge, S. Activation of protic, hydridic and apolar E–H bonds by a boryl-substituted GeII cation. Chem. Eur. J. 2020, 26, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Mangan, R.J.; Davies, A.R.; Hicks, J.; Sindlinger, C.P.; Thompson, A.L.; Aldridge, S. Synthesis, structure and reactivity of terphenyl-substituted germylium-ylidene cations. Polyhedron 2021, 196, 115006. [Google Scholar] [CrossRef]
- Klemmer, L.; Thömmes, A.-L.; Zimmer, M.; Huch, V.; Morgenstern, B.; Scheschkewitz, D. Metathesis of Ge=Ge double bonds. Nature Chem. 2021, 13, 373–377. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Yamazaki, H.; Kabuto, C.; Sakurai, H.; Nagase, S. Cyclotrigermenes: A new unsaturated ring system. J. Am. Chem. Soc. 1995, 117, 8025–8026. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Tsukamoto, M.; Ichinohe, M. A Free Cyclotrigermenium cation with a 2π-electron system. Science 1997, 275, 60–61. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Fukaya, N.; Ichinohe, M.; Takagi, N.; Nagase, S. Synthesis of unsymmetrically substituted cyclotrigermenes and the first example of cis configuration around the Ge=Ge double Bond. J. Am. Chem. Soc. 1999, 121, 11587–11588. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Ishida, Y.; Fukaya, N.; Ichinohe, M.; Takagi, N.; Nagase, S. The first halogen-substituted cyclotrigermenes: A unique halogen walk over the three-membered ring skeleton and facial stereoselectivity in the Diels-Alder reaction. J. Am. Chem. Soc. 2002, 124, 1158–1159. [Google Scholar] [CrossRef]
- Lee, V.Y.; Yasuda, H.; Ichinohe, M.; Sekiguchi, A. SiGe2 and Ge3: Cyclic digermenes that undergo unexpected ring-expansion reactions. Angew. Chem. Int. Ed. 2005, 44, 6378–6381. [Google Scholar] [CrossRef]
- Lee, V.Y.; Yasuda, H.; Ichinohe, M.; Sekiguchi, A. Heavy cyclopropene analogues R4SiGe2 and R4Ge3 (R = SiMetBu2)—New members of the cyclic digermenes family. J. Organomet. Chem. 2007, 692, 10–19. [Google Scholar] [CrossRef]
- McNeice, K.; Lee, V.Y.; Sekiguchi, A. Making a cyclotrigermene from a digermene. Organometallics 2011, 30, 4796–4797. [Google Scholar] [CrossRef]
- Lee, V.Y.; Yasuda, H.; Sekiguchi, A. Interplay of EnE′3–nC valence isomers (E, E′ = Si, Ge): Bicyclo[1.1.0]butanes with very short bridging bonds and their isomerization to alkyl-substituted cyclopropenes. J. Am. Chem. Soc. 2007, 129, 2436–2437. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A.; Takagi, N.; Nagase, S. The first three-membered unsaturated rings consisting of different heavier group 14 elements: 1-disilagermirene with a Si=Si double bond and its isomerization to a 2-disilagermirene with a Si=Ge double bond. J. Am. Chem. Soc. 2000, 122, 9034–9035. [Google Scholar] [CrossRef]
