
Analytical Investigation of Iron-Based Stains on Carbonate Stones: Rust Formation, Diffusion Mechanisms, and Speciation


Abstract
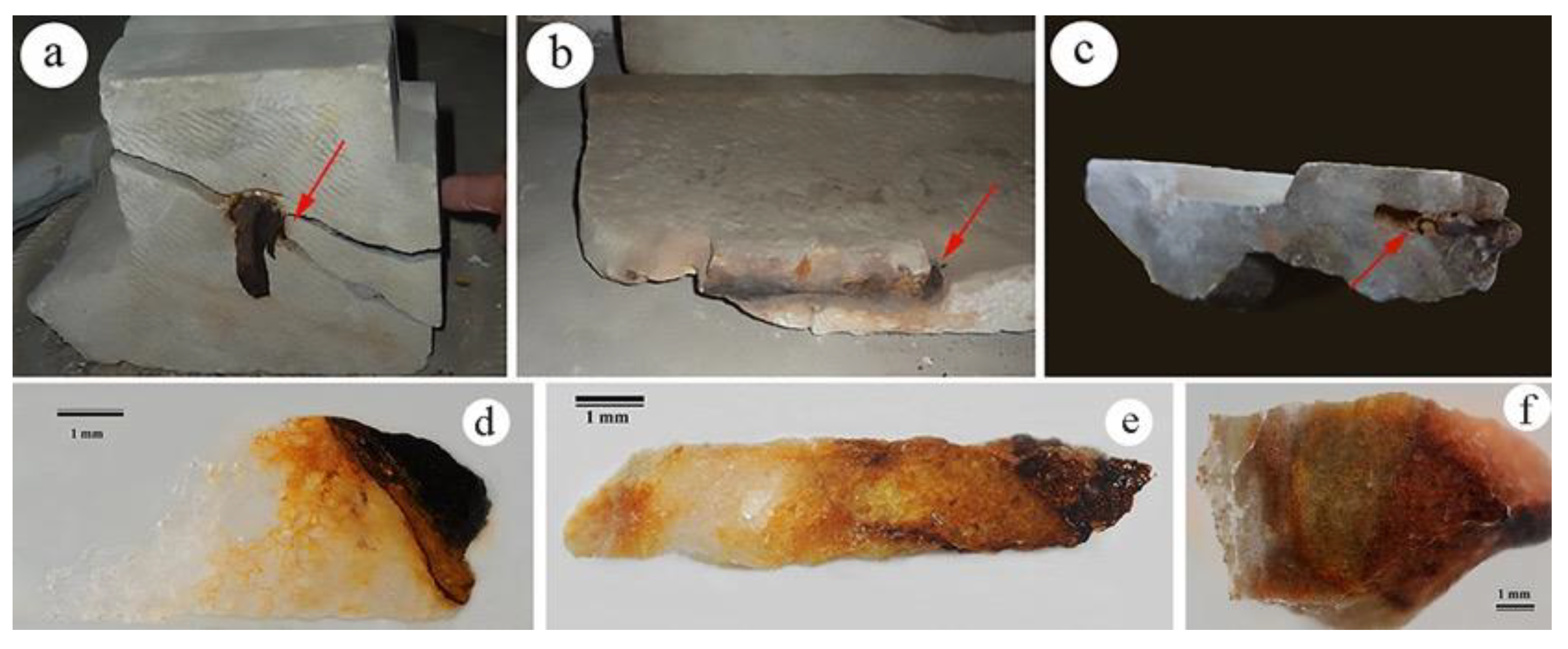
:1. Introduction
2. Results
2.1. Optical Microscopy (OM) and Electron Scanning Microscopy Coupled with X-ray Microanalysis (SEM/EDS)
2.2. XPS Analysis
2.3. Mössbauer Analysis
3. Discussion
4. Materials and Methods
4.1. Sampling
4.2. Analytical Techniques
4.2.1. Optical Microscopy
4.2.2. Scanning Electron Microscopy with EDS (Energy-Dispersive Spectroscopy)
4.2.3. X-ray Photoelectron Spectroscopy (XPS)
4.2.4. Mössbauer Spectroscopy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vidorni, G.; Sardella, A.; De Nuntiis, P.; Volpi, F.; Dinoi, A.; Contini, D.; Bonazza, A. Air pollution impact on carbonate building stones in Italian urban sites. Eur. Phys. J. Plus 2019, 134, 1–10. [Google Scholar] [CrossRef]
- appitelli, F.; Cattò, C.; Villa, F. The control of cultural heritage microbial deterioration. Microorganisms 2020, 8, 1542. [Google Scholar] [CrossRef]
- Salvi, A.M.; Langerame, F.; Macchia, A.; Sammartino, M.P.; Tabasso, M.L. XPS characterization of (copperbased) ed stains formed on limestone surfaces of outdoor Roman monuments. Chem. Cent. J. 2012, 6, S10. [Google Scholar] [CrossRef]
- Winkler, E.M. Iron in Minerals and the Formation of Rust in Stone. In Stone Archit; Springer: Berlin/Heidelberg, Germany, 1997; pp. 233–240. [Google Scholar]
- Macchia, A.; Ruffolo, S.A.; Rivaroli, L.; La Russa, M.F. The treatment of iron-stained marble: Toward a “green” solution. Int. J. Conserv. Sci. 2016, 7, 323–332. [Google Scholar]
- Schwertmann, U. Solubility and dissolution of iron oxides, Iron nutrition and interactions in plants. Springer Neth. 1991, 130, 1–25. [Google Scholar]
- Mohapatra, M.; Anand, S. Synthesis and applications of nano-structured iron oxides/hydroxides—A review. Int. J. Eng. Sci. Technol. 2010, 2, 127–146. [Google Scholar] [CrossRef]
- Dillmann, P.; Mazaudier, F.; Hoerlé, S. Advances in understanding atmospheric corrosion of iron. I. Rust characterisation of ancient ferrous artefacts exposed to indoor atmospheric corrosion. Corros. Sci. 2004, 46, 1401–1429. [Google Scholar] [CrossRef]
- Cudennec, Y.; Lecerf, A. The transformation of ferrihydrite into goethite or hematite, revisited. J. Solid State Chem. 2006, 179, 716–722. [Google Scholar] [CrossRef]
- Bams, V.; Dewaelem, S. Staining of white marble. Mater. Charact. 2007, 58, 1052–1062. [Google Scholar] [CrossRef]
- Anstice, C.; Alonso, C.; Molina, F.J. Cover cracking as a function of bar corrosion: Part I-experimental test. Mater. Struct. 1993, 26, 453–464. [Google Scholar]
- Caré, S.; Nguyen, Q.T.; l’Hostis, V.; Berthaud, Y. Mechanical properties of the rust layer induced by impressed current method in reinforced mortar. Cem. Concr. Res. 2008, 38, 1079–1091. [Google Scholar] [CrossRef]
