
Comparative Studies on Resampling Techniques in Machine Learning and Deep Learning Models for Drug-Target Interaction Prediction


Abstract
:1. Introduction
2. Related Works
2.1. Machine Learning Methods in DTI Prediction
2.1.1. Support Vector Machine
2.1.2. Naïve Bayes
2.1.3. Random Forest
2.1.4. Decision Tree
2.2. Deep Learning Methods in DTI Prediction
2.2.1. Convolutional Neural Networks
2.2.2. Multilayer Perceptrons
3. Results and Discussion
3.1. Machine Learning vs Machine Learning with Sampling
3.2. Deep Learning with No Resampling
3.2.1. Convolutional Neural Network (CNN)
3.2.2. Multilayer Perceptron (MLP)
4. Materials and Methods
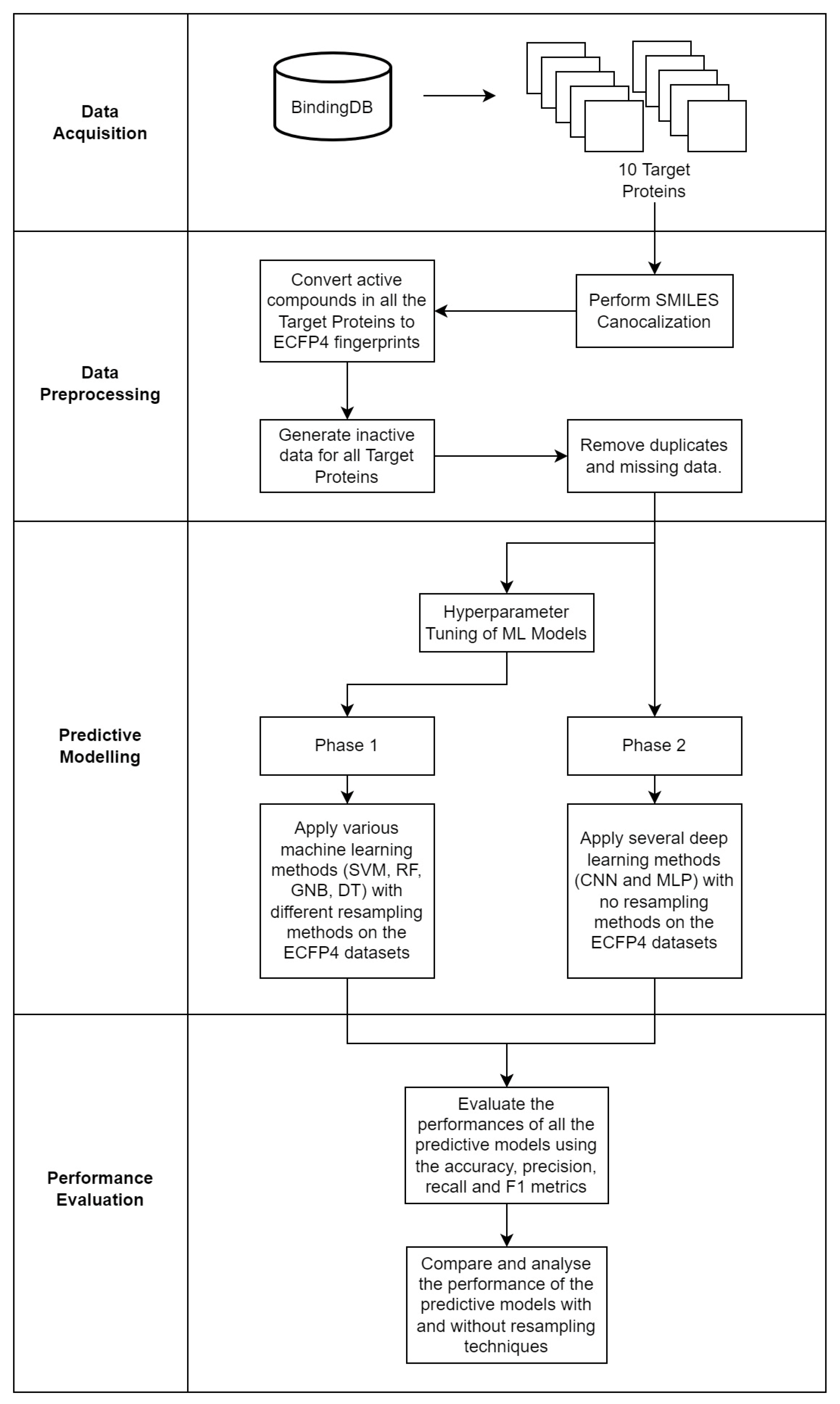
4.1. Data Acquisition
4.2. Data Preprocessing
4.3. Predictive Modeling
4.3.1. Machine Learning vs Machine Learning with Resampling
4.3.2. Deep Learning with No Sampling
4.4. Performance Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, K.Y.; Fokoue, A.; Luo, H.; Iyengar, A.; Dey, S.; Zhang, P. Interpretable Drug Target Prediction Using Deep Neural Representation. In Proceedings of the Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence, Stockholm, Sweden, 13–19 July 2018; pp. 3371–3377. [Google Scholar]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine Learning in Chemoinformatics and Drug Discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Mitchell, B.O.J. Machine Learning Methods in Chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep Learning in Drug Discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef]
- Unterthiner, T.; Mayr, A.; Klambauer, G.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Hochreiter, S. Deep Learning for Drug Target Prediction. In Proceedings of the Conference Neural Information Processing Systems Foundation, Montreal, QC, Canada, 8–13 December 2014. [Google Scholar]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-Scale Comparison of Machine Learning Methods for Drug Target Prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef]
- Molinaro, A.M.; Simon, R.; Pfeiffer, R.M. Prediction Error Estimation: A Comparison of Resampling Methods. Bioinformatics 2005, 21, 3301–3307. [Google Scholar] [CrossRef]
- Khaldy, M.A.; Kambhampati, C. Resampling Imbalanced Class and the Effectiveness of Feature Selection Methods for Heart Failure Dataset. Int. Robot. Autom. J. 2018, 4, 37–45. [Google Scholar] [CrossRef]
- Poolsawad, N.; Kambhampati, C.; Cleland, J.G.F. Balancing Class for Performance of Classification with a Clinical Dataset. In Proceedings of the Proceedings of the World Congress on Engineering, London, UK, 2–4 July 2014; Volume 1. [Google Scholar]
- Pliakos, K.; Vens, C. Drug-Target Interaction Prediction with Tree-Ensemble Learning and Output Space Reconstruction. BMC Bioinform. 2020, 21, 49. [Google Scholar] [CrossRef]