- Lee, V.Y.; Takanashi, K.; Ichinohe, M.; Sekiguchi, A. A chemical trick: How to make a digermene from a disilene, formation of 3Δ-1,2,3,4-disiladigermetene. J. Am. Chem. Soc. 2003, 125, 6012–6013. [Google Scholar] [CrossRef]
- Lee, V.Y.; Ito, Y.; Yasuda, H.; Takanashi, K.; Sekiguchi, A. From tetragermacyclobutene to tetragermacyclobutadiene dianion to tetragermacyclobutadiene transition metal complexes. J. Am. Chem. Soc. 2011, 133, 5103–5108. [Google Scholar] [CrossRef]
- Ramaker, G.; Saak, W.; Haase, D.; Weidenbruch, M. Reaction modes of a tetragermabutadiene: Cycloadditions versus Ge–Ge bond cleavages. Organometallics 2003, 22, 5212–5216. [Google Scholar] [CrossRef]
- Sasamori, T.; Sugahara, T.; Agou, T.; Sugamata, K.; Guo, J.-D.; Nagase, S.; Tokitoh, N. Reaction of a diaryldigermyne with ethylene. Chem. Sci. 2015, 6, 5526–5530. [Google Scholar] [CrossRef]
- Schäfer, H.; Saak, W.; Weidenbruch, M. Hexaaryltetragermabuta-1,3-diene: A molecule with conjugated Ge–Ge double bonds. Angew. Chem. Int. Ed. 2000, 39, 3703–3705. [Google Scholar] [CrossRef]
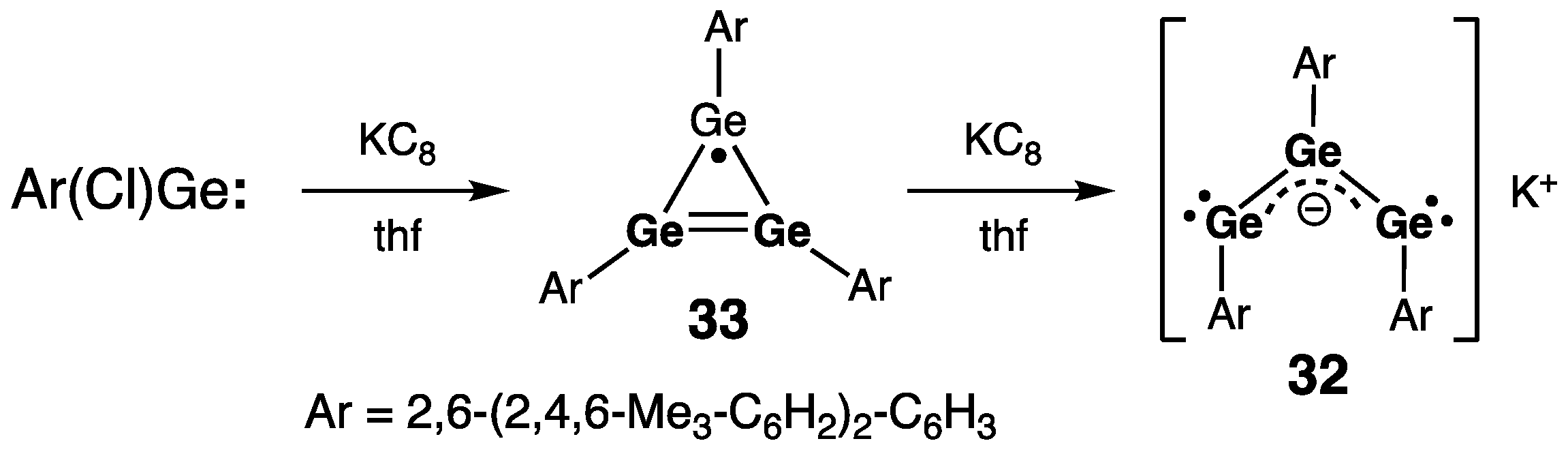
- Olmstead, M.M.; Pu, L.; Simons, R.S.; Power, P.P. Reduction of Ge(Cl)C6H3mes2-2,6 to give the cyclotrigermenyl radical (GeC6H3mes2-2,6)3∙ and the trigermenyl anion salt K(GeC6H3mes2-2,6)3. Chem. Commun. 1997, 1595–1596. [Google Scholar] [CrossRef]
- Lee, V.Y.; Ito, Y.; Gapurenko, O.A.; Minyaev, R.M.; Gornitzka, H.; Sekiguchi, A. From a (silatrigerma)cyclobutenylium ion to a (silatrigerma)cyclobutenyl radical and back. J. Am. Chem. Soc. 2020, 142, 16455–16460. [Google Scholar] [CrossRef] [PubMed]
- Ramaker, G.; Schäfer, A.; Saak, W.; Weidenbruch, M. One-pot synthesis of a tetragermabutadiene and its reactions with oxygen and sulfur. Organometallics 2003, 22, 1302–1304. [Google Scholar] [CrossRef]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Albers, L.; Müller, T.; Jouikov, V.V. Formation and properties of a bicyclic silylated digermene. Chem. Eur. J. 2014, 20, 9357–9366. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Baum, G.; Massa, W.; Berndt, A. Stable germaethenes. Angew. Chem., Int. Ed. Engl. 1987, 26, 798–799. [Google Scholar] [CrossRef]
- Couret, C.; Escudié, J.; Satgé, J.; Lazraq, M. The first stable germene: A compound with a germanium–carbon double bond. J. Am. Chem. Soc. 1987, 109, 4411–4412. [Google Scholar] [CrossRef]
- Lazraq, M.; Escudié, J.; Couret, C.; Satgé, J.; Dräger, M.; Dammel, R. (Mesityl)2Ge(fluorenylidene)–stabilization of a Ge–C double bond by charge transfer into an aromatic system. Angew. Chem. Int. Ed. Engl. 1988, 27, 828–829. [Google Scholar] [CrossRef]
