
Green Drug Discovery: Novel Fragment Space from the Biomass-Derived Molecule Dihydrolevoglucosenone (CyreneTM)
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
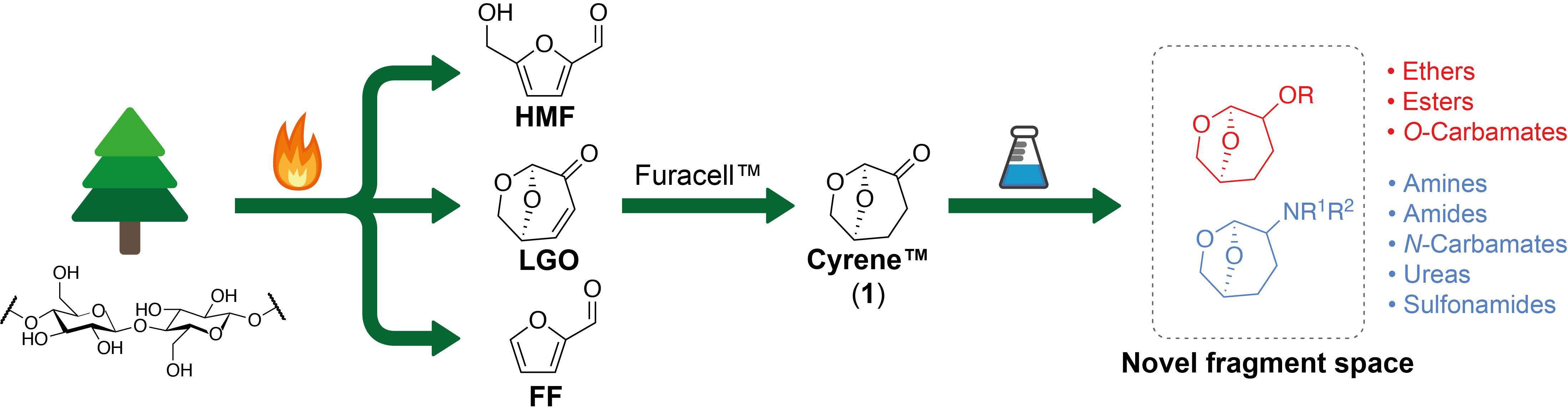
2.1. Selection of Precursor
2.2. Design of Fragments
2.3. Synthetic Routes
2.4. Stereochemistry
2.5. Physicochemical Properties
2.6. Solubility and Stability
2.7. Three-Dimensionality
3. Discussion
4. Materials and Methods
4.1. Nephelometry
4.2. Incubation Experiments
4.3. General Experimental
- Multiplicity is not solely reported based on peak shapes, but also distinguishes the coupling to all non-equivalent protons that have similar J values;
- If additional smaller couplings are observed or expected but are too small for accurate quantitation because the precision is smaller than the digital resolution, a symbol ∆ will be used;
- The notation “m” is used in case of obscured accurate interpretation as a result of:
- Overlapping signals for different protons, or;
- A result of overlapping signal lines within the same proton signal;
- For compounds that were isolated as mixtures of diastereomers with a d.r. < 9:1, signals were listed separately if possible. Signals were annotated with the corresponding diastereomer as follows: a signal(s) assigned to diastereomer 1; b signal(s) assigned to diastereomer 2; a/b signal(s) could not be assigned with certainty to either diastereomer 1 or 2; a,b signal(s) assigned to both diastereomer 1 and 2 (only applies to multiplets). Diastereomer 1 indicates the major diastereomer or an arbitrarily assigned diastereomer in the case of a d.r. of 1:1. The number of protons that cause a signal is corrected for the d.r. in the 1H NMR listings, i.e., in the example “3.77 (d, J = 6.1 Hz, 0.9H)a, 3.73 (d, J = 6.4 Hz, 0.1H)b”, the d.r. ≅ 9:1 and one proton of major diastereomer 1 gives a doublet at 3.77 ppm; the same proton in diastereomer 2 gives a doublet at 3.73 ppm. The corrected number of protons was summed in the case of overlapping signals. In the case of d.r. > 9:1 and/or in the case of extensive overlap of signals of both diastereomers, only the major signals were listed. For any fumaric acid salts, the CH protons of fumaric acid counterion (HOOC–CH=CH–COOH) were listed as “6.xx (m, 2H)a,b”, irrespective of the acid/base ratio;
- NMR signals that could only be detected with HSQC analysis are denoted with a # symbol;
- NMR signals that could only be detected with HMBC analysis are denoted with a * symbol;
- If one or more signals remain undetected after extensive 1D and 2D NMR analyses, this will be mentioned;
- Signals for exchangeable proton atoms (such as NH and OH groups) are only listed if clearly visible (excluding e.g., the use of D2O or CD3OD) and if confirmed by a D2O shake and/or HSQC.