- Johnson, J.M.; Khoshgoftaar, T.M. Survey on Deep Learning with Class Imbalance. J. Big Data 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Hasanin, T.; Khoshgoftaar, T.M.; Leevy, J.L.; Bauder, R.A. Severely Imbalanced Big Data Challenges: Investigating Data Sampling Approaches. J. Big Data 2019, 6, 107. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Blood–Brain Barrier Permeability of Compounds by Machine Learning and Resampling Methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, D.F. Rules of Evidence for Cancer Molecular-Marker Discovery and Validation. Nat. Rev. Cancer 2004, 4, 309–314. [Google Scholar] [CrossRef]
- Korotcov, A.; Tkachenko, V.; Russo, D.P.; Ekins, S. Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol. Pharm. 2017, 14, 4462–4475. [Google Scholar] [CrossRef]
- Ezzat, A.; Wu, M.; Li, X.-L.; Kwoh, C.-K. Drug-Target Interaction Prediction via Class Imbalance-Aware Ensemble Learning. BMC Bioinform. 2016, 17, 509. [Google Scholar] [CrossRef]
- Yaseen, B.T.; Kurnaz, S. Drug–Target Interaction Prediction Using Artificial Intelligence. Appl. Nanosci. 2021. [Google Scholar] [CrossRef]
- Gao, D.; Chen, Q.; Zeng, Y.; Jiang, M.; Zhang, Y. Applications of Machine Learning in Drug Target Discovery. Curr. Drug Metab. 2020, 21, 790–803. [Google Scholar] [CrossRef]
- Carracedo-Reboredo, P.; Liñares-Blanco, J.; Rodríguez-Fernández, N.; Cedrón, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A Review on Machine Learning Approaches and Trends in Drug Discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Xu, L.; Ru, X.; Song, R. Application of Machine Learning for Drug–Target Interaction Prediction. Front Genet 2021, 12. [Google Scholar] [CrossRef]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine Learning Approaches and Databases for Prediction of Drug–Target Interaction: A Survey Paper. Brief Bioinform. 2021, 22, 247–269. [Google Scholar] [CrossRef]
- Faulon, J.-L.; Misra, M.; Martin, S.; Sale, K.; Sapra, R. Genome Scale Enzyme–Metabolite and Drug–Target Interaction Predictions Using the Signature Molecular Descriptor. Bioinformatics 2008, 24, 225–233. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F. Identification of Drug-Target Interactions via Multiple Information Integration. Inf. Sci. (N.Y.) 2017, 418–419, 546–560. [Google Scholar] [CrossRef]
- Lavecchia, A. Machine-Learning Approaches in Drug Discovery: Methods and Applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian Machine Learning Approach for Drug Target Identification Using Diverse Data Types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef]
- Yao, Z.-J.; Dong, J.; Che, Y.-J.; Zhu, M.-F.; Wen, M.; Wang, N.-N.; Wang, S.; Lu, A.-P.; Cao, D.-S. TargetNet: A Web Service for Predicting Potential Drug–Target Interaction Profiling via Multi-Target SAR Models. J. Comput. Aided Mol. Des. 2016, 30, 413–424. [Google Scholar] [CrossRef]