- Lazraq, M.; Couret, C.; Escudié, J.; Satgé, J.; Soufiaoui, M. New stable germenes. Polyhedron 1991, 10, 1153–1161. [Google Scholar] [CrossRef]
- Anselme, G.; Escudié, J.; Couret, C.; Satgé, J. Difluorenyl- and tert-butylfluorenyl(fluorenylidene)germenes: Synthesis, stabilization and first aspects of their reactivity. J. Organomet. Chem. 1991, 403, 93–100. [Google Scholar] [CrossRef]
- Chaubon, M.-A.; Escudié, J.; Ranaivonjatovo, H.; Satgé, J. Halogenogermenes: Evidence for the formation of the chloro- or fluoro-germenes (Me5C5)(X)Ge–CR2 (CR2 = fluorenylidene). J. Chem. Soc. Dalton Trans. 1996, 893–897. [Google Scholar] [CrossRef]
- Couret, C.; Escudié, J.; Delpon-Lacaze, G.; Satgé, J. Dimesitylneopentylgermene, a new stable germene. Organometallics 1992, 11, 3176–3177. [Google Scholar] [CrossRef]
- Stürmann, M.; Saak, W.; Weidenbruch, M.; Berndt, A.; Scheschkewitz, D. Molecular structures of new compounds with Ge=C and Sn=C double bonds. Heteroatom Chem. 1999, 10, 554–558. [Google Scholar] [CrossRef]
- Meiners, F.; Saak, W.; Weidenbruch, M. Reaction of a diarylgermylene with a phosphaalkyne: Formation of a germadiphosphacyclobutene with an exocyclic C=Ge double bond. Chem. Commun. 2001, 215–216. [Google Scholar] [CrossRef]
- Meiners, F.; Saak, W.; Weidenbruch, M. Acetylene-linked bis(germaethenes): The first molecules with conjugated germanium–carbon double bonds. Organometallics 2000, 19, 2835–2836. [Google Scholar] [CrossRef]
- Meiners, F.; Haase, D.; Koch, R.; Saak, W.; Weidenbruch, M. Cycloaddition reactions of an acetylene-linked bis(germaethene). Organometallics 2002, 21, 3990–3995. [Google Scholar] [CrossRef]
- Tokitoh, N.; Kishikawa, K.; Okazaki, R. Synthesis and structure of the first germaketenedithioacetal. J. Chem. Soc. Chem. Commun. 1995, 1425–1426. [Google Scholar] [CrossRef]
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A. 2,4-Disila-1-germatricyclo[2.1.0.02,5]pentane: A new type of cage compound of group 14 elements with an extremely long Ge–C bridge bond and an “umbrella”-type configuration of a Ge atom. J. Am. Chem. Soc. 2002, 124, 9962–9963. [Google Scholar] [CrossRef]
- Haas, M.; Leypold, M.; Schnalzer, D.; Torvisco, A.; Stueger, H. Stable germenolates and germenes with exocyclic structures. Organometallics 2015, 34, 5291–5297. [Google Scholar] [CrossRef]
- Baines, K.M.; Cooke, J.A. Tetramesitylgermasilene: The first relatively stable germasilene and its rearrangement to a silylgermylene. Organometallics 1992, 11, 3487–3488. [Google Scholar] [CrossRef]