- Campanella, L.; Dell’Aglio, E.; Reale, R.; Cardellicchio, F.; Salvi, A.M.; Casieri, C.; Cerichelli, G.; Gabriele, F.; Spreti, N.; Bernardo, G.; et al. Development of natural gels for cleaning the stone materials of cultural heritage from iron stains and biodeteriogenic microorganisms. In Proceedings of the XII International Conference Diagnosis, Conservation and Enhancement of the Cultural Heritage, Naples, Italy, 9–10 December 2021. [Google Scholar]
- Bernardo, G.; Guida, A.; Porcari, V.; Campanella, L.; Dell’Aglio, E.; Reale, R.; Cardellicchio, F.; Salvi, A.M.; Casieri, C.; Cerichelli, G.; et al. New materials and diagnostic techniques to prevent and control calcarenite degradation. In Proceedings of the XII International Conference Diagnosis, Conservation and Enhancement of the Cultural Heritage, Naples, Italy, 9–10 December 2021. [Google Scholar]
- Campanella, L.; Cardellicchio, F.; Dell’Aglio, E.; Reale, R.; Salvi, A.M. A green approach to clean iron stains from marble surfaces. Herit. Sci. 2022, 10, 14. [Google Scholar] [CrossRef]
- Little, B.J.; Gerke, T.L.; Lee, J.S. Mini-review: The morphology, mineralogy and microbiology of accumulated iron corrosion products. Biofouling 2014, 30, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Nabika, H. Liesegang phenomena: Spontaneous pattern formation engineered by chemical reactions. Curr. Phys. Chem. 2015, 5, 5–20. [Google Scholar] [CrossRef]
- Reale, R.; Campanella, L.; Sammartino, M.P.; Visco, G.; Bretti, G.; Ceseri, M.; Notarnicola, F. A mathematical, experimental study on iron rings formation in porous stones. J. Cult. Herit. 2019, 38, 158–166. [Google Scholar] [CrossRef]
- Merino, E.; Banerjee, A. Terra Rossa Genesis, Implications for Karst, and Eolian Dust: A Geodynamic Thread. J. Geol. 2008, 116, 62–75. [Google Scholar] [CrossRef]
- Loeppert, R.H.; Hossner, L.R. Reaction of Fe2+ and Fe3+ with Calcite. Clays Clay Miner. 1984, 32, 213–222. [Google Scholar] [CrossRef]
- Magalhaes, M.S.; Brandao, P.R.G.; Tavares, R.P. Types of goethite from Quadrilátero Ferrífero’s iron ores and their implications in the sintering process. Miner. Process. Extr. Metall. 2007, 116, 54–64. [Google Scholar] [CrossRef]
- Castle, J.E.; Chapman-Kpodo, H.; Proctor, A.; Salvi, A.M. Curve-fitting in XPS using extrinsic and intrinsic background structure. J. Electron Spectrosc. Relat. Phenom. 2000, 106, 65–80. [Google Scholar] [CrossRef]
- Castle, J.E.; Salvi, A.M. Chemical state information from the near-peak region of the X-ray photoelectron background. J. Electron Spectrosc. Relat. Phenom. 2001, 114–116, 1103–1113. [Google Scholar] [CrossRef]
- Graat, P.C.J.; Somers, M.A.J. Simultaneous determination of composition and thickness of thin iron oxide films from XPS Fe2p spectra. Appl. Surf. Sci. 1996, 100–101, 36–40. [Google Scholar] [CrossRef]
- Briggs, D.; Grant, J.T. Surface Analysis by Auger and X-ray Photoelectron Spectroscopy; IM Publications: Chichester, UK, 2003. [Google Scholar]