- Li, Z.-C.; Huang, M.-H.; Zhong, W.-Q.; Liu, Z.-Q.; Xie, Y.; Dai, Z.; Zou, X.-Y. Identification of Drug–Target Interaction from Interactome Network with ‘Guilt-by-Association’ Principle and Topology Features. Bioinformatics 2016, 32, 1057–1064. [Google Scholar] [CrossRef]
- Yu, H.; Chen, J.; Xu, X.; Li, Y.; Zhao, H.; Fang, Y.; Li, X.; Zhou, W.; Wang, W.; Wang, Y. A Systematic Prediction of Multiple Drug-Target Interactions from Chemical, Genomic, and Pharmacological Data. PLoS ONE 2012, 7, e37608. [Google Scholar] [CrossRef]
- Ezzat, A.; Wu, M.; Li, X.; Kwoh, C.-K. Computational Prediction of Drug-Target Interactions via Ensemble Learning. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1903, pp. 239–254. [Google Scholar]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The Rise of Deep Learning in Drug Discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Lavecchia, A. Deep Learning in Drug Discovery: Opportunities, Challenges and Future Prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef]
- Lipinski, C.F.; Maltarollo, V.G.; Oliveira, P.R.; da Silva, A.B.F.; Honorio, K.M. Advances and Perspectives in Applying Deep Learning for Drug Design and Discovery. Front. Robot. AI 2019, 6. [Google Scholar] [CrossRef]
- Abbasi, K.; Razzaghi, P.; Poso, A.; Ghanbari-Ara, S.; Masoudi-Nejad, A. Deep Learning in Drug Target Interaction Prediction: Current and Future Perspectives. Curr. Med. Chem. 2021, 28, 2100–2113. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent Applications of Deep Learning and Machine Intelligence on in Silico Drug Discovery: Methods, Tools and Databases. Brief Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Hasan Mahmud, S.M.; Chen, W.; Jahan, H.; Dai, B.; Din, S.U.; Dzisoo, A.M. DeepACTION: A Deep Learning-Based Method for Predicting Novel Drug-Target Interactions. Anal. Biochem. 2020, 610, 113978. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of Drug-Target Interactions via Deep Learning with Convolution on Protein Sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, G.; Liu, S.; Jiang, J.-Y.; Wang, W. Drug-Target Interaction Prediction with Graph Attention Networks. arXiv 2021, arXiv:2107.06099. [Google Scholar]
- Tayebi, A.; Yousefi, N.; Yazdani-Jahromi, M.; Kolanthai, E.; Neal, C.; Seal, S.; Garibay, O. UnbiasedDTI: Mitigating Real-World Bias of Drug-Target Interaction Prediction by Using Deep Ensemble-Balanced Learning. Molecules 2022, 27, 2980. [Google Scholar] [CrossRef]
- Google Developers Imbalanced Data | Data Preparation and Feature Engineering for Machine Learning | Google Developers. Available online: https://developers.google.com/machine-learning/data-prep/construct/sampling-splitting/imbalanced-data (accessed on 12 July 2022).