4.4. Computational Methods and Figures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fitzgerald, N.D. Chemistry Challenges to Enable a Sustainable Bioeconomy. Nat. Rev. Chem. 2017, 1, 0080. [Google Scholar] [CrossRef]
- Tursi, A. A Review on Biomass: Importance, Chemistry, Classification, and Conversion. Biofuel Res. J. 2019, 6, 962–979. [Google Scholar] [CrossRef]
- Li, Y.C.E. Sustainable Biomass Materials for Biomedical Applications. ACS Biomater. Sci. Eng. 2019, 5, 2079–2092. [Google Scholar] [CrossRef]
- Chafran, L.; Carfagno, A.; Altalhi, A.; Bishop, B. Green Hydrogel Synthesis: Emphasis on Proteomics and Polymer Particle-Protein Interaction. Polymers 2022, 14, 4755. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.; Zhong, L.; Chua, W.; Xiang, Z.; Sun, R. Lignocellulosic Biomass Derived Functional Materials: Synthesis and Applications in Biomedical Engineering. Curr. Med. Chem. 2019, 26, 2456–2474. [Google Scholar] [CrossRef] [PubMed]
- What Is the Bioeconomy. Available online: https://ec.europa.eu/research/bioeconomy/ (accessed on 9 January 2022).
- PGEU Best Practice Paper on Green and Sustainable Pharmacy in Europe. Available online: https://www.pgeu.eu/publications/pgeu-best-practice-paper-on-green-and-sustainable-pharmacy-in-europe/ (accessed on 9 January 2022).
- Advancing Sustainability in the Pharmaceutical Industry. Available online: https://cen.acs.org/acs-news/comment/Advancing-sustainability-pharmaceutical-industry/98/i17 (accessed on 9 January 2022).
- Wang, G.; Dai, Y.; Yang, H.; Xiong, Q.; Wang, K.; Zhou, J.; Li, Y.; Wang, S. A Review of Recent Advances in Biomass Pyrolysis. Energy Fuels 2020, 34, 15557–15578. [Google Scholar] [CrossRef]
- Leng, L.; Yang, L.; Chen, J.; Leng, S.; Li, H.; Li, H.; Yuan, X.; Zhou, W.; Huang, H. A Review on Pyrolysis of Protein-Rich Biomass: Nitrogen Transformation. Biores. Techn. 2020, 315, 123801. [Google Scholar] [CrossRef] [PubMed]
- Hert, J.; Irwin, J.J.; Laggner, C.; Keiser, M.J.; Shoichet, B.K. Quantifying Biogenic Bias in Screening Libraries. Nat. Chem. Biol. 2009, 5, 479–483. [Google Scholar] [CrossRef]
- Nelson, A.; Karageorgis, G. Natural Product-Informed Exploration of Chemical Space to Enable Bioactive Molecular Discovery. RSC Med. Chem. 2021, 12, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Flitsch, S.L.; Grigalunas, M.; Leeson, P.D.; Quinn, R.J.; Turner, N.J.; Waldmann, H. The Time and Place for Nature in Drug Discovery. JACS Au 2022, 2, 2400–2416. [Google Scholar] [CrossRef]
- Espro, C.; Paone, E.; Mauriello, F.; Gotti, R.; Uliassi, E.; Bolognesi, M.L.; Rodríguez-Padrón, D.; Luque, R. Sustainable Production of Pharmaceutical, Nutraceutical and Bioactive Compounds from Biomass and Waste. Chem. Soc. Rev. 2021, 50, 11191–11207. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty Years on: The Impact of Fragments on Drug Discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- de Esch, I.J.P.; Erlanson, D.A.; Jahnke, W.; Johnson, C.N.; Walsh, L. Fragment-to-Lead Medicinal Chemistry Publications in 2020. J. Med. Chem. 2022, 65, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Lamoree, B.; Hubbard, R.E. Current Perspectives in Fragment-Based Lead Discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-Based Drug Discovery—The Importance of High Quality Molecule Libraries. Mol. