
Structure-Based Virtual Screening and Molecular Dynamics Simulation Assessments of Depsidones as Possible Selective Cannabinoid Receptor Type 2 Agonists

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Protein and Ligand Preparation
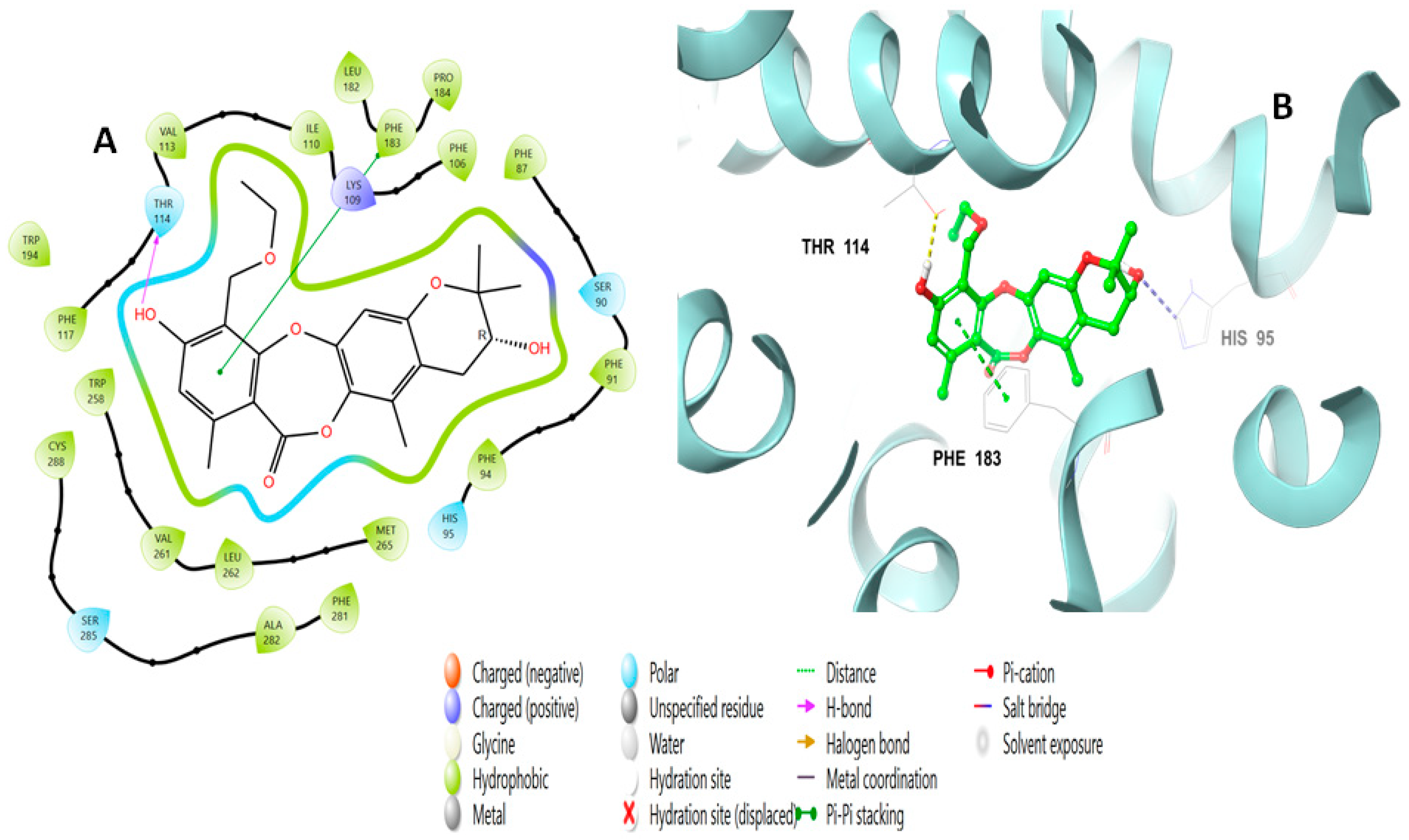
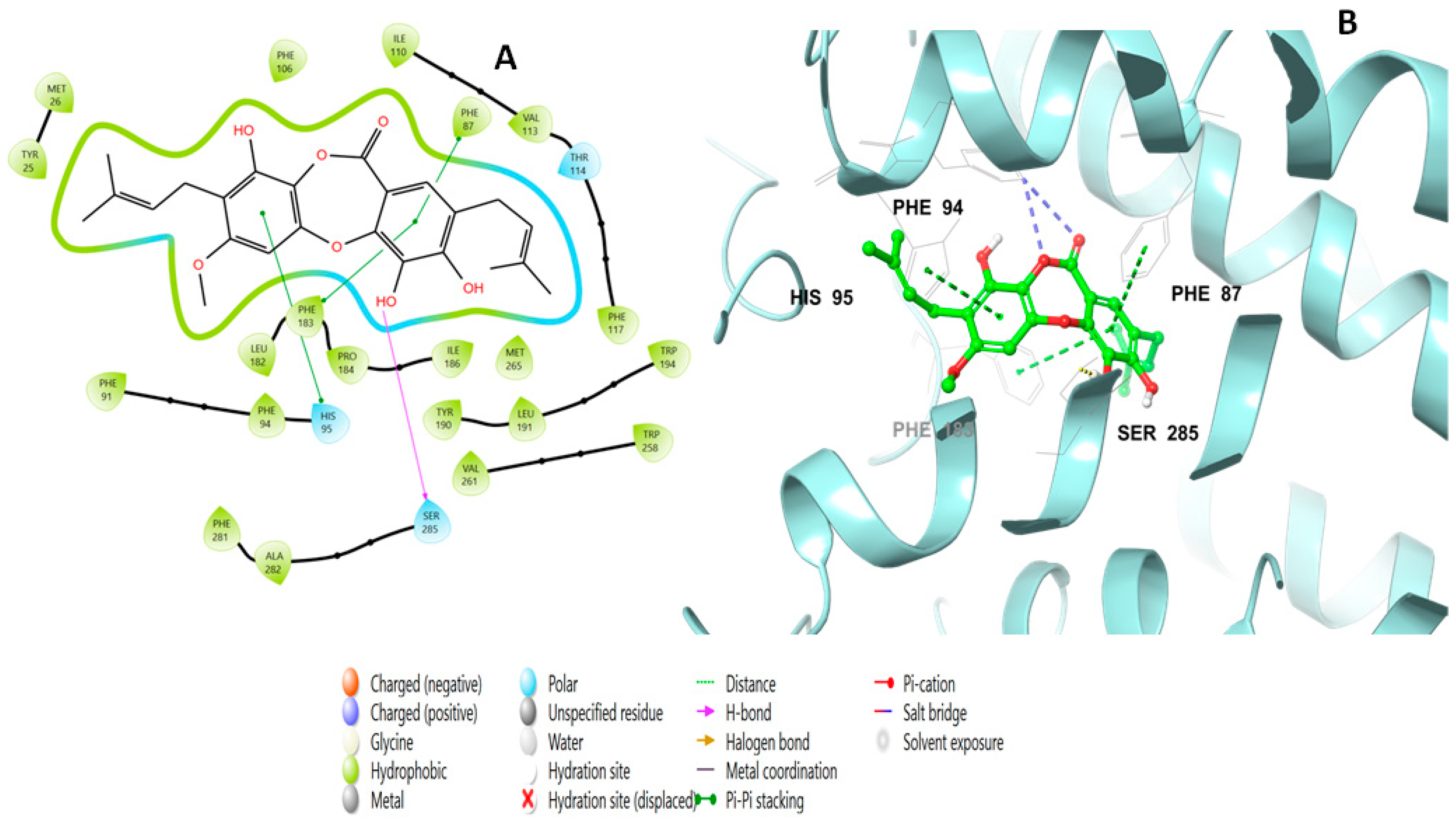
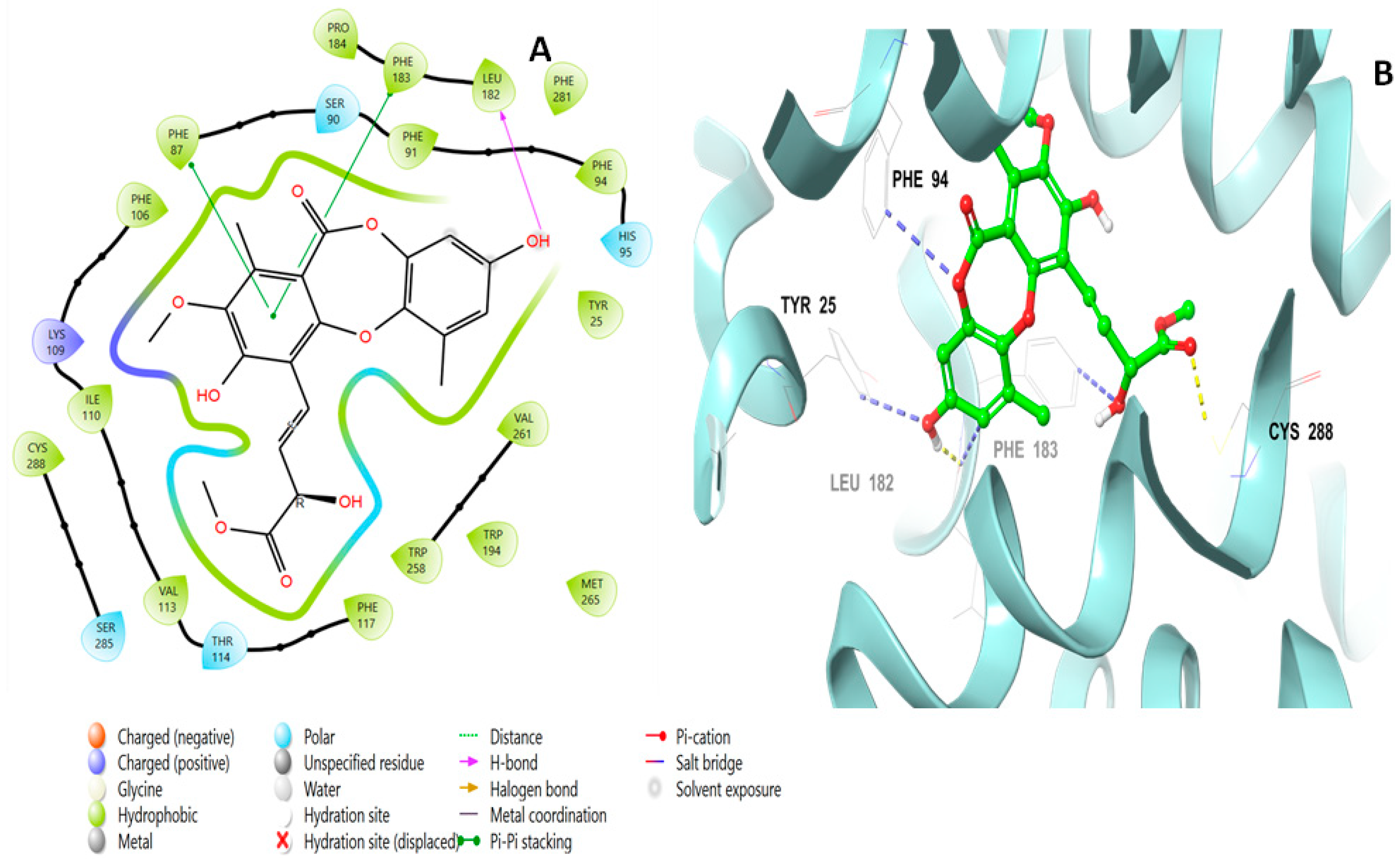