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A Public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Charlton, P.; Spicer, J. Targeted Therapy in Cancer. Medicine 2016, 44, 34–38. [Google Scholar] [CrossRef]
- Mohamed, A.; Krajewski, K.; Cakar, B.; Ma, C.X. Targeted Therapy for Breast Cancer. Am. J. Pathol. 2013, 183, 1096–1112. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G.M. Targeted Therapy for Non-Small Cell Lung Cancer: Current Standards and the Promise of the Future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef] [PubMed]
- P. Mazanetz, M.; J. Marmon, R.; B. T. Reisser, C.; Morao, I. Drug Discovery Applications for KNIME: An Open Source Data Mining Platform. Curr. Top Med. Chem. 2012, 12, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G.; Tosco, P.; Kelley, B.; Sriniker; Gedeck; Vianello, R.; Nadine, S.; Kawashima; Dalkel. RDKit: Open-Source Chemoinformatics. 2021. Available online: https://zenodo.org/record/5773460#.Y-Sf3HbMJPY (accessed on 8 April 2022).
- Ismail, H.; Ahamed Hassain Malim, N.H.; Mohamad Zobir, S.Z.; Wahab, H.A. Comparative Studies On Drug-Target Interaction Prediction Using Machine Learning and Deep Learning Methods With Different Molecular Descriptors. In Proceedings of the 2021 International Conference of Women in Data Science at Taif University (WiDSTaif ), Taif, Saudi Arabia, 30–31 March 2021; pp. 1–6. [Google Scholar]
- Feng, Q.; Dueva, E.; Cherkasov, A.; Ester, M. PADME: A Deep Learning-Based Framework for Drug-Target Interaction Prediction. 2018. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Over-Sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Lemaitre, G.; Nogueira, F.; Aridas, C.K. Imbalanced-Learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. 2016, 18, 559–563. [Google Scholar]
- He, H.; Bai, Y.; Garcia, E.A.; Li, S. ADASYN: Adaptive Synthetic Sampling Approach for Imbalanced Learning. In Proceedings of the 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence), Hong Kong, China, 1–8 June 2008; pp. 1322–1328. [Google Scholar]
- Han, H.; Wang, W.-Y.; Mao, B.-H. Borderline-SMOTE: A New Over-Sampling Method in Imbalanced Data Sets Learning. In Proceedings of the International Conference on Intelligent Computing, Hefei, China, 23–26 August 2005; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3644, pp. 878–887. [Google Scholar]
- Nguyen, H.M.; Cooper, E.W.; Kamei, K. Borderline Over-Sampling for Imbalanced Data Classification. Int. J. Knowl. Eng. Soft Data Paradig. 2011, 3, 4. [Google Scholar] [CrossRef]
- Batista, G.E.A.P.A.; Bazzan, A.L.C.; Monard, M.C. Balancing Training Data for Automated Annotation of Keywords: A Case Study. Second Brazilian Workshop on Bioinformatics 2003, 2, 10–18. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- Agrawal, T. Hyperparameter Optimization in Machine Learning; Apress: Berkeley, CA, USA, 2021; ISBN 978-1-4842-6578-9. [Google Scholar]
- Wang, C.; Wang, W.; Lu, K.; Zhang, J.; Chen, P.; Wang, B. Predicting Drug-Target Interactions with Electrotopological State Fingerprints and Amphiphilic Pseudo Amino Acid Composition. Int. J. Mol. Sci. 2020, 21, 5694. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Degree of Data Imbalance | Proportion of Minority Class (%) |
|---|---|
| Severe | <1% of the dataset |
| Moderate | 1–20% of the dataset |