- Briggs, D.; Seah, M.P. Practical Surface Analysis; John Wiley & Sons: Chichester, UK, 1990; ISBN 0-471-26279-X. [Google Scholar]
- Yamashita, T.; Hayes, P. Analysis of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- NIST X-ray Photoelectron Spectroscopy Database 20, Version 4.1. Available online: https://srdata.nist.gov/xps/default.aspx (accessed on 30 September 2022).
- Kloprogge, J.T.; Ponce, C.P.; Ortillo, D.O. X-ray Photoelectron Spectroscopy Study of some Organic and Inorganic Modified Clay Minerals. Materials 2021, 14, 7115. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Li, H.; Fan, X.; Yan, H.; Cai, M.; Zhu, M. Insights into the tribological behavior of choline chloride—Urea and choline chloride—Thiourea deep eutectic solvents. Friction 2021, 11, 76–92. [Google Scholar] [CrossRef]
- Ciotonea, C.; Averlant, R.; Rochard, G.; Mamede, A.S.; Giraudon, J.M.; Alamdari, H.; Lamonier, J.F.; Royer, S. A simple and green procedure to prepare efficient manganese oxide nanopowder for the low temperature removal of formaldehyde. ChemCatChem 2017, 9, 2366–2376. [Google Scholar] [CrossRef]
- Wang, H.; Liang, Y.; Gong, M.; Li, Y.; Chang, W.; Mefford, T.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; et al. An ultrafast nickel–iron battery from strongly coupled inorganic nanoparticle/nanocarbon hybrid materials. Nat. Commun. 2012, 3, 917. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, C.; Yue, J.; Zhang, X.; Ma, L. Effect of a potassium promoter on the Fischer–Tropsch synthesis of light olefins over iron carbide catalysts encapsulated in graphene-like carbon. Catal. Sci. Technol. 2019, 9, 2728–2741. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Lin, H.; Gupta, S.; Wang, C.; Tao, Z.; Fu, H.; Wang, T.; Zheng, J.; Wu, G.; et al. Directly converting Fe-doped metal- organic frameworks into highly active and stable Fe-N-C catalysts for oxygen reduction in acid. Nano Energy 2016, 25, 110–119. [Google Scholar] [CrossRef]
- Martín-García, L.; Bernal-Villamil, I.; Oujja, M.; Carrasco, E.; Gargallo-Caballero, R.; Castillejo, M.; Marco, J.F.; Gallego, S.; de la Figuera, J. Unconventional properties of nanometric FeO (111) films on Ru (0001): Stoichiometry and surface structure. J. Mater. Chem. C 2016, 4, 1850–1859. [Google Scholar] [CrossRef]
- Reale, R. Alterazioni Cromatiche di Materiali Lapidei Carbonatici: Studio Delle Macchie Indotte Dalla Coesistenza Con Materiali Ferrosi e Metodi Innovativi per la Loro Rimozione. Ph.D. Thesis, Sapienza University, Rome, Italy, 2017. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences, and Uses; Wiley-Vch: Weinheim, Germany, 2003; Volume 664. [Google Scholar]
- Leidheiser, H., Jr.; Czakó-Nagy, I. A Mössbauer spectroscopic study of rust formed during simulated atmospheric corrosion. Corros. Sci. 1984, 24, 569–577. [Google Scholar] [CrossRef]
- Joos, A.; Rumenapp, C.; Wagner, F.; Gleich, B. Characterisation of iron oxide nanoparticles by Mossbauer spectroscopy at ambient temperature? J. Magn. Magn. Mater. 2016, 399, 123–129. [Google Scholar] [CrossRef]
- Beltran, M.; Playà, E.; Artigau, M.; Arroyo, P.; Guinea, A. Iron patinas on alabaster surfaces (Santa Maria de Poblet Monastery, Tarragona, NE Spain). J. Cult. Herit. 2016, 18, 370–374. [Google Scholar] [CrossRef]
- Grassini, S.; Angelini, E.; Parvis, M.; Bouchar, M.; Dillmann, P.; Neff, D. An in situ corrosion study of Middle Ages wrought iron bar chains in the Amiens Cathedral. Appl. Phys. A 2013, 113, 971–979. [Google Scholar] [CrossRef]
- Fassina, V. Environmental pollution in relation to stone decay. Durab. Build. Mater. 1988, 5, 317–358. [Google Scholar]
- Gaylarde, C.; Little, B. Biodeterioration of stone and metal- Fundamental microbial cycling processes with spatial and temporal scale differences. Sci. Total Environ. 2022, 823, 153193. [Google Scholar] [CrossRef] [PubMed]
- Lagarec, K.; Rancourt, D.G. Recoil-Mössbauer Spectral Analysis Software for Windows; University of Ottawa: Ottawa, ON, USA, 1998. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spectrum | O | Na | Mg | Al | Si | P | S | Cl | K | Ca | Fe |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 43.3 | 0.1 | 0.7 | 0.2 | 0.3 | 0.1 | 0.1 | 0.1 | 0.0 | 54.8 | 0.3 |
| 2 | 33.0 | 0.7 | 0.7 | 0.4 | 4.3 | 0.6 | 0.2 | 0.1 | 0.2 | 4.3 | 55.6 |
| (a) | ||||
|---|---|---|---|---|
| Element, Orbital Chemical State | Carrara Surface | Carrara Interface | ||
| Corrected BE (eV) | Normalized Area | Corrected BE (eV) | Normalized Area | |
| C1s → lower BE carbons * | 283.5 | 122.8 | 283.3 | 247.1 |
| C-C (IS) | 285.0 | 1267.8 | 285.0 | 1887.4 |
| C-O/C-O-C | 286.0 | 946.9 | 286.5 | 328.9 |
| O-C-O/C=O/COO | 288.7 | 341.5 | 288.6 | 517.7 |
| CO32−/COOR | 290.5 | 723.1 | 290.1 | 3075.1 |
| * Typical of carbides, polycyclic compounds etc. | ||||
| Ca 2p3/2→Ca | --- | --- | 344.8 | 130.1 |
| nnaCaO/ | 345.9 | 44.8 | 346.7 | 692.3 |
| CaCO3 | 347.9 | 441.7 | 347.9 | 2437.6 |
| Ca 2p1/2→ Ca | --- | --- | 348.3 | 65.0 |
| CaO | 349.5 | 22.4 | 350.2 | 346.1 |
| CaCO3 | 351.5 | 220.9 | 351.4 | 1194.4 |
| Mg KL1L2 * | --- | --- | 352.8 | --- |
| SU1 CaCO3 | --- | --- | 355.9 | 100.1 |
| SU2 CaCO3 | --- | --- | 359.7 | 121.4 |
| * Auger signal | ||||
| O1s-peak 1 (metal oxides) | 530.4 | 2422.3 | 530.0 | 1275.8 |
| O1s-peak 2 (calcite matrix) | 532.3 | 4337.8 | 532.1 | 11610.0 |
| Mg1s→ MgO | 1304.8 | 51.8 | 1305.4 | 504.8 |
| (b) | ||||
| Fe2p3/2 Fe |→ Metallic FeO ↔ Fe(II) compounds | --- | --- | 704.6 708.2 | 30.0 28.7 |
| Fe2O3main peak | 710.7 | 387.1 | 710.2 | 67.0 |
| Fe2O3 MS(I) | 711.9 | 290.3 | 711.5 | 53.6 |
| Fe2O3 MS(II) | 713.8 | 130.6 | 713.5 | 21.4 |
| Fe2O3 SU | 720.6 | 185.6 | 719.6 | 43.5 |
| Fe2p1/2 Fe |→ Metallic FeO ↔ Fe(II) compounds Fe2O3 main peak | --- --- 724.1 | --- --- 193.5 | 717.6 721.2 723.5 | 15.0 14.4 26.1 |