- Ichinohe, M.; Arai, Y.; Sekiguchi, A.; Takagi, N.; Nagase, S. A new approach to the synthesis of unsymmetrical disilenes and germasilene: Unusual 29Si NMR chemical shifts and regiospecific methanol addition. Organometallics 2001, 20, 4141–4143. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Izumi, R.; Ihara, S.; Ichinohe, M.; Lee, V.Y. The first isolable 1,1-dilithiogermane and its unusual dimeric structure—An effective reagent for the preparation of double-bonded derivatives of group 14 elements. Angew. Chem. Int. Ed. 2002, 41, 1598–1600. [Google Scholar] [CrossRef]
- Igarashi, M.; Ichinohe, M.; Sekiguchi, A. A stable silagermene (tBu3Si)2Si=Ge(Mes)2 (Mes = 2,4,6-trimethylphenyl): Synthesis, X-ray crystal structure, and thermal isomerization. Heteroatom Chem. 2008, 19, 649–653. [Google Scholar] [CrossRef]
- Al-Rafia, S.M.I.; Malcolm, A.C.; McDonald, R.; Ferguson, M.J.; Rivard, E. Trapping the parent inorganic ethylenes H2SiGeH2 and H2SiSnH2 in the form of stable adducts at ambient temperature. Angew. Chem. Int. Ed. 2011, 50, 8354–8357. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Roesky, H.W.; Merkel, S.; Henn, J.; Stalke, D. Lewis base stabilized dichlorosilylene. Angew. Chem. Int. Ed. 2009, 48, 5683–5686. [Google Scholar] [CrossRef]
- Majhi, P.K.; Huch, V.; Scheschkewitz, D. A mixed heavier Si=Ge analogue of a Vinyl Anion. Angew. Chem. Int. Ed. 2021, 60, 242–246. [Google Scholar] [CrossRef]
- Chaubon, M.-A.; Escudié, J.; Ranaivonjatovo, H.; Satgé, J. First characterization of a compound with a tin-germanium double bond: The dimesityl(diisitylstanna)germene (Is)2Sn=Ge(Mes)2. Chem. Commun. 1996, 2621–2622. [Google Scholar] [CrossRef]
- Schäfer, A.; Saak, W.; Weidenbruch, M. Tetraarylstannagermene: A molecule with a Ge=Sn double bond. Organometallics 2003, 22, 215–217. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Izumi, R.; Lee, V.Y.; Ichinohe, M. R2Ge=SnR′2 and RR′Ge=SnRR′ (R = SiMetBu2, R′ = 2,4,6-iPr3C6H2): The new stable germastannenes. Organometallics 2003, 22, 1483–1486. [Google Scholar] [CrossRef]
- Lee, V.Y.; Takanashi, K.; Nakamoto, M.; Sekiguchi, A. 3Δ-1,2,3,4-Disilagermastannetene: The first cyclic germastannene. Russ. Chem. Bull. Int. Ed. 2004, 53, 1102–1104. [Google Scholar] [CrossRef]
- Stender, M.; Phillips, A.D.; Wright, R.J.; Power, P.P. Synthesis and characterization of a digermanium analogue of an alkyne. Angew. Chem. Int. Ed. 2002, 41, 1785–1787. [Google Scholar] [CrossRef]