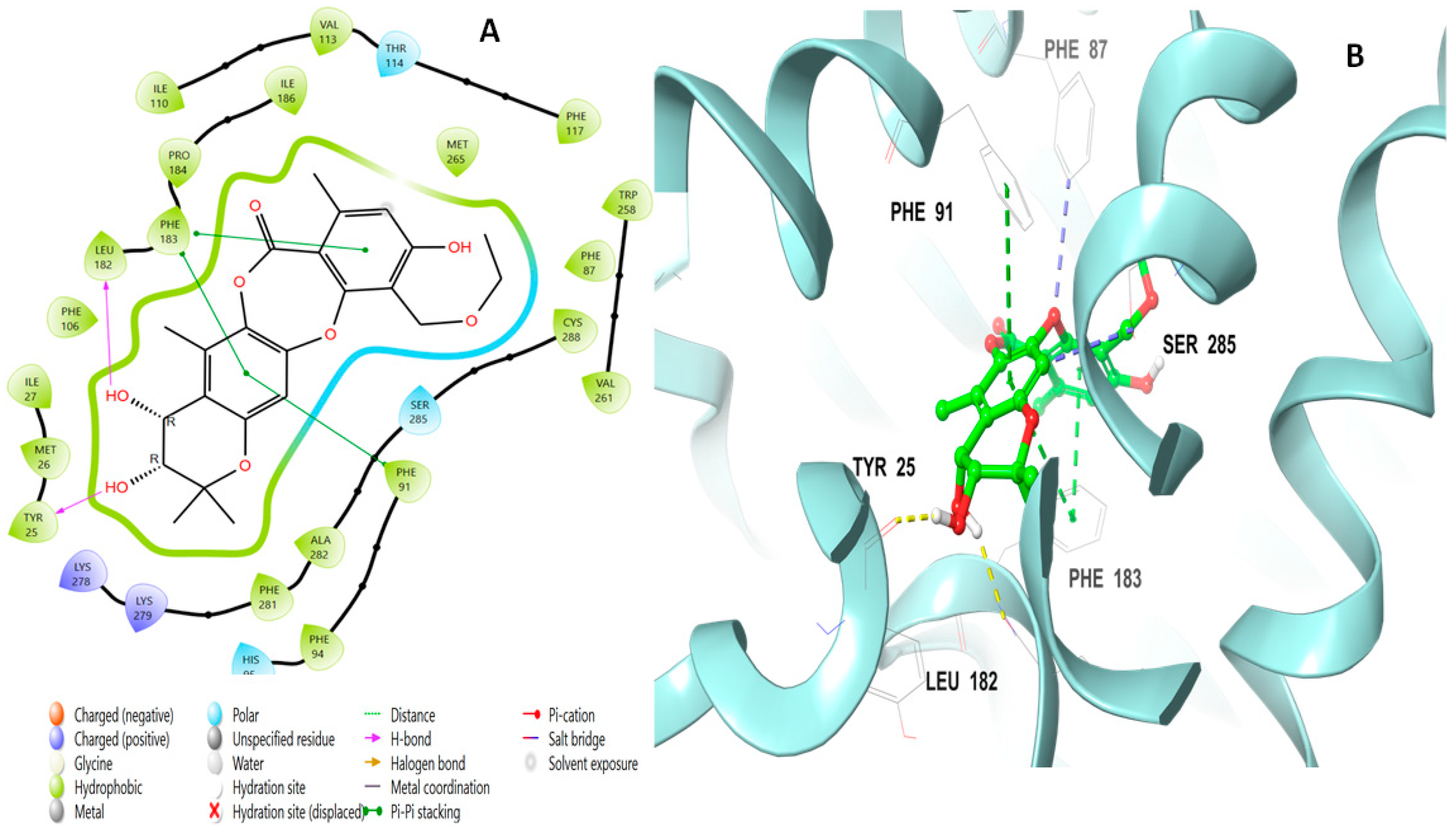
2.2. Molecular Docking Studies Analysis
2.3. Induced Fit Docking Analysis
2.4. QM/MM (Quantum Mechanics/Molecular Mechanics) Analysis
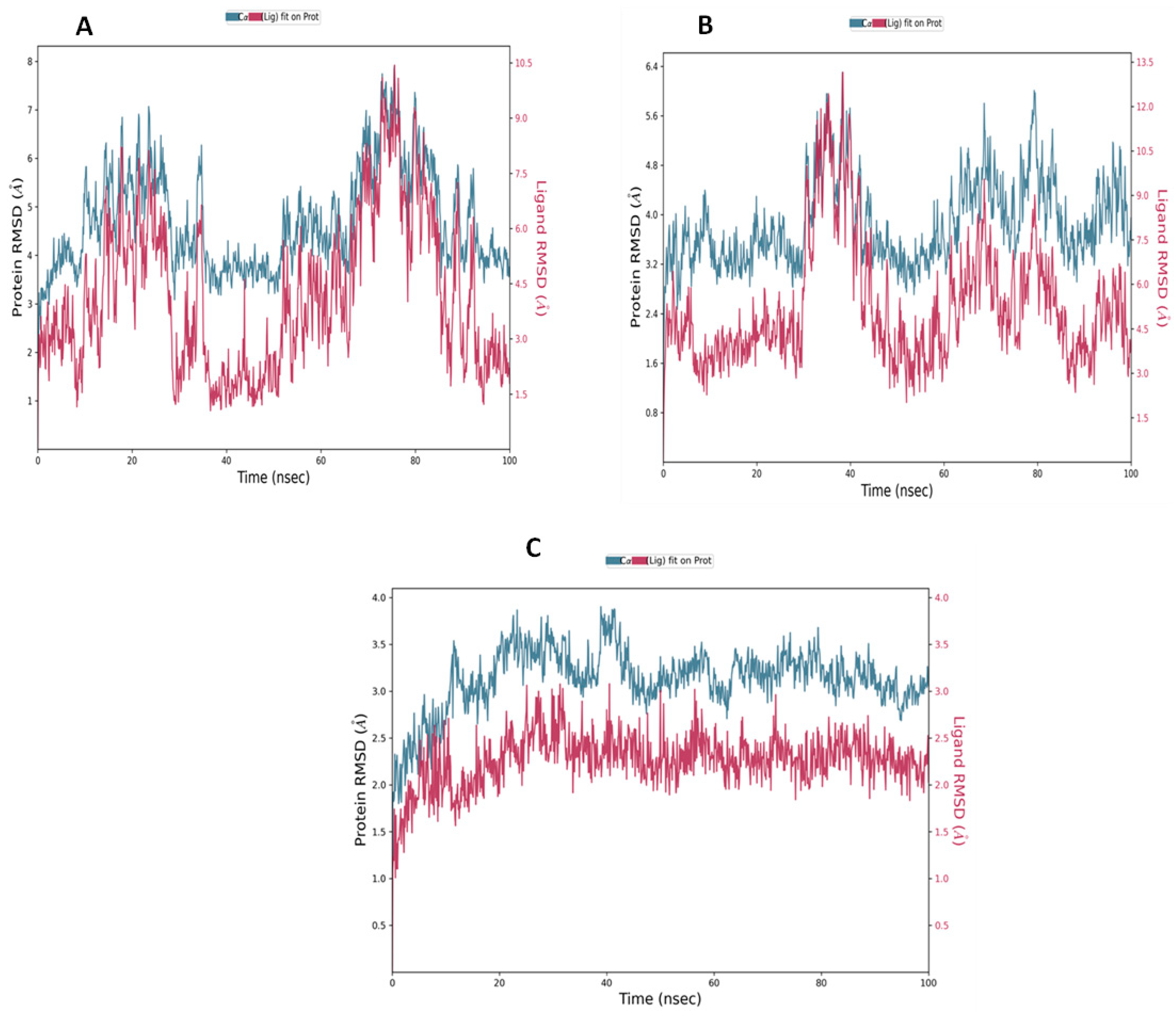
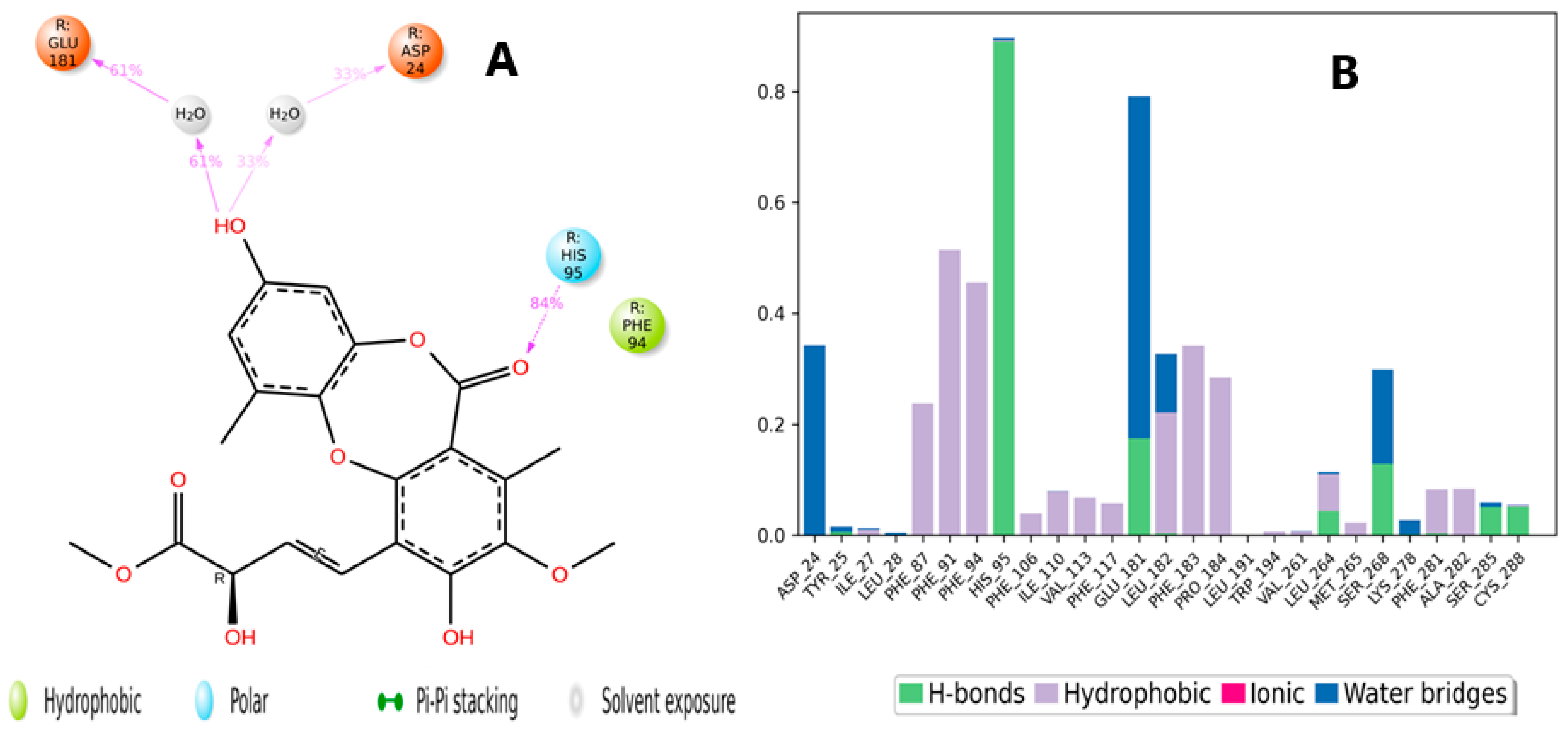
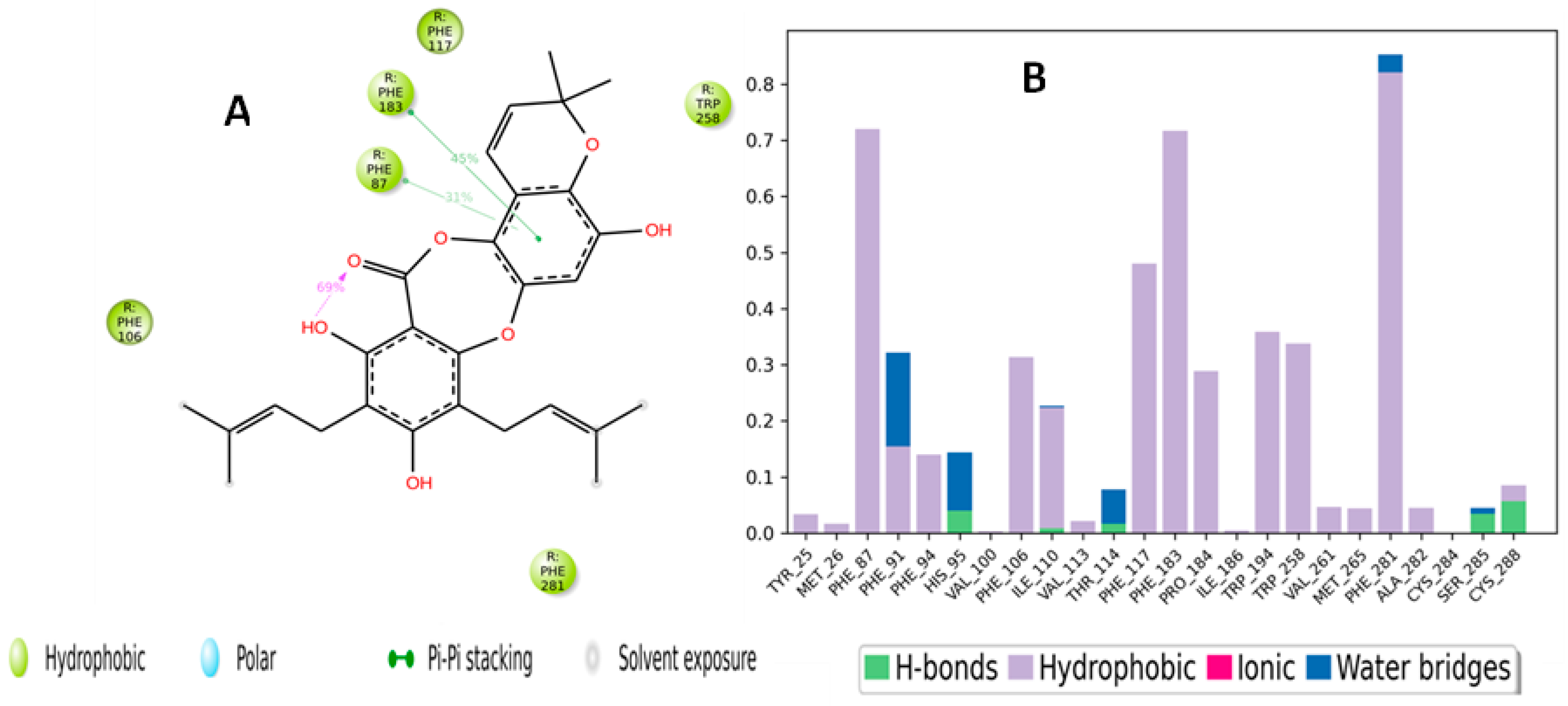
2.5. Molecular Dynamic Simulation
2.6. ADMET Properties
3. Materials and Methods
3.1. Preparation of Protein
3.2. Ligand Preparation
3.3. Grid Generation and Molecular Docking
3.4. Induced Fit Docking
3.5. Molecular Dynamic Simulation (MD)
3.6. Quantum Mechanics/Molecular Mechanics (QM/MM) Calculations
3.7. Prediction of ADMET Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brahmachari, G. Natural Products in Drug Discovery: Impacts and Opportunities ? An Assessment. In Bioactive Natural Products; World Scientific: Singapore, 2011; pp. 1–199. [Google Scholar] [CrossRef]
- Natural Products in Drug Discovery: Advances and Opportunities|Nature Reviews Drug Discovery. Available online: https://www.nature.com/articles/s41573-020-00114-z (accessed on 29 December 2022).