| Fe2O3 MS(I) | 725.3 | 145.1 | 724.8 | 19.6 |
| Fe2O3 MS(II) | 727.2 | 65.3 | 726.6 | 9.1 |
| Fe2O3 SU | 734.2 | 92.8 | 732.6 | 16.0 |
| Sample | IS (mm/s) | QS (mm/s) | HF (T) | A (%) | Attribution |
|---|---|---|---|---|---|
| Sp | 0.34 | 0.57 | – | 100 | Nanogoethite/lepidocrocite/ferrihydrite/nanohematite |
| Sp/S | 0.37 | 0.59 | – | 100 | Nanogoethite/lepidocrocite/ferrihydrite/nanohematite |
| Sp/S/A | 0.36 | 0.61 | – | 100 | Nanogoethite/lepidocrocite/ferrihydrite/nanohematite |
| Sp/S/A/W | 0.37 | 0.61 | – | 89 | Nanogoethite/lepidocrocite/ferrihydrite/nanohematite |
| 0.33 | – | 49 | 11 | Maghemite/hematite | |
| AS25 | 0.35 | 0.60 | – | 14 | Nanogoethite/lepidocrocite/ferrihydrite/nanohematite |
| 0.30 | – | 48 | 54 | Maghemite/hematite | |
| 0.34 | – | 50 | 32 | Maghemite/hematite | |
| NS1 | 0.37 | – | 36 | 17 | Goethite |
| 0.41 | – | 31 | 5 | Nanomagnetite | |
| 0.59. | – | 20 | 24 | Nanomagnetite | |
| 0.31 | 0.75 | – | 21 | Ferrihydrite | |
| 0.76 | 1.18 | – | 33 | Fe2.5+ (IVCT) | |
| NS3 | 0.27 | – | 49 | 37 | Maghemite/hematite |
| 0.35 | – | 33 | 5 | Nanomagnetite | |
| 0.67 | – | 20 | 6 | Nanomagnetite | |
| 0.37 | 0.55 | – | 27 | Lepidocrocite | |
| 0.67 | 1.15 | – | 25 | Fe2.5+ (IVCT) | |
| NS8 | 0.35 | – | 50 | 10 | Maghemite/hematite |
| 0.35 | – | 36 | 15 | Goethite | |
| 0.35 | – | 28 | 22 | Nanohematite | |
| 0.35 | 0.67 | – | 18 | Ferrihydrite | |
| 0.67 | 1.44 | – | 35 | Fe2.5+ (IVCT) |
| (a) | |||
| Sample | Description | Provenience | Date |
| NS1 | Base of bust | Vatican Museum | 19th century AD |
| NS3 | Sarcophagus’ fragment | Vatican Museum | 4th century AD |
| NS8 | Fragment of marble slab | Roman bath Palazzo Valentini (Room 5) | 4th century AD |
| (b) | |||
| Samples | Description | ||
| AS25 | Travertine stained indoor for three months | ||
| Sp | Marble stained outdoor during spring | ||
| Sp/S | Marble stained during spring/summer | ||
| Sp/S/A | Marble stained during spring/summer/autumn | ||
| Sp/S/A/W | Marble stained during spring/summer/autumn/winter | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reale, R.; Andreozzi, G.B.; Sammartino, M.P.; Salvi, A.M. Analytical Investigation of Iron-Based Stains on Carbonate Stones: Rust Formation, Diffusion Mechanisms, and Speciation. Molecules 2023, 28, 1582. https://doi.org/10.3390/molecules28041582
Reale R, Andreozzi GB, Sammartino MP, Salvi AM. Analytical Investigation of Iron-Based Stains on Carbonate Stones: Rust Formation, Diffusion Mechanisms, and Speciation. Molecules. 2023; 28(4):1582. https://doi.org/10.3390/molecules28041582
Chicago/Turabian StyleReale, Rita, Giovanni Battista Andreozzi, Maria Pia Sammartino, and Anna Maria Salvi. 2023. "Analytical Investigation of Iron-Based Stains on Carbonate Stones: Rust Formation, Diffusion Mechanisms, and Speciation" Molecules 28, no. 4: 1582. https://doi.org/10.3390/molecules28041582