- Stender, M.; Phillips, A.D.; Power, P.P. Formation of [Ar*Ge{CH2C(Me)C(Me)CH2}CH2C(Me)=]2 (Ar* = C6H3-2,6-Trip2; Trip = C6H2-2,4,6-i-Pr3) via reaction of Ar*GeGeAr* with 2,3-dimethyl-1,3-butadiene: Evidence for the existence of a germanium analogue of an alkyne. Chem. Commun. 2002, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Phillips, A.D.; Richards, A.F.; Stender, M.; Simons, R.S.; Olmstead, M.M.; Power, P.P. Germanium and tin analogues of alkynes and their reduction products. J. Am. Chem. Soc. 2003, 125, 11626–11636. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Fischer, R.C.; Merrill, W.A.; Fischer, J.; Pu, L.; Ellis, B.D.; Fettinger, J.C.; Herber, R.H.; Power, P.P. Substituent effects in ditetrel alkyne analogues: Multiple vs. single bonded isomers. Chem. Sci. 2010, 1, 461–468. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Sasamori, T.; Hosoi, Y.; Furukawa, Y.; Takagi, N.; Nagase, S.; Tokitoh, N. Synthesis and properties of a new kinetically stabilized digermyne: New insights for a germanium analogue of an alkyne. J. Am. Chem. Soc. 2006, 128, 1023–1031. [Google Scholar] [CrossRef]
- Wu, P.-C.; Su, M.-D. Theoretical design for germaacetylene (RC≡GeR′): A new target for synthesis. Dalton Trans. 2011, 40, 4253–4259. [Google Scholar] [CrossRef]
- Bibal, C.; Mazières, S.; Gornitzka, H.; Couret, C. A route to a germanium–carbon triple bond: First chemical evidence for a germyne. Angew. Chem. Int. Ed. 2001, 40, 952–954. [Google Scholar] [CrossRef]
- Bonnefille, E.; Mazières, S.; Saffon, N.; Couret, C. Reactivity of a germa-alkyne: Evidence for a germanone intermediate in the hydrolysis and alcoholysis processes. J. Organomet. Chem. 2009, 694, 2246–2251. [Google Scholar] [CrossRef]
- Berthe, J.; Garcia, J.M.; Ocando, E.; Kato, T.; Saffon-Merceron, N.; De Cózar, A.; Cossío, F.P.; Baceiredo, A. Synthesis and reactivity of a phosphine-stabilized monogermanium analogue of alkynes. J. Am. Chem. Soc. 2011, 133, 15930–15933. [Google Scholar] [CrossRef]
- Schäfer, A.; Weidenbruch, M.; Müller, T.; Pravinkumar, K.; Becker, J.Y. Electrochemical properties of a disiliene, a tetrasila-1,3-butadiene, and their germanium analogues. Chem. Eur. J. 2009, 15, 8424–8428. [Google Scholar] [CrossRef]
- Suzuki, K.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. A planar rhombic charge-separated tetrasilacyclobutadiene. Science 2011, 331, 1306–1309. [Google Scholar] [CrossRef]
- Suzuki, K.; Numata, Y.; Fujita, N.; Hayakawa, N.; Tanikawa, T.; Hashizume, D.; Tamao, K.; Fueno, H.; Tanaka, K.; Matsuo, T. A stable free tetragermacyclobutadiene incorporating fused-ring bulky EMind groups. Chem. Commun. 2018, 54, 2200–2203. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A. The first metalladiene of group 14 elements with a silole-type structure with Si=Ge and C=C double bonds. J. Am. Chem. Soc. 2000, 122, 12604–12605. [Google Scholar] [CrossRef]
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A. Reaction of disilagermirenes with phenylacetylene: From a germasilene –Ge=Si– to a metalladiene of the type –Si=Ge–C=C–. J. Organomet. Chem. 2001, 636, 41–48. [Google Scholar] [CrossRef]
- Lee, V.Y.; Kato, R.; Aoki, S.; Sekiguchi, A. 1,3-Disila-3-germacyclopentadienes: Cyclopentadiene analogs based on heavier group 14 elements. Russ. Chem. Bull., Int. Ed. 2011, 60, 2434–2435. [Google Scholar] [CrossRef]
- Cui, C.; Olmstead, M.M.; Power, P.P. Reaction of Ar′GeGeAr′ (Ar′ = C6H3-2,6-Dipp2, Dipp = C6H3-2,6-iPr2) toward alkynes: Isolation of a stable digermacyclobutadiene. J. Am. Chem. Soc. 2004, 126, 5062–5063. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Olmstead, M.M.; Fettinger, J.C.; Spikes, G.H.; Power, P.P. Reactions of the heavier alkyne analogues Ar′GeGeAr′ (Ar′ = C6H3-2,6-(C6H3-2,6-Pri2)2; E = Ge, Sn) with unsaturated molecules: Probing the character of the EE multiple bonds. J. Am. Chem. Soc. 2005, 127, 17530–17541. [Google Scholar] [CrossRef]
- Sugahara, T.; Guo, J.-D.; Sasamori, T.; Karatsu, Y.; Furukawa, Y.; Ferao, A.E.; Nagase, S.; Tokitoh, N. Reaction of a stable digermyne with acetylenes: Synthesis of a 1,2-digermabenzene and a 1,4-digermabarrelene. Bull. Chem. Soc. Jpn. 2016, 89, 1375–1384. [Google Scholar] [CrossRef]
- Sugahara, T.; Sasamori, T.; Tokitoh, N. 2,5-Digermaselenophenes: Germanium analogues of selenophenes. J. Am. Chem. Soc. 2018, 140, 11206–11209. [Google Scholar] [CrossRef]
- Ishida, S.; Iwamoto, T.; Kabuto, C.; Kira, M. A stable silicon-based allene analogue with a formally sp-hybridized silicon atom. Nature 2003, 421, 725–727. [Google Scholar] [CrossRef]
- Iwamoto, T.; Masuda, H.; Kabuto, C.; Kira, M. Trigermaallene and 1,3-digermasilaallene. Organometallics 2005, 24, 197–199. [Google Scholar] [CrossRef]
- Iwamoto, T.; Abe, T.; Kabuto, C.; Kira, M. A missing allene of heavy group 14 elements: 2-germadisilaallene. Chem. Commun. 2005, 5190–5192. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, T.; Sasamori, T.; Tokitoh, N. Highly bent 1,3-digerma-2-silaallene. Angew. Chem. Int. Ed. 2017, 56, 9920–9923. [Google Scholar] [CrossRef]
- Eichler, B.E.; Powell, D.R.; West, R. Synthesis and structure of a 1-germapropadiene. Organometallics 1998, 17, 2147–2148. [Google Scholar] [CrossRef]
- Tokitoh, N.; Kishikawa, K.; Okazaki, R. Synthesis and reactions of the first stable 1-germaallene. Chem. Lett. 1998, 27, 811–812. [Google Scholar] [CrossRef]
- Rit, A.; Campos, J.; Niu, H.; Aldridge, S. A stable heavier group 14 analogue of vinylidene. Nature Chem. 2016, 8, 1022–1026. [Google Scholar] [CrossRef]
- Krebs, K.M.; Hanselmann, D.; Schubert, H.; Wurst, K.; Scheele, M.; Wesemann, L. Phosphine-stabilized digermavinylidene. J. Am. Chem. Soc. 2019, 141, 3424–3429. [Google Scholar] [CrossRef]
- Jana, A.; Huch, V.; Scheschkewitz, D. NHC-stabilized silagermenylidene: A heavier analogue of vinylidene. Angew. Chem. Int. Ed. 2013, 52, 12179–12182. [Google Scholar] [CrossRef]
- Jana, A.; Majumdar, M.; Huch, V.; Zimmer, M.; Scheschkewitz, D. NHC-coordinated silagermenylidene functionalized in allylic position and its behaviour as a ligand. Dalton Trans. 2014, 43, 5175–5181. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Y.; Wei, P.; King, R.B.; Schaefer, H.F., III; Schleyer, P.; von, R.; Robinson, G.H. A stable silicon(0) compound with a Si=Si double bond. Science 2008, 321, 1069–1071. [Google Scholar] [CrossRef]
- Sidiropoulos, A.; Jones, C.; Stasch, A.; Klein, S.; Frenking, G. N-heterocyclic carbene stabilized digermanium(0). Angew. Chem. Int. Ed. 2009, 48, 9701–9704. [Google Scholar] [CrossRef]
- Jones, C.; Sidiropoulos, A.; Holzmann, N.; Frenking, G.; Stasch, A. An N-heterocyclic carbene adduct of diatomic tin: Sn=Sn. Chem. Commun. 2012, 48, 9855–9857. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.-L.; Yim, W.-L.; So, C.-W. An N-heterocyclic silylene-stabilized digermanium(0) complex. Angew. Chem. Int. Ed. 2014, 53, 13155–13158. [Google Scholar] [CrossRef] [PubMed]
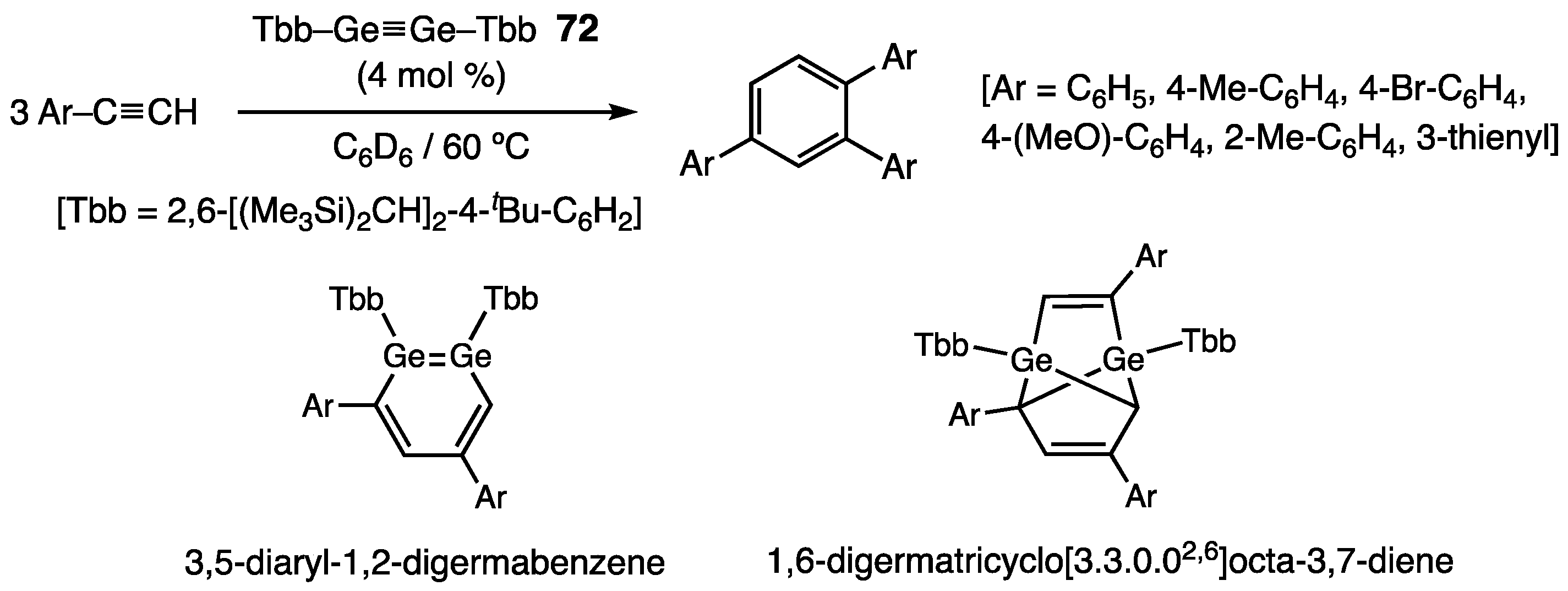
- Sugahara, T.; Guo, J.-D.; Sasamori, T.; Nagase, S.; Tokitoh, N. Regioselective cyclotrimerization of terminal alkynes using a digermyne. Angew. Chem. Int. Ed. 2018, 57, 3499–3503. [Google Scholar] [CrossRef] [PubMed]
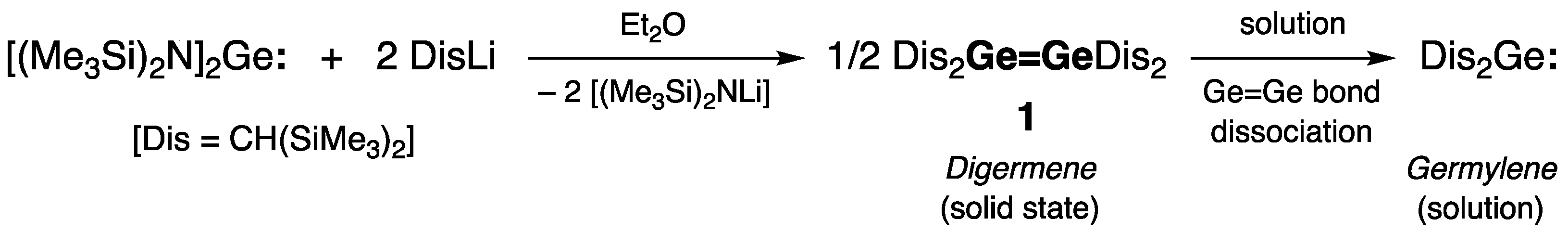

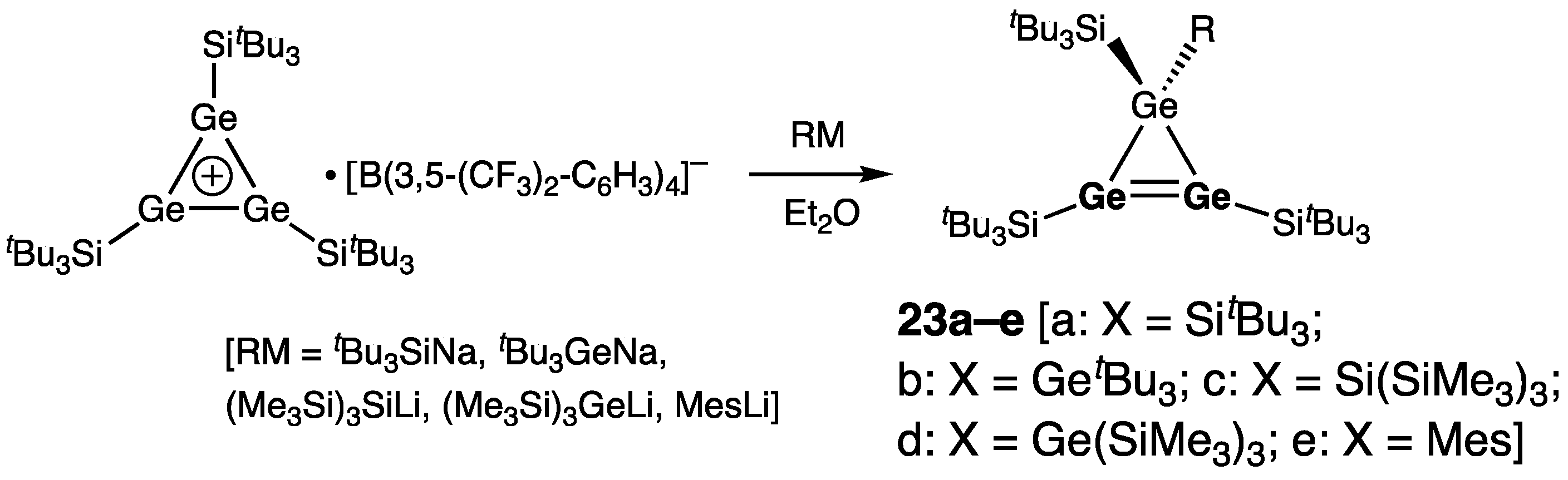
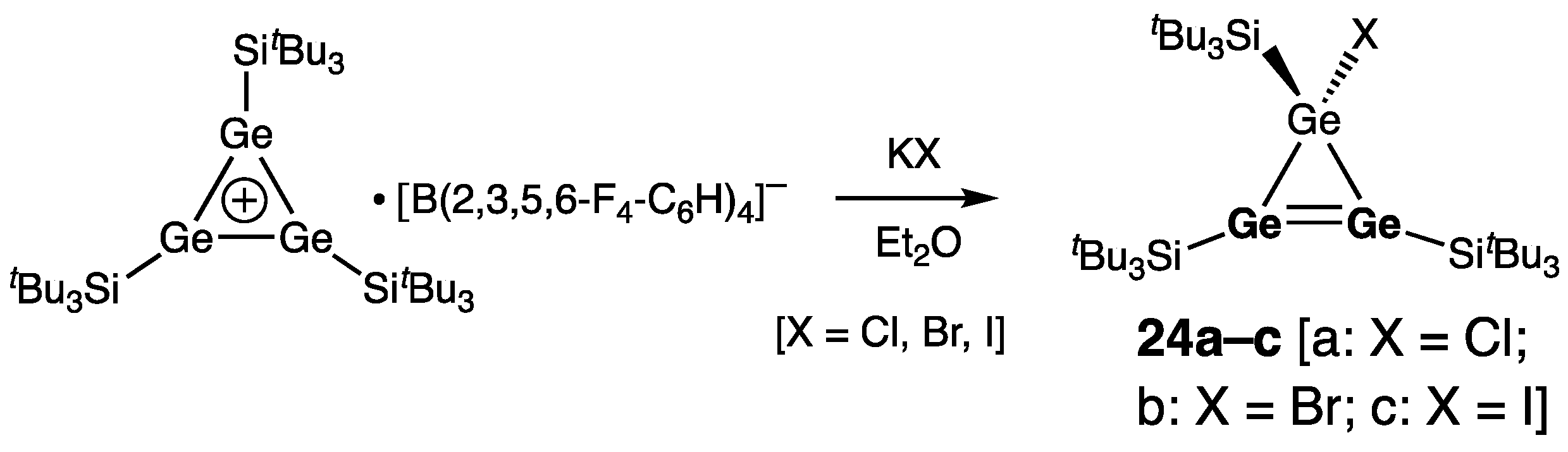

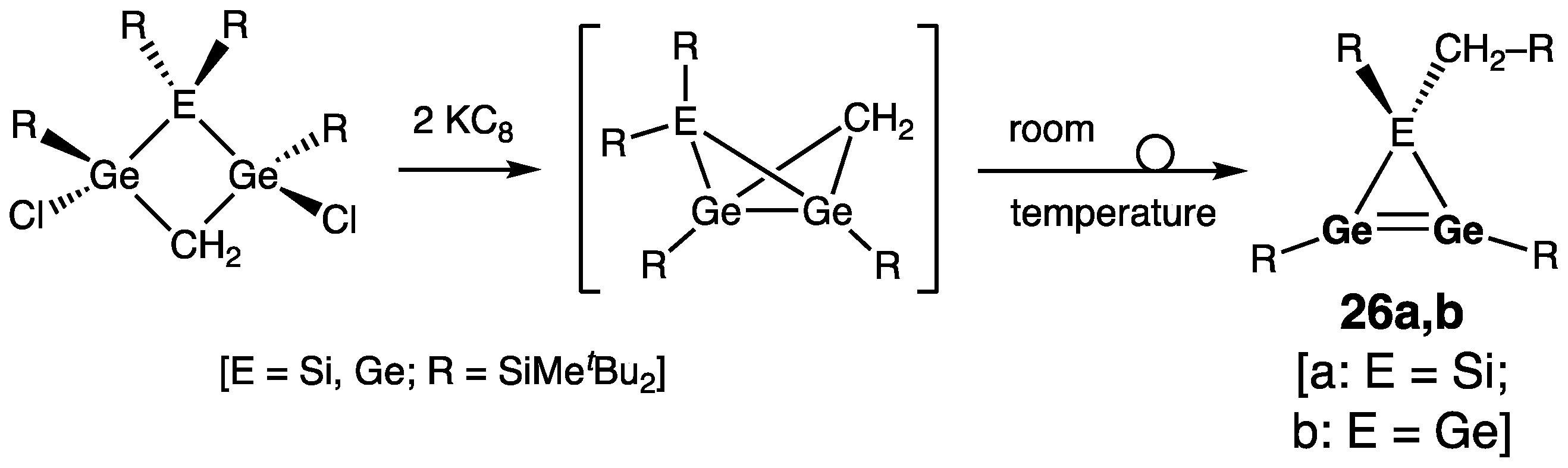
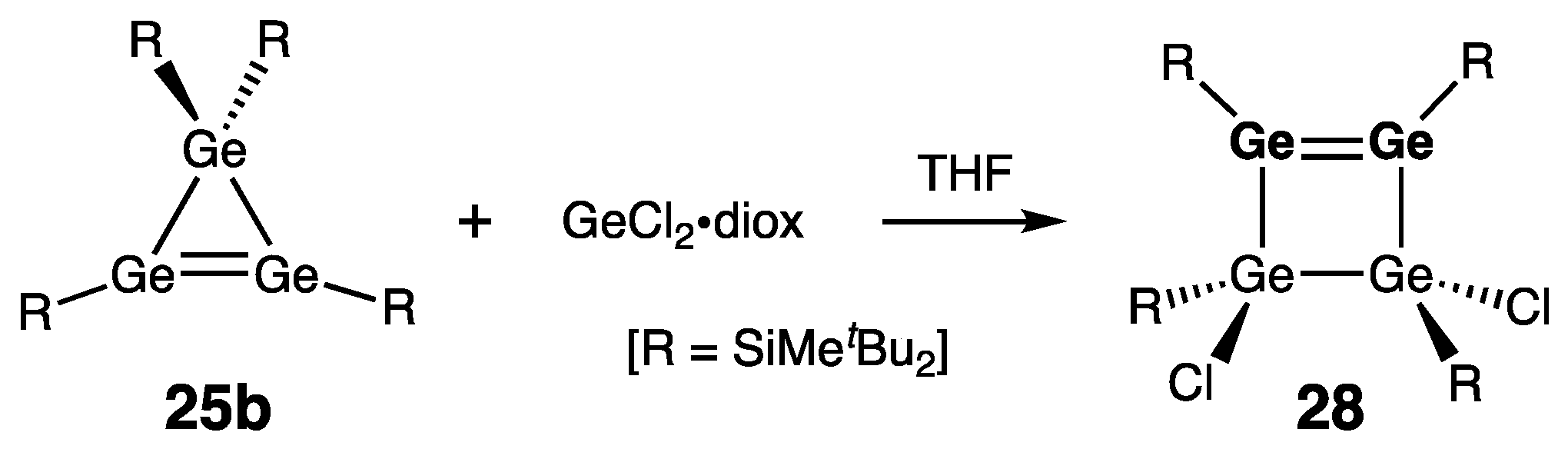
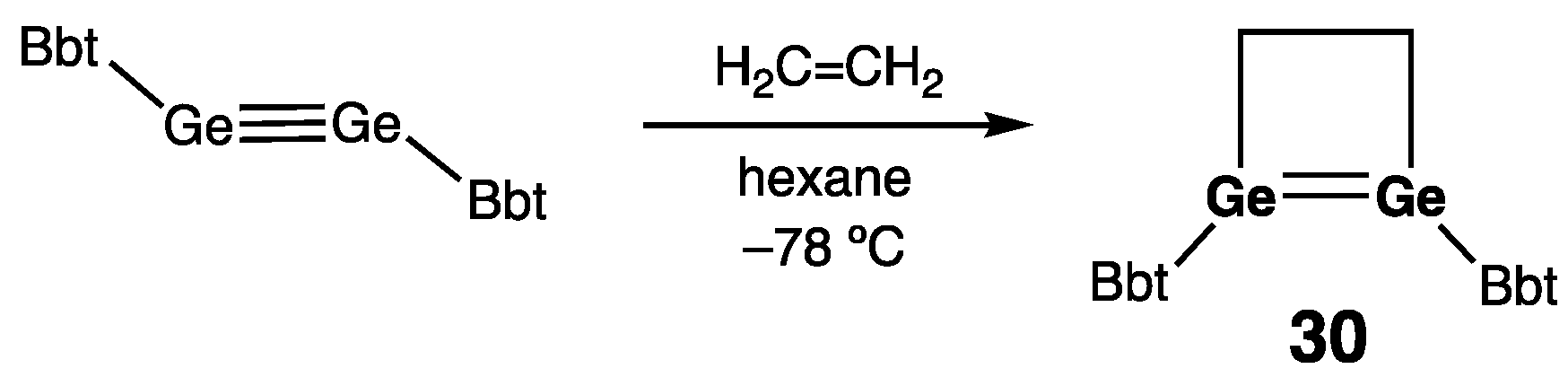
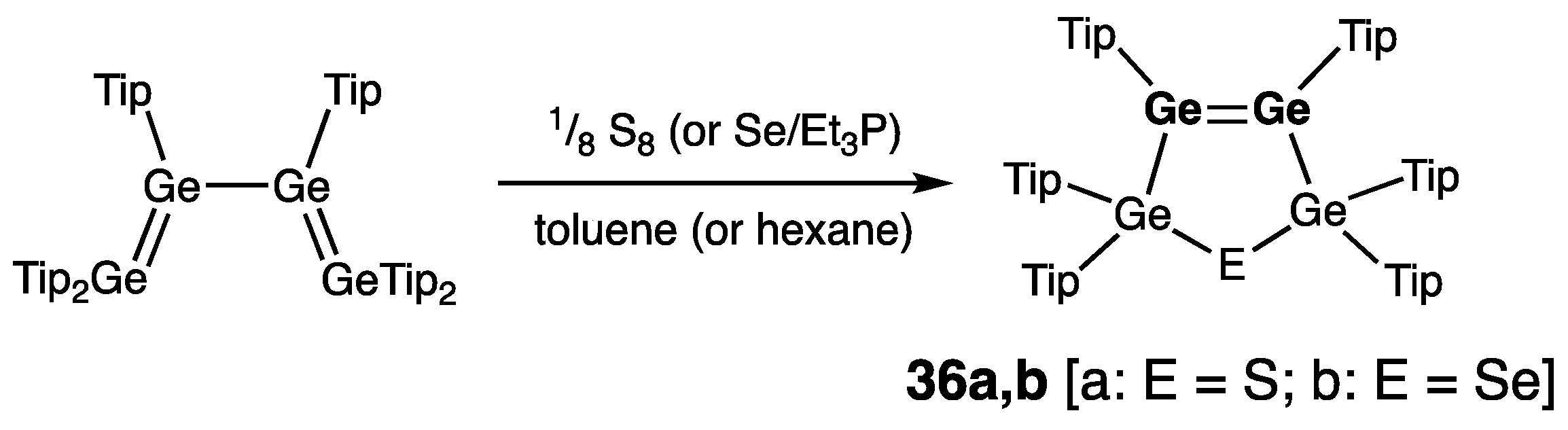

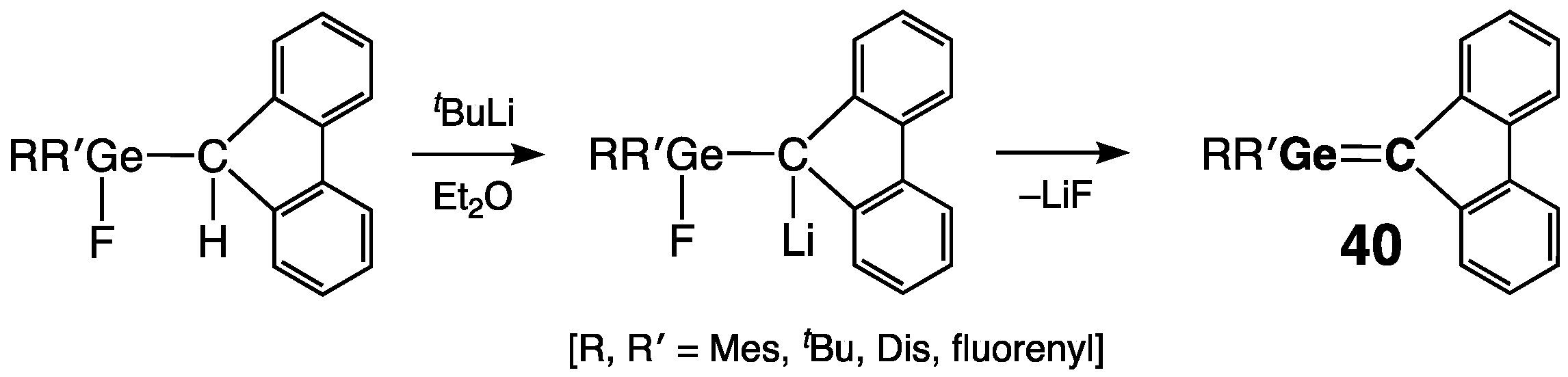

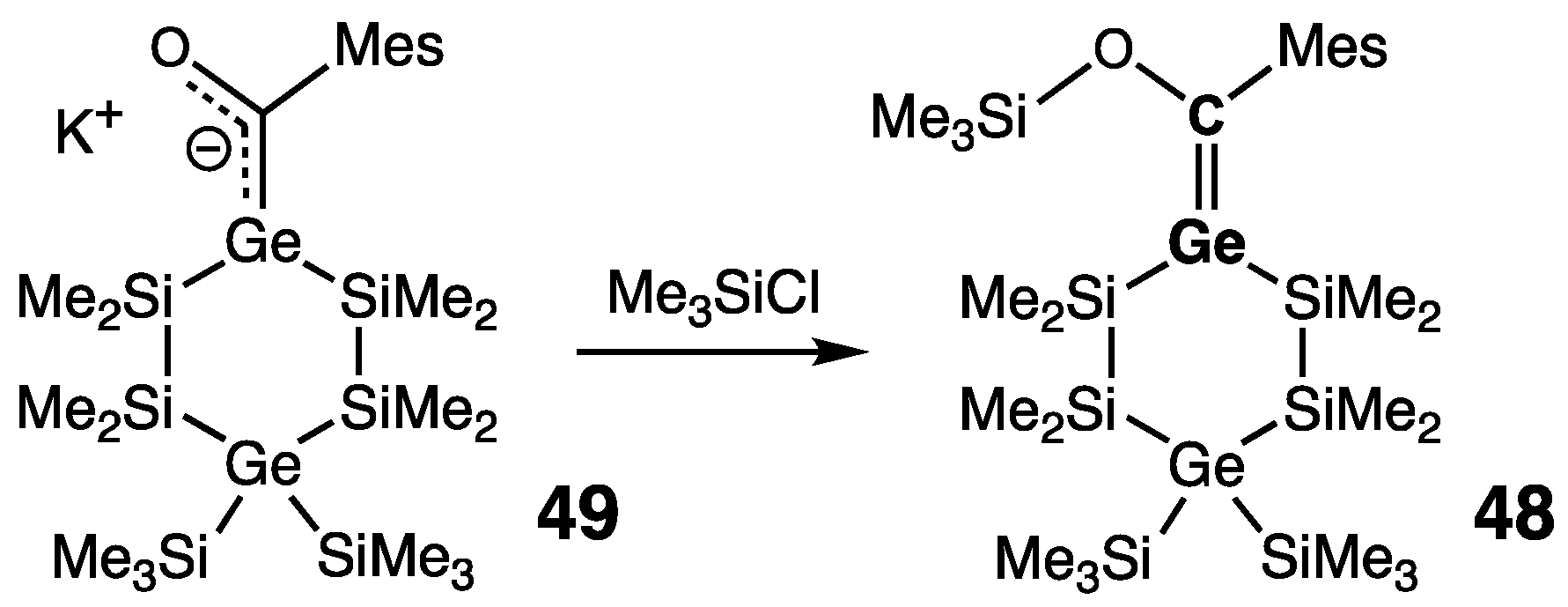

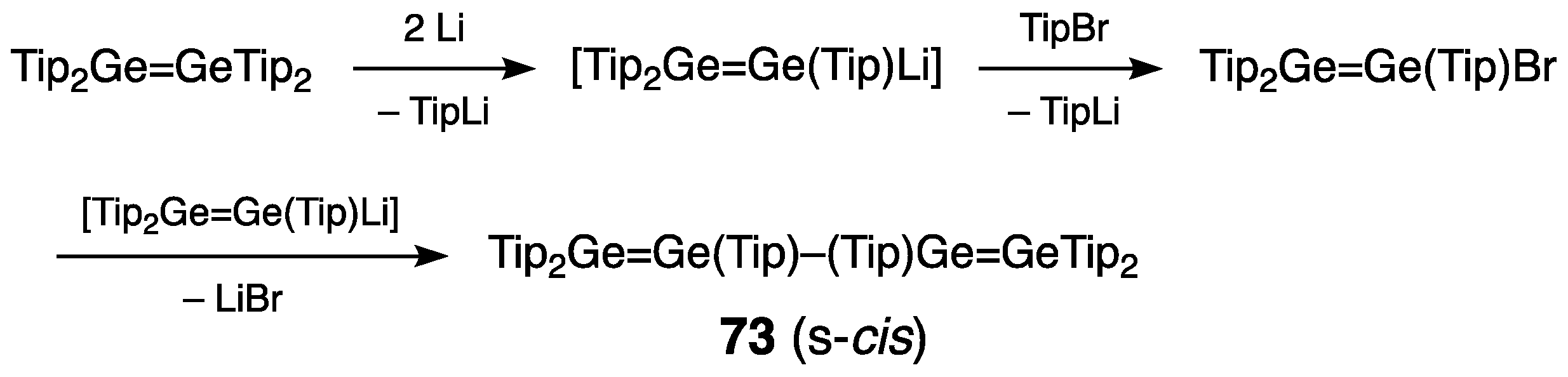

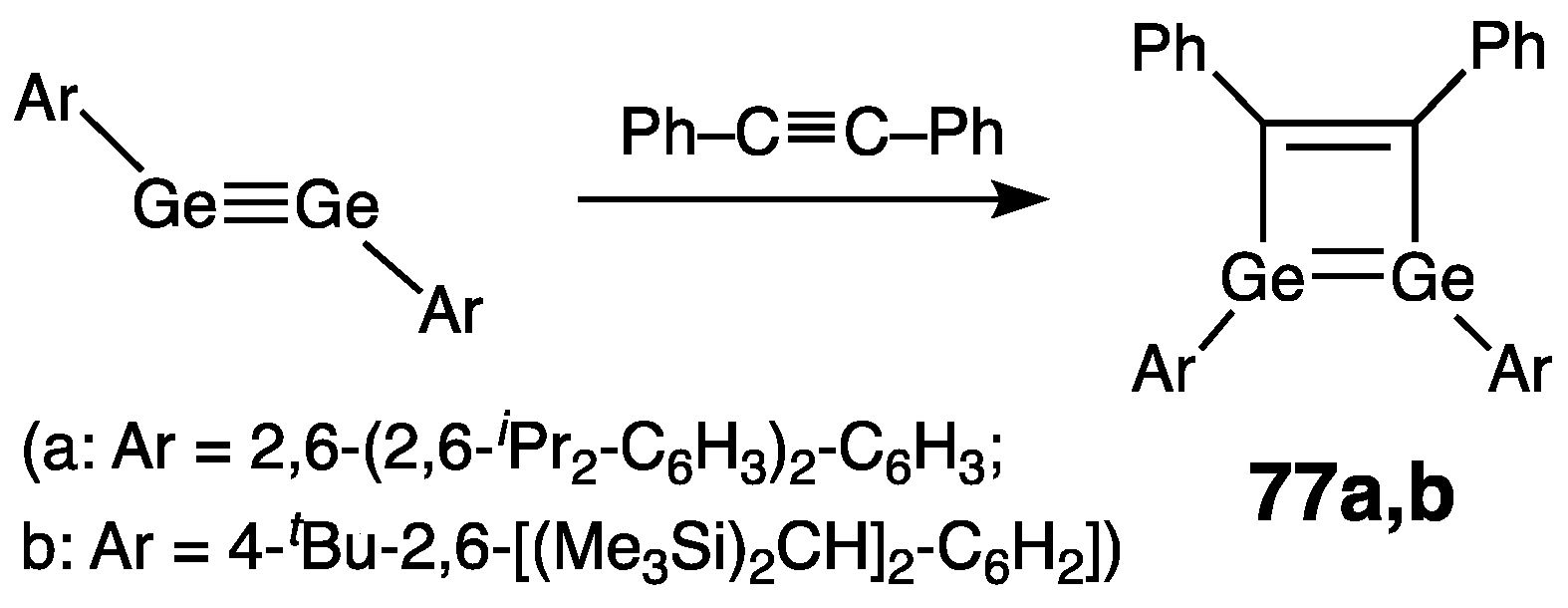


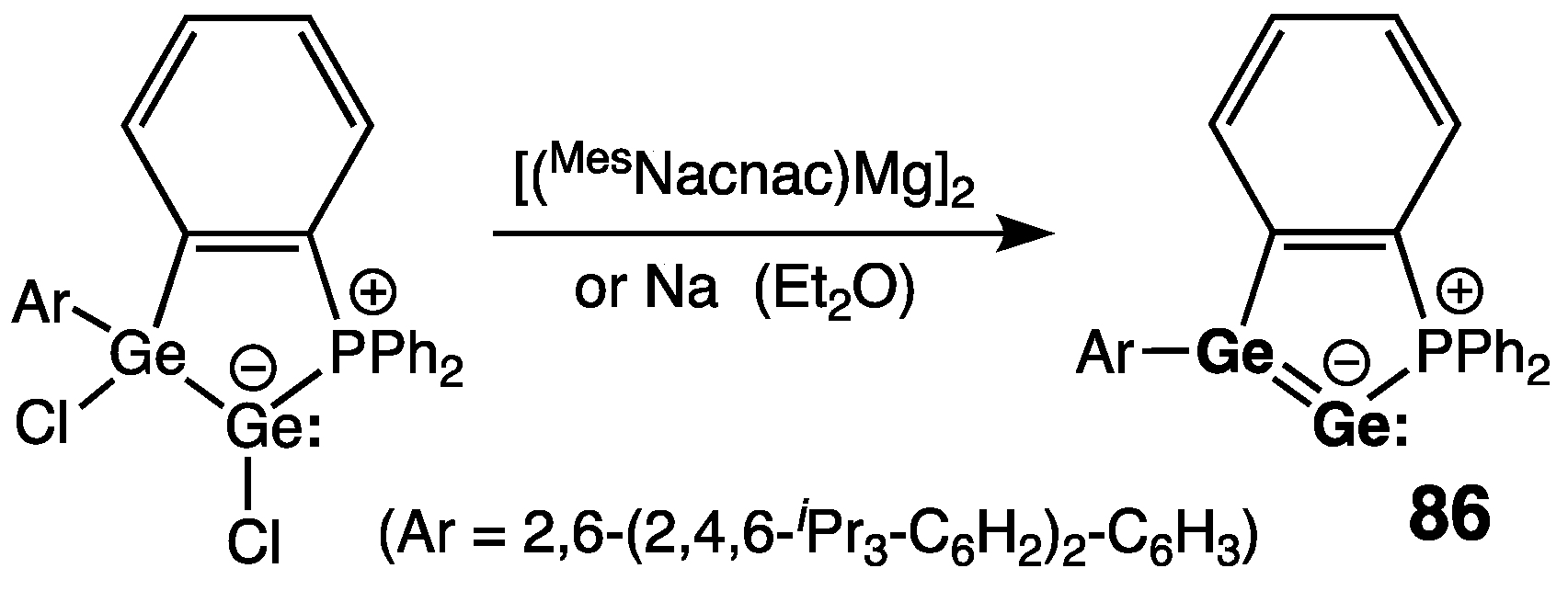

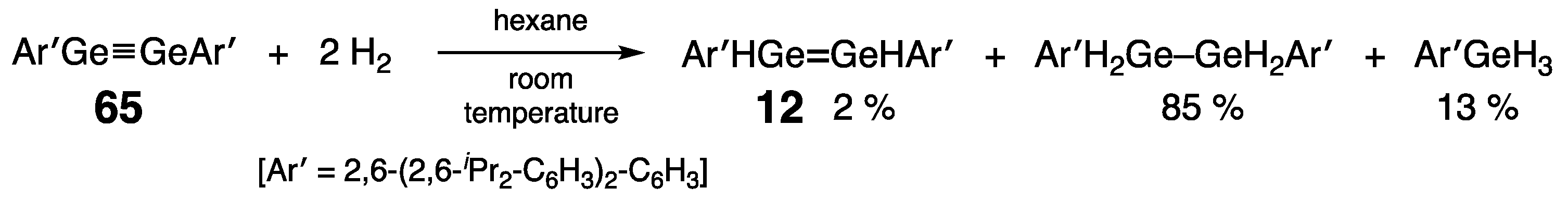
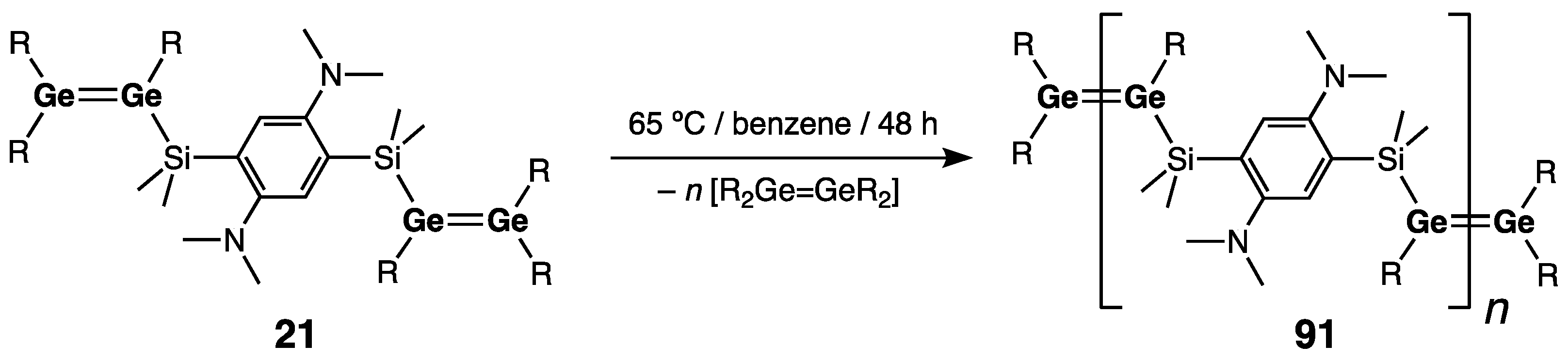

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
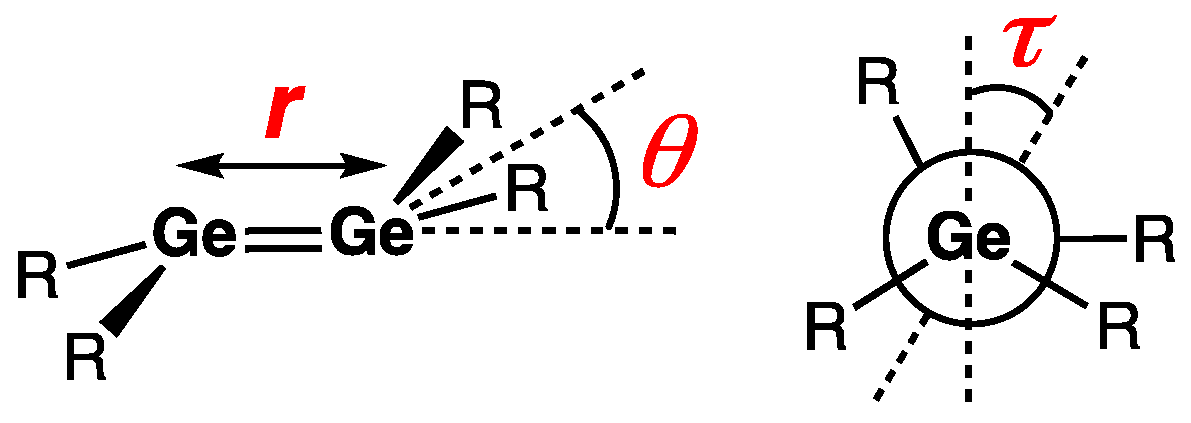
| Digermene (Preparation Method) | rGe=Ge (Å) | θ (°) | ΣGe (°) | τGe=Ge (°) | Reference |
|---|---|---|---|---|---|
| 1 (B) | 2.347(2) | 32.0 | 348.5 | 0.0 | [12] |
| 2 (A) | 2.2856(8) | 33.4 | 348.1 | 2.9 | [14] |
| 3a (B) | 2.3173(3) | 39.7 | 342.9 | — | [15] |
| 3b (B) | 2.347(3) | 37.9 | 343.0 | — | [15] |
| 3c (B) | 2.3183(5) | 33.7 | 348.4 | — | [15] |
| 4 (B) | 2.3224(4) | 41.7 | 341.9 | — | [16] |
| 5 (B) | 2.3643(4) | 37.2/42.6 | — | — | [17] |
| 6 (B) | 2.5087(7) | 44.6 | — | — | [18] |
| 7 (B) | 2.4065(6)/ 2.3970(6) | — | 335.5/336.3 | — | [19] |
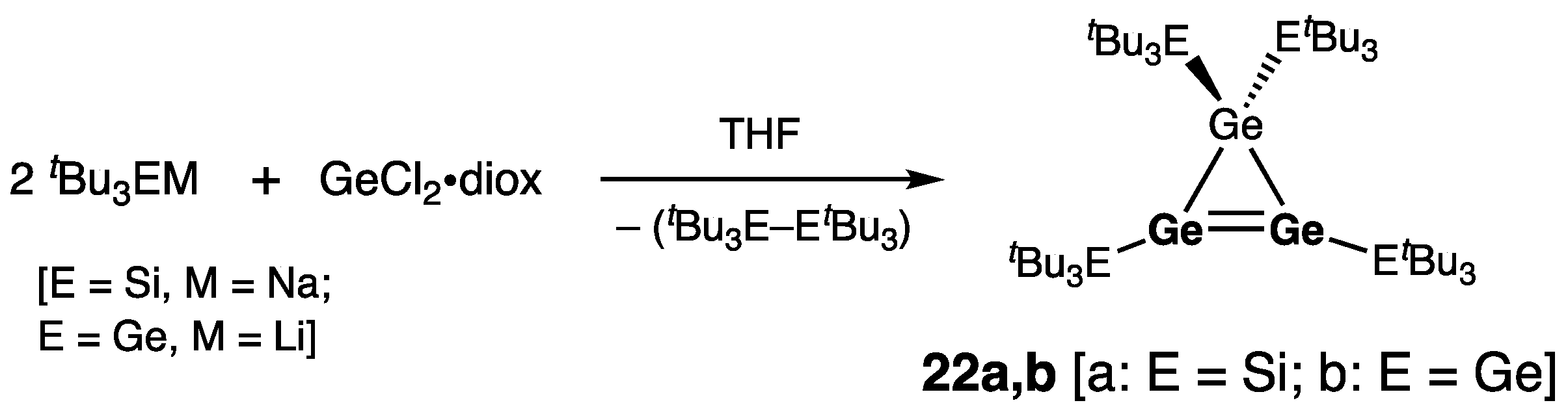
| 9 (C) | 2.2703(8) | 0.3 | 360 | 7.5 | [22] |
| 10 (C) | 2.416(2) | — | 356.6/355.3 | 16.5/34.6 | [23] |
| 11 (C) | 2.3320(5) | 43.7 | 337.8 | — | [24] |
| 12 (D) | 2.3026(3) | 45.0 | — | — | [25] |
| 13 (D) | 2.3098(5) | 36.8 [GeAr′R] | 345.3 [GeAr′R] | — | [27] |
| 14 (C) | 2.346(2) | — | 358.8/359.2 | 52.8 | [31,32] |
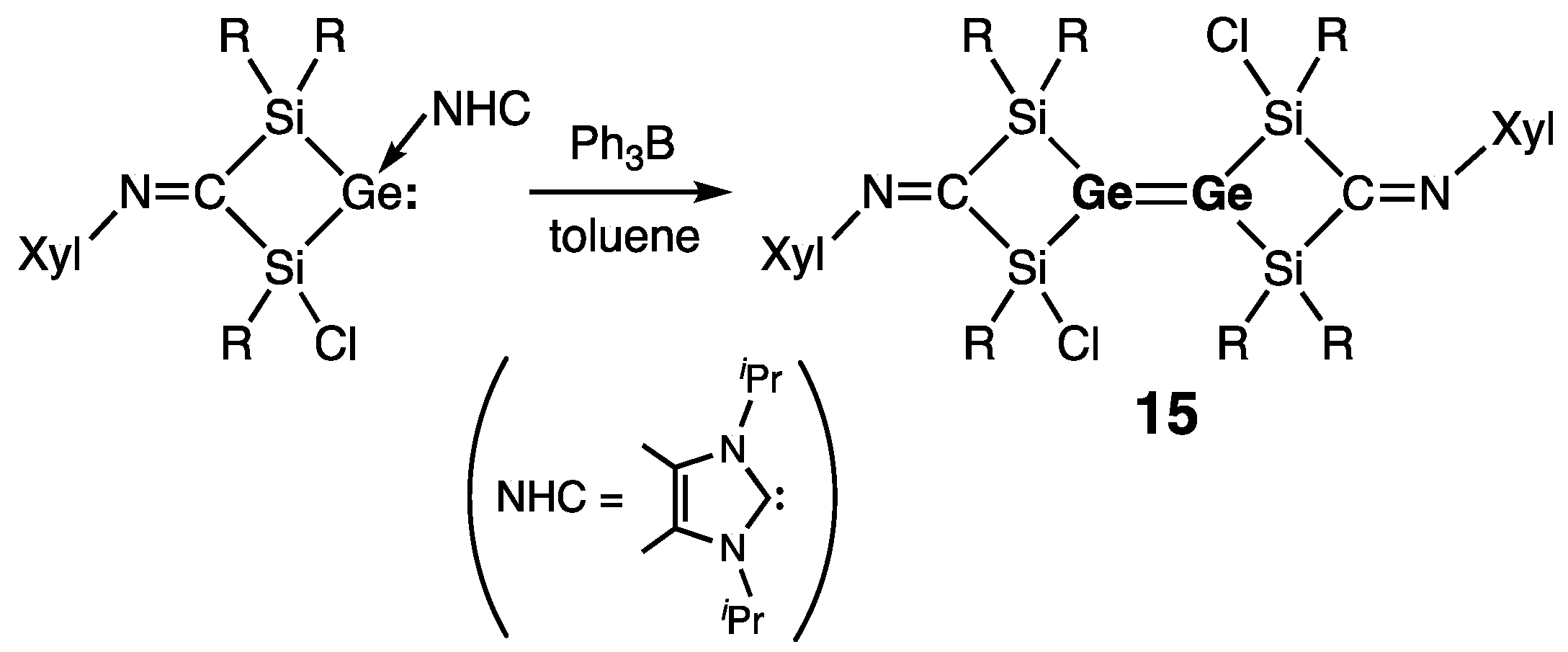
| 15 (special) | 2.2944(4) | 37.7 | 334.5 | — | [34] |
| 16 (special) | 2.284(6) | 7.1/12.8 | — | 19.9 | [35] |
| 17a (special) | 2.4119(5) | 44.3 | 335.9 | — | [37] |
| 17b (special) | 2.4145(3) | 43.3 | 337.1 | — | [37] |
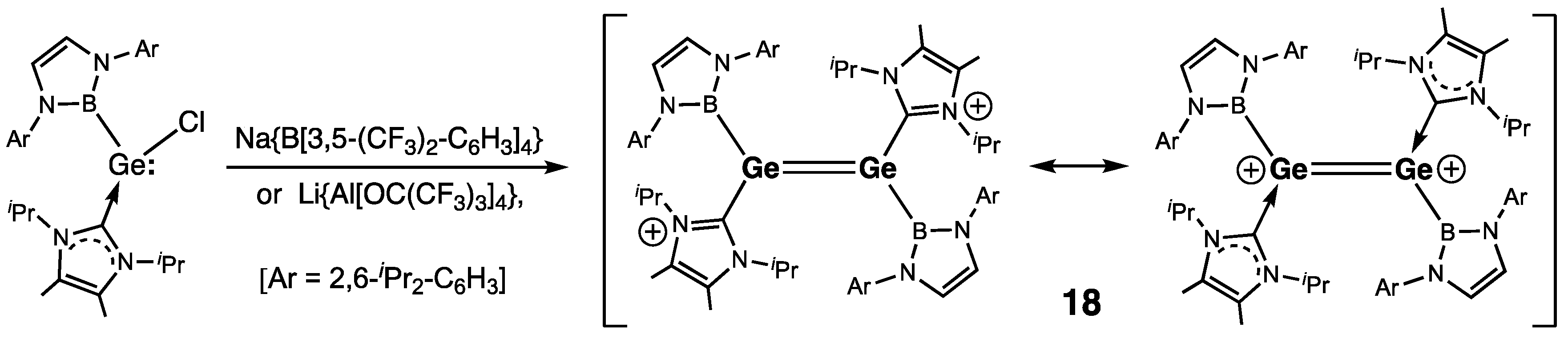
| 18 (B) | 2.300(2) | — | 353.1/353.6 | — | [38] |
| 19 (B) | 2.380(1) | — | — | — | [39] |
| 20 (special) | 2.2576(5) | 21.5 | — | 0 | [40] |
| 21 (special) | 2.3038(4) | 24.9/31.9 | — | 18.0 | [40] |
| Cyclic Digermene | rGe=Ge (Å) | ΣGe (°) | τGe=Ge (°) | Ring Folding (°) | Reference |
|---|---|---|---|---|---|
| Heavy cyclopropenes | |||||
| 22a | 2.239(4) | 359.9 | — | [41] | |
| 23c | 2.264(2) | 358.7/359.8 | 8.1 | [43] | |
| 24a | 2.2723(8) | 357.2/359.3 | 29.7 | [44] | |
| 24b | 2.2743(8) | 343.7/359.4 | 34.9 | [44] | |
| 24c | 2.2721(6) | 344.8/359.6 | 35.3 | [44] | |
| 25a | 2.2429(6) | 353.8/354.2 | 51.0 | [45,46] | |
| Heavy cyclobutenes | |||||
| 27 | 2.2911(4) | 357.2/358.6 | — | 28.3 | [50] |
| 28 | 2.2993(5) | 354.4/356.4 | — | 24.4/24.6 | [51] |
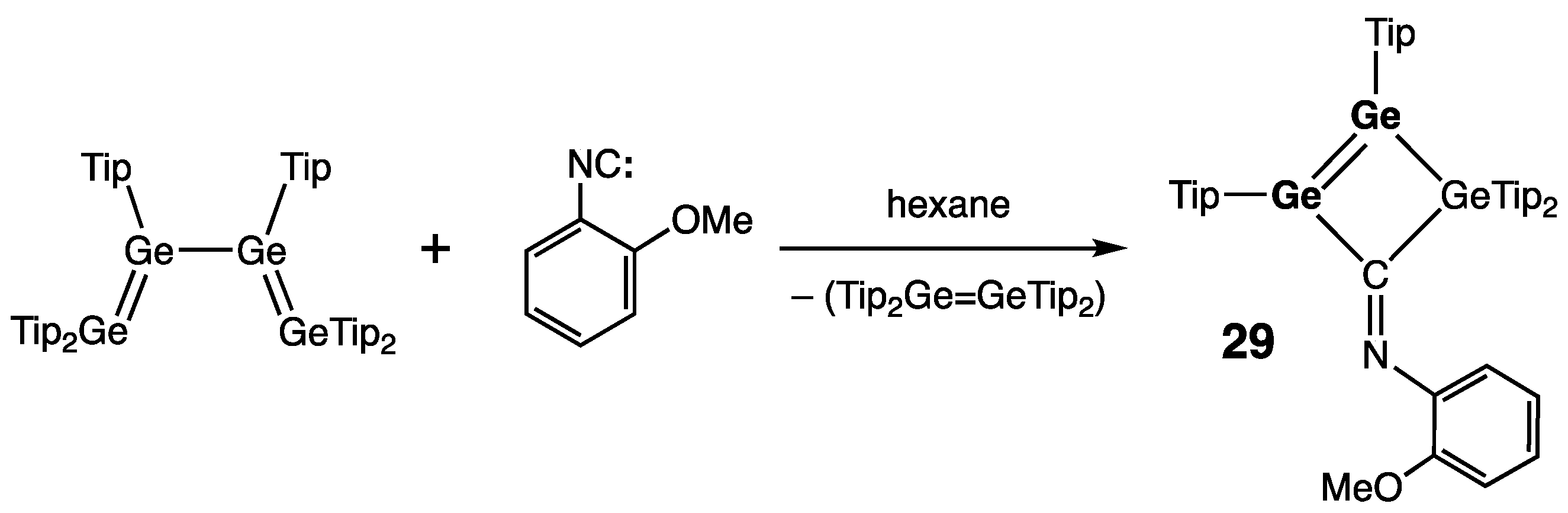
| 29 | 2.2808(7) | — | — | — | [52] |
| 30 | 2.4132(5) | — | — | 11.1 | [53] |
| Heavy cyclopentenes | |||||
| 36a | 2.2841(5) | 348.4/351.4 | — | — | [57] |
| 36b | 2.2975(5) | 349.2/349.9 | — | — | [52] |
| 37 | 2.2663(9) | 360.0/360.0 | 16.2 | — | [58] |
| Heavy cyclohexene | |||||
| 38 | 2.2896(6) | 359.5/360.0 | 5.2 | — | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, V.Y. Organogermanium Analogues of Alkenes, Alkynes, 1,3-Dienes, Allenes, and Vinylidenes. Molecules 2023, 28, 1558. https://doi.org/10.3390/molecules28041558
Lee VY. Organogermanium Analogues of Alkenes, Alkynes, 1,3-Dienes, Allenes, and Vinylidenes. Molecules. 2023; 28(4):1558. https://doi.org/10.3390/molecules28041558
Chicago/Turabian StyleLee, Vladimir Ya. 2023. "Organogermanium Analogues of Alkenes, Alkynes, 1,3-Dienes, Allenes, and Vinylidenes" Molecules 28, no. 4: 1558. https://doi.org/10.3390/molecules28041558
APA StyleLee, V. Y. (2023). Organogermanium Analogues of Alkenes, Alkynes, 1,3-Dienes, Allenes, and Vinylidenes. Molecules, 28(4), 1558. https://doi.org/10.3390/molecules28041558