| Mild | 20–40% of the dataset |
| Activity Classes | Number of Active Compounds | Percentage of Minority Class (%) | Degree of Data Imbalance |
|---|---|---|---|
| MEK1 | 36 | 0.23% | Severe |
| PDGFR-B | 41 | 0.26% | Severe |
| KRAS | 191 | 1.22% | Moderate |
| PD-1 | 470 | 3.00% | Moderate |
| CDK-6 | 581 | 3.71% | Moderate |
| PDGFR-A | 724 | 4.62% | Moderate |
| VEGFR-1 | 1373 | 8.77% | Moderate |
| HER2 | 2426 | 15.50% | Moderate |
| BRAF | 3071 | 19.62% | Moderate |
| VEGFR-2 | 9241 | 40.97% | Mild |
| Activity Classes | Best Machine Learning and Resampling Method Pair | ||
|---|---|---|---|
| Accuracy | Precision | Recall | |
| BRAF | RF + SMOTETomek | RF + None | GNB + SVM-SMOTE |
| CDK-6 | RF + SVM-SMOTE | RF + None | GNB + SVM-SMOTE |
| HER2 | RF + SMOTETomek | RF + None | GNB + SVM-SMOTE |
| KRAS | RF + SMOTETomek | RF + SMOTETomek | GNB + SMOTE |
| MEK1 | RF + ADASYN | RF + ADASYN | GNB + SMOTETomek |
| PD-1 | RF + ADAYSN | RF + ADAYSN | GNB + SVM-SMOTE |
| PDGFR-A | RF + SMOTE | RF + None | RF + BorderlineSMOTE |
| PDGFR-B | RF + BorderlineSMOTE | RF + BorderlineSMOTE | RF + RUS |
| VEGFR-1 | RF + SMOTETomek | RF + None | RF + None |
| VEGFR-2 | RF + SVM-SMOTE | RF + BorderlineSMOTE | RF + None |
| Activity Class | ML + Resampling Pair | F1 Score |
|---|---|---|
| BRAF | RF + SMOTE | 85.41% |
| CDK-6 | RF + ADAYSN | 96.00% |
| HER2 | RF + SVM-SMOTE | 89.71% |
| KRAS | RF + SVM-SMOTE | 97.34% |
| MEK1 | GNB + SVM-SMOTE | 93.87% |
| PD-1 | RF + ADASYN | 99.36% |
| PDGFR-A | GNB + SVM-SMOTE | 86.19% |
| PDGFR-B | GNB + SVM-SMOTE | 85.68% |
| VEGFR-1 | RF + None | 93.50% |
| VEGFR-2 | RF + BorderlineSMOTE | 91.67% |
| Activity Class | Machine Learning with Resampling Pair | F1 Score of Machine Learning with Resampling Pair | F1 Score of MLP |
|---|---|---|---|
| BRAF | RF + SMOTE | 85.41% | 95.19% |
| CDK-6 | RF + ADAYSN | 96.00% | 97.20% |
| HER2 | RF + SVM-SMOTE | 89.71% | 90.41% |
| KRAS | RF + SVM-SMOTE | 97.34% | 98.59% |
| MEK1 | GNB + SVM-SMOTE | 93.87% | 100.00% |
| PD-1 | RF + ADASYN | 99.36% | 99.03% |
| PDGFR-A | GNB + SVM-SMOTE | 86.19% | 68.97% |
| PDGFR-B | GNB + SVM-SMOTE | 85.68% | 100.00% |
| VEGFR-1 | RF + None | 93.50% | 79.70% |
| VEGFR-2 | RF + BorderlineSMOTE | 91.67% | 97.26% |
| Average F1 Score | 91.87% | 92.36% | |
| Target Proteins (Activity Classes) | Abbreviation | Number of Active Compounds |
|---|---|---|
| B-Raf Proto-Oncogene | BRAF | 4389 |
| Cyclin-Dependent Kinase 6 | CDK-6 | 726 |
| Human Epidermal Growth Factor Receptor 2 | HER2 | 3046 |
| Kirsten Rat Sarcoma 2 Viral Oncogene Homolog | KRAS | 261 |
| Dual Specificity Mitogen-Activated ProteinKinase Kinase 1 | MEK1 | 38 |
| Programmed Cell Death Protein 1 | PD-1 | 479 |
| Platelet-Derived Growth Factor Receptor A | PDGFR-A | 864 |
| Platelet-Derived Growth Factor Receptor B | PDGFR-B | 41 |
| Vascular Endothelial Growth Factor Receptor 1 | VEGFR-1 | 1616 |
| Vascular Endothelial Growth Factor Receptor 2 | VEGFR-2 | 12,036 |
| Target Protein (Activity Classes) | Abbreviation | Number of Active Compounds | Number of Inactive Compounds |
|---|---|---|---|
| B-Raf Proto-Oncogene | BRAF | 3071 | 12,585 |
| Cyclin-Dependent Kinase 6 | CDK-6 | 581 | 15,075 |
| Human Epidermal Growth Factor Receptor 2 | HER2 | 2426 | 13,230 |
| Kirsten Rat Sarcoma 2 Viral Oncogene Homolog | KRAS | 191 | 15,465 |