- Kiriiri, G.K.; Njogu, P.M.; Mwangi, A.N. Exploring different approaches to improve the success of drug discovery and development projects: A review. Future J. Pharm. Sci. 2020, 6, 27. [Google Scholar] [CrossRef]
- Contemporary Computational Applications and Tools in Drug Discovery|ACS Medicinal Chemistry Letters. Available online: https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00662 (accessed on 29 December 2022).
- An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. Int. J. Mol. Sci. 2020, 21, 5064. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed]
- Proietto, J.; Rissanen, A.; Harp, J.B.; Erondu, N.; Yu, Q.; Suryawanshi, S.; Jones, M.E.; Johnson-Levonas, A.O.; Heymsfield, S.B.; Kaufman, K.D.; et al. A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: Low-dose study. Int. J. Obes. 2010, 34, 1243–1254. [Google Scholar] [CrossRef]
- Morales, P.; Hernandez-Folgado, L.; Goya, P.; Jagerovic, N. Cannabinoid receptor 2 (CB2) agonists and antagonists: A patent update. Expert Opin. Ther. Pat. 2016, 26, 843–856. [Google Scholar] [CrossRef]
- Mannino, F.; Pallio, G.; Corsaro, R.; Minutoli, L.; Altavilla, D.; Vermiglio, G.; Allegra, A.; Eid, A.H.; Bitto, A.; Squadrito, F.; et al. Beta-Caryophyllene Exhibits Anti-Proliferative Effects through Apoptosis Induction and Cell Cycle Modulation in Multiple Myeloma Cells. Cancers 2021, 13, 5741. [Google Scholar] [CrossRef]
- Fuchs, A.; Rempel, V.; Müller, C.E. The natural product magnolol as a lead structure for the development of potent cannabinoid receptor agonists. PLoS ONE 2013, 8, e77739. [Google Scholar] [CrossRef]
- Schuehly, W.; Paredes, J.M.V.; Kleyer, J.; Huefner, A.; Anavi-Goffer, S.; Raduner, S.; Altmann, K.H.; Gertsch, J. Mechanisms of osteoclastogenesis inhibition by a novel class of biphenyl-type cannabinoid CB(2) receptor inverse agonists. Chem. Biol. 2011, 18, 1053–1064. [Google Scholar] [CrossRef]
- Chianese, G.; Fattorusso, E.; Taglialatela-Scafati, O.; Bavestrello, G.; Calcinai, B.; Dien, H.A.; Ligresti, A.; Di Marzo, V. Desulfohaplosamate, a new phosphate-containing steroid from Dasychalina sp., is a selective cannabinoid CB2 receptor ligand. Steroids 2011, 76, 998–1002. [Google Scholar] [CrossRef]
- Thadhani, V.M.; Karunaratne, V. Potential of Lichen Compounds as Antidiabetic Agents with Antioxidative Properties: A Review. Oxid. Med. Cell. Longev. 2017, 2017, 2079697. [Google Scholar] [CrossRef]
- White, P.A.S.; Oliveira, R.; Oliveira, A.P.; Serafini, M.R.; Araújo, A.A.; Gelain, D.P.; Moreira, J.C.; Almeida, J.R.; Quintans, J.S.; Quintans-Junior, L.J.; et al. Antioxidant activity and mechanisms of action of natural compounds isolated from lichens: A systematic review. Molecules 2014, 19, 14496–14527. [Google Scholar] [CrossRef] [PubMed]
- Lichens as a Potential Natural Source of Bioactive Compounds: A Review|SpringerLink. Available online: https://link.springer.com/article/10.1007/s11101-010-9189-6 (accessed on 29 December 2022).
- Ibrahim, S.R.M.; Mohamed, G.A.; Al Haidari, R.A.; El-Kholy, A.A.; Zayed, M.F.; Khayat, M.T. Biologically active fungal depsidones: Chemistry, biosynthesis, structural characterization, and bioactivities. Fitoterapia 2018, 129, 317–365. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Armaleo, D.; Dal Grande, F.; Schmitt, I. Depside and Depsidone Synthesis in Lichenized Fungi Comes into Focus through a Genome-Wide Comparison of the Olivetoric Acid and Physodic Acid Chemotypes of Pseudevernia furfuracea. Biomolecules 2021, 11, 1445. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Structure-Based Virtual Screening, Molecular Docking and Dynamics Studies of Natural Product and Classical Inhibitors against Human Dihydrofolate Reductase|SpringerLink. Available online: https://link.springer.com/article/10.1007/s13721-020-00244-9 (accessed on 29 December 2022).
- Philipp, D.M.; Friesner, R.A. Mixed ab initio QM/MM modeling using frozen orbitals and tests with alanine dipeptide and tetrapeptide. J. Comput. Chem. 1999, 20, 1468–1494. [Google Scholar] [CrossRef]
- Charanya, C.; Sampathkrishnan, S.; Balamurugan, N. Quantum mechanical analysis, spectroscopic (FT-IR, FT-Raman, UV-Visible) study, and HOMO-LUMO analysis of (1S,2R)-2-amino-1-phenylpropan-1-ol using Density Functional Theory. J. Mol. Liq. 2017, 231, 116–125. [Google Scholar] [CrossRef]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Schrödinger, LigPrep. Schrödinger Release 2021-4; Schrödinger LLC: New York, NY, USA, 2021.
- Glide. Schrödinger Release 2021-4: Glide; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prime. Schrödinger Release 2022–3: Prime; Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]
- Induced Fit Docking. Schrödinger Suite 2009 Induced Fit Docking Protocol; Prime Version; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger Release 2021-4: Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, USA; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2021.
- Qsite. Schrödinger Release 2022-4; Schrödinger LLC: New York, NY, USA, 2021. [Google Scholar]
- QikProp. Schrödinger Release 2021-4: QikProp; Schrödinger LLC: New York, NY, USA, 2021. [Google Scholar]
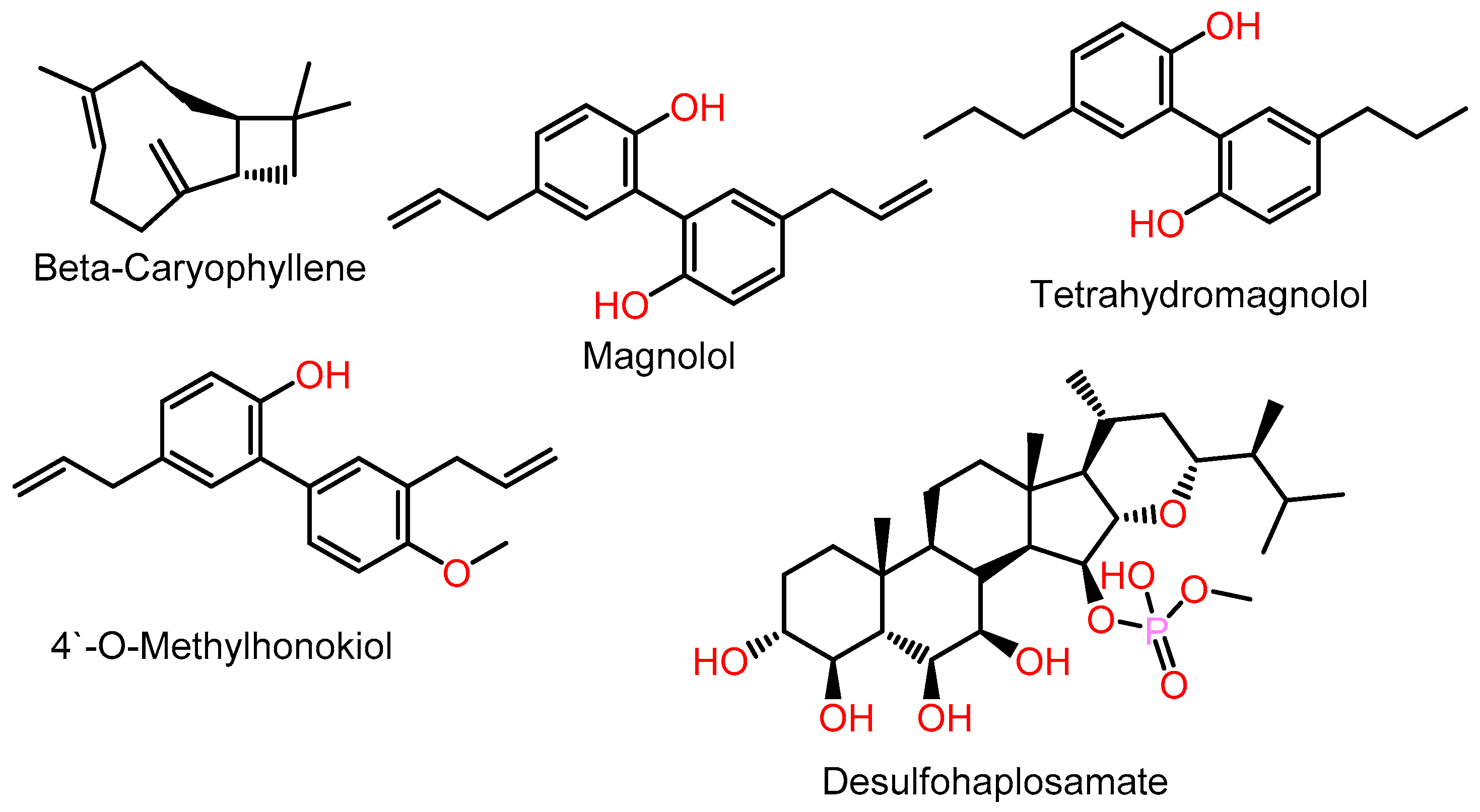
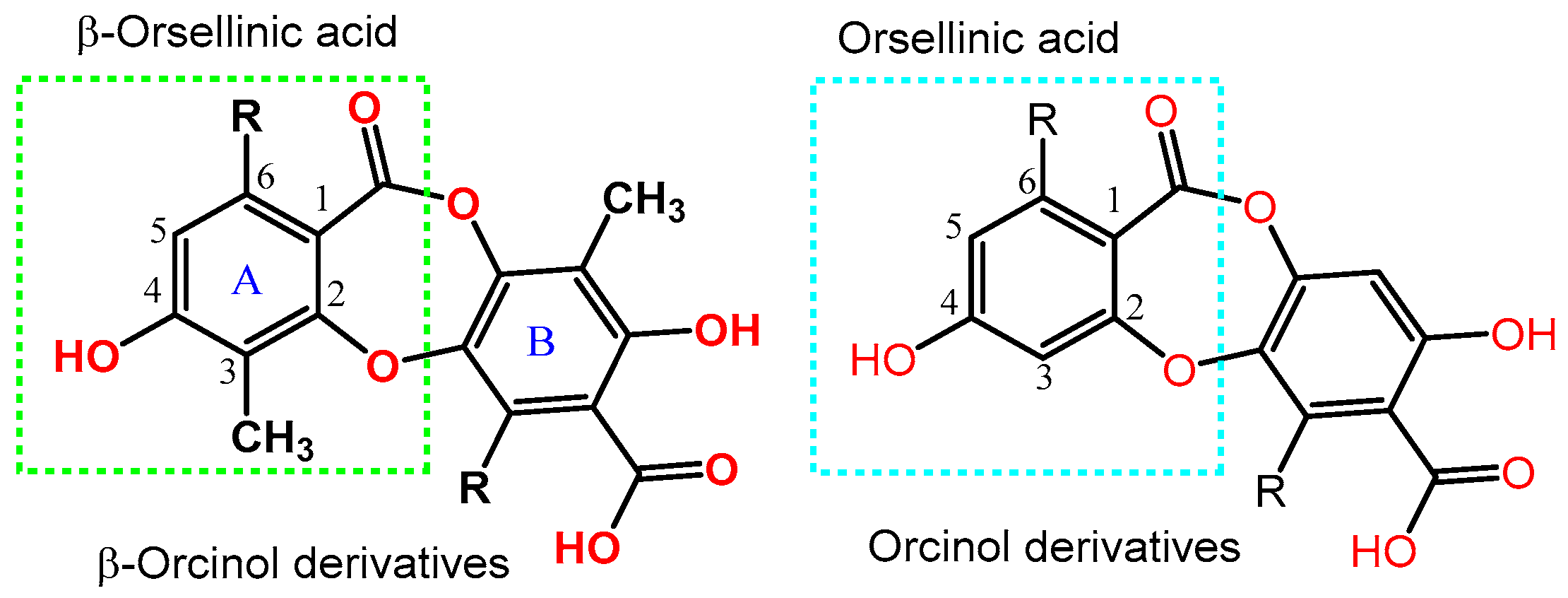
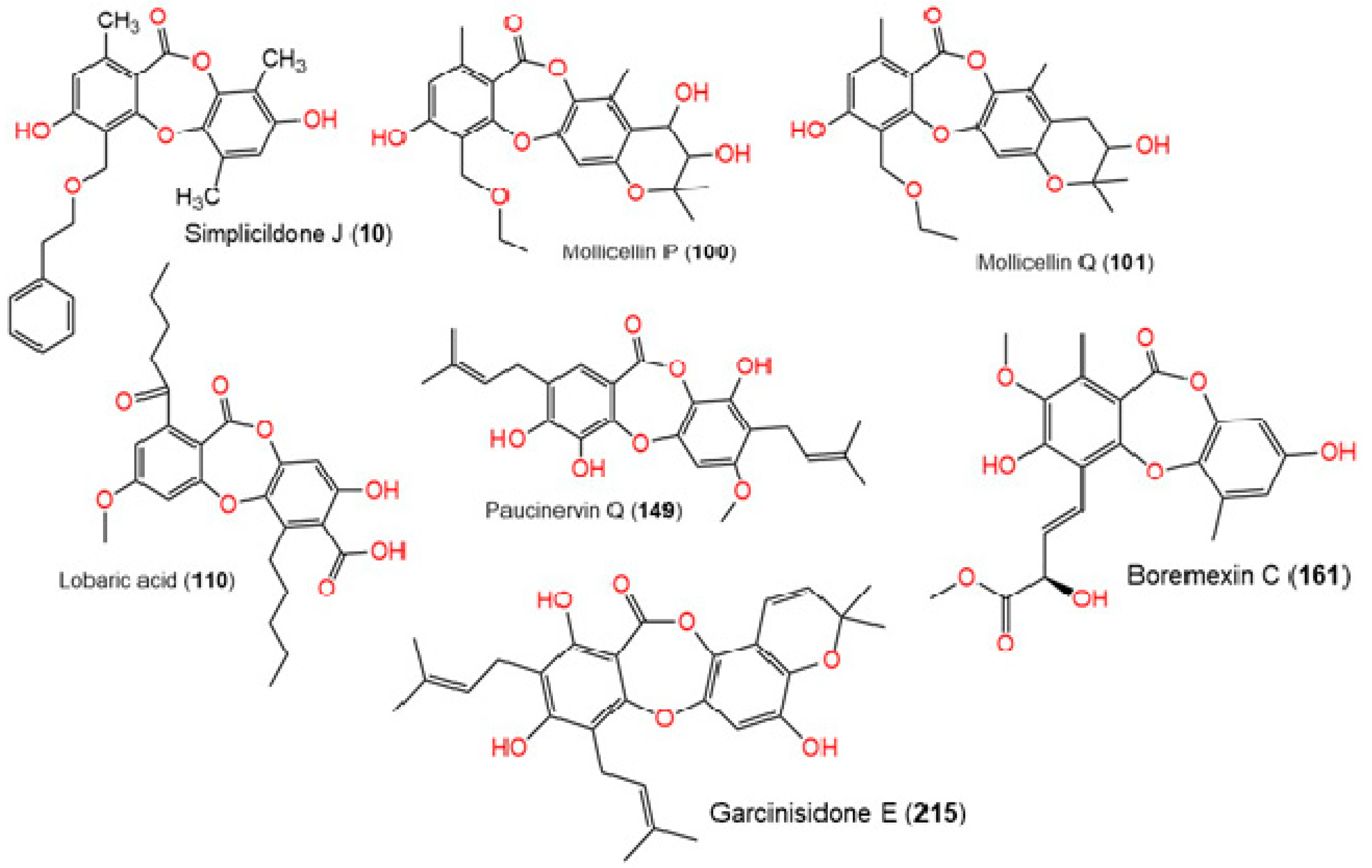
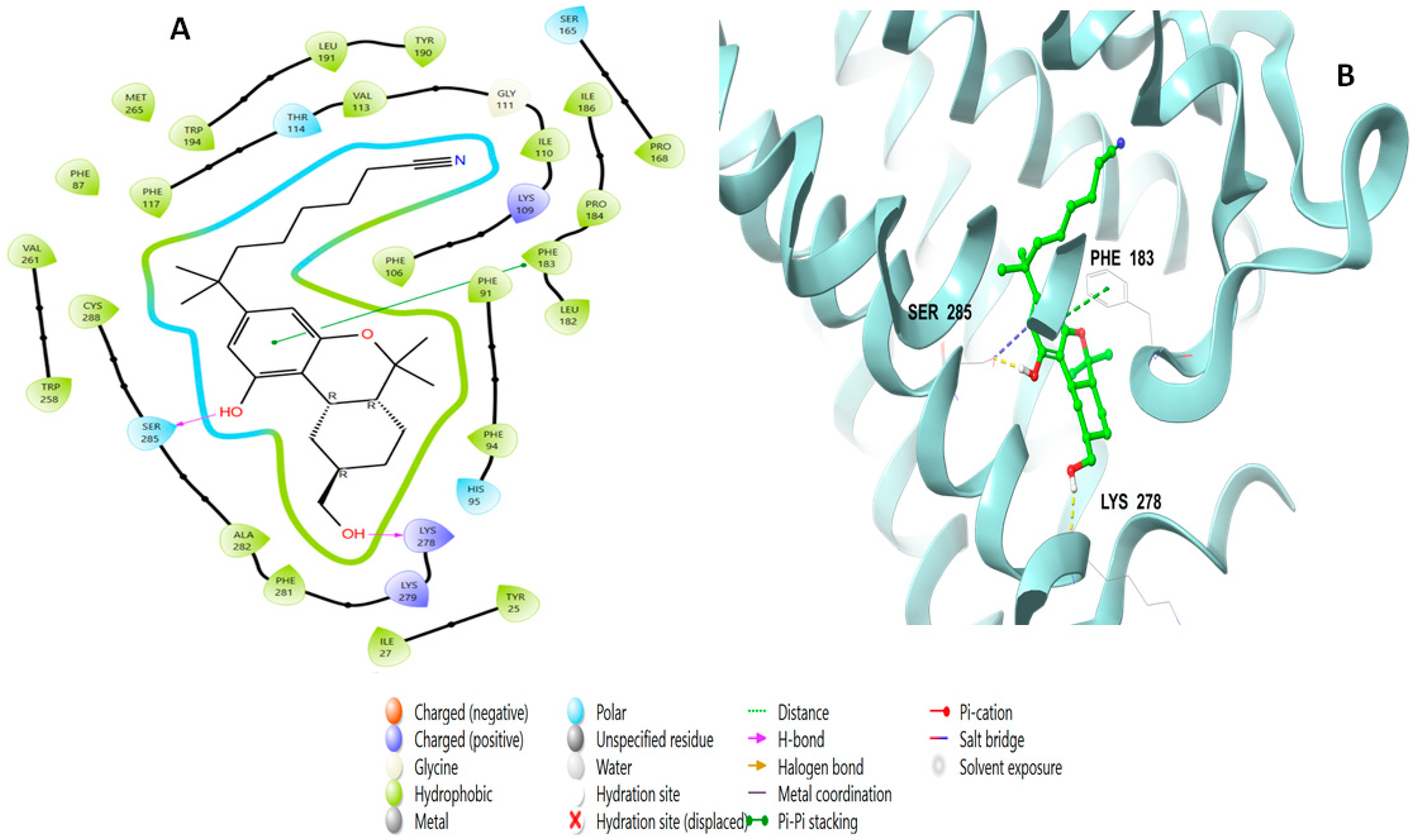




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title | Docking Score | XP GScore | GlideScore | Glide Emodel | Prime Energy | MMGBSA dG Bind | IFD Score |
|---|---|---|---|---|---|---|---|
| 6KPF–prepared _ligand | −12.240 | −12.240 | −12.240 | −79.066 | −43,026.5 | −86.26 | −532.50 |
| Simplicildone J (10) | −12.134 | −12.174 | −12.174 | −17.822 | −43,002.1 | −52.76 | −530.54 |
| Lobaric acid (110) | −11.944 | −11.944 | −11.944 | −37.511 | −42,921.3 | −20.56 | −533.43 |
| Mollicellin Q (101) | −11.479 | −11.513 | −11.513 | 55.427 | −42,995.8 | −56.65 | −531.30 |
| Garcinisidone E (215) | −11.394 | −11.633 | −11.633 | 15.908 | −43,035.0 | −54.14 | −534.60 |
| Mollicellin P (100) | −11.322 | −11.356 | −11.356 | −16.759 | −42,996.9 | −65.65 | −531.21 |
| Paucinervin Q (149) | −11.305 | −11.542 | −11.542 | −23.918 | −42,958.9 | −66.13 | −536.89 |
| Boremexin C (161) | −11.254 | −11.376 | −11.376 | −12.854 | −42,950.3 | −61.63 | −531.22 |
| Compound | Number of Canonical Orbitals | QM/MM Energy | HOMO | LUMO | Energy Gap |
|---|---|---|---|---|---|
| Paucinervin Q (149) | 1018 | −2655.605583 | −0.437907 | −0.278268 | 0.716175 |
| Garcinisidone_E (215) | 848 | −2278.252156 | −0.428884 | −0.263329 | 0.692213 |
| Lobaric_acid (110) | 980 | −2599.618583 | −0.442702 | −0.261762 | 0.708782 |
| 6KPF_Native Agonist | 759 | −1865.074602 | −0.418376 | −0.216867 | 0.635243 |
| Molecule | #stars | #rtvFG | CNS | mol_MW | SASA | donorHB | accptHB | QPlogPo/w | QPlogHERG | QPPCaco | QPlogBB | #metab | QPlogKhsa | Percent Human Oral Absorption |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recommended Range | (0.0–5.0) | (0–2) | (−2 inactive) (+2 active) | (130–725) | (300–1000) | (0–6) | (2.0–20.0) | (−2–6.5) | concen below −5 | <25 poor, >500 great) | (−3–1.2) | (1–8) | (−1.5–1.5) | (<25% poor; >80% high) |
| Boremexin C (161) | 0 | 2 | −2 | 416.384 | 650.358 | 3 | 8.95 | 1.511 | −4.765 | 85.053 | −1.935 | 6 | −0.131 | 70.328 |
| Garcinisidone E (215) | 2 | 1 | −2 | 478.541 | 778.681 | 2 | 5 | 5.471 | −5.428 | 603.731 | −1.194 | 9 | 1.25 | 95.791 |
| Lobaric acid (110) | 0 | 1 | −2 | 456.491 | 782.184 | 1 | 7.5 | 4.226 | −3.811 | 28.355 | −2.352 | 4 | 0.325 | 77.691 |
| Mollicellin P (100) | 0 | 1 | −2 | 430.454 | 702.313 | 3 | 9.6 | 2.213 | −5.061 | 403.047 | −1.284 | 7 | −0.021 | 86.531 |
| Mollicellin Q (101) | 0 | 1 | −2 | 414.454 | 698.882 | 2 | 7.9 | 3.048 | −5.079 | 657.833 | −1.004 | 7 | 0.283 | 95.231 |
| Paucinervin Q (149) | 1 | 1 | −2 | 426.465 | 733.431 | 3 | 6 | 3.82 | −5.24 | 307.837 | −1.578 | 10 | 0.607 | 93.85 |
| Simplicildone J (10) | 0 | 1 | −2 | 420.461 | 708.462 | 2 | 6.2 | 4.133 | −6.007 | 660.471 | −1.12 | 8 | 0.578 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, G.A.; Omar, A.M.; AlKharboush, D.F.; Fallatah, M.A.; Sindi, I.A.; El-Agamy, D.S.; Ibrahim, S.R.M. Structure-Based Virtual Screening and Molecular Dynamics Simulation Assessments of Depsidones as Possible Selective Cannabinoid Receptor Type 2 Agonists. Molecules 2023, 28, 1761. https://doi.org/10.3390/molecules28041761
Mohamed GA, Omar AM, AlKharboush DF, Fallatah MA, Sindi IA, El-Agamy DS, Ibrahim SRM. Structure-Based Virtual Screening and Molecular Dynamics Simulation Assessments of Depsidones as Possible Selective Cannabinoid Receptor Type 2 Agonists. Molecules. 2023; 28(4):1761. https://doi.org/10.3390/molecules28041761
Chicago/Turabian StyleMohamed, Gamal A., Abdelsattar M. Omar, Dana F. AlKharboush, Mona A. Fallatah, Ikhlas A. Sindi, Dina S. El-Agamy, and Sabrin R. M. Ibrahim. 2023. "Structure-Based Virtual Screening and Molecular Dynamics Simulation Assessments of Depsidones as Possible Selective Cannabinoid Receptor Type 2 Agonists" Molecules 28, no. 4: 1761. https://doi.org/10.3390/molecules28041761
APA StyleMohamed, G. A., Omar, A. M., AlKharboush, D. F., Fallatah, M. A., Sindi, I. A., El-Agamy, D. S., & Ibrahim, S. R. M. (2023). Structure-Based Virtual Screening and Molecular Dynamics Simulation Assessments of Depsidones as Possible Selective Cannabinoid Receptor Type 2 Agonists. Molecules, 28(4), 1761. https://doi.org/10.3390/molecules28041761