| Dual Specificity Mitogen-Activated Protein Kinase Kinase 1 | MEK1 | 36 | 15,620 |
| Programmed Cell Death Protein 1 | PD-1 | 470 | 15,186 |
| Platelet-Derived Growth Factor Receptor A | PDGFR-A | 724 | 14,932 |
| Platelet-Derived Growth Factor Receptor B | PDGFR-B | 41 | 15,615 |
| Vascular Endothelial Growth Factor Receptor 1 | VEGFR-1 | 1373 | 14,283 |
| Vascular Endothelial Growth Factor Receptor 2 | VEGFR-2 | 9241 | 6415 |
| Resampling Method | Brief Description of Resampling Method | Authors & Year |
|---|---|---|
| SMOTE | Synthetic Minority Oversampling Technique or SMOTE was introduced by the authors in [54]. SMOTE is an oversampling technique that oversamples the minority class in the dataset by synthesizing fake minority data into the original dataset so that the minority and majority classes are balanced. | Chawla et al. (2022) [54] |
| RUS | Random Undersampling or RUS is an undersampling technique that randomly discards instances from the majority class so that the majority class is balanced with the minority classes [55]. | Lemaitre et al. (2016) [55] |
| ADASYN | ADASYN or Adaptive Synthetic, introduced by the authors in [56] is a data resampling technique that is similar to SMOTE, but in ADASYN, synthetic data are generated for minority classes that are difficult to learn. | Haibo et al. (2008) [56] |
| BorderlineSMOTE | BorderlineSMOTE is a resampling method that was developed by the authors in [57]. BorderlineSMOTE is an oversampling method where only minority samples that are near the borderline are oversampled. | Han et al. (2005) [57] |
| SVM-SMOTE | SVM-SMOTE is a resampling method that was developed by [58] that is similar to BorderlineSMOTE. The difference is that, in this method, SVM is used to help generate new minority instances near the borderline in order to establish boundaries between the classes in the dataset. | Nguyen et al. (2011) [58] |
| SMOTETomek | SMOTE with Tomek Links or SMOTETomek is a hybrid resampling technique that combines oversampling using SMOTE and undersampling using Tomek links and it was introduced by [59]. | Batista et al. (2003) [59] |
| Machine Learning Classifier | Best Parameters |
|---|---|
| SVM | C = 100, gamma = 0.1, kernel = sigmoid |
| RF | criterion = entropy, max_depth = 8, min_samples_leaf = 2, min_samples_split = 5, n_estimators = 100 |
| GNB | var_smoothing = 0.1 |
| DT | ccp_alpha = 0.001, criterion = entropy, max_depth = 4, max_features = auto, min_samples_leaf = 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azlim Khan, A.K.; Ahamed Hassain Malim, N.H. Comparative Studies on Resampling Techniques in Machine Learning and Deep Learning Models for Drug-Target Interaction Prediction. Molecules 2023, 28, 1663. https://doi.org/10.3390/molecules28041663
Azlim Khan AK, Ahamed Hassain Malim NH. Comparative Studies on Resampling Techniques in Machine Learning and Deep Learning Models for Drug-Target Interaction Prediction. Molecules. 2023; 28(4):1663. https://doi.org/10.3390/molecules28041663
Chicago/Turabian StyleAzlim Khan, Azwaar Khan, and Nurul Hashimah Ahamed Hassain Malim. 2023. "Comparative Studies on Resampling Techniques in Machine Learning and Deep Learning Models for Drug-Target Interaction Prediction" Molecules 28, no. 4: 1663. https://doi.org/10.3390/molecules28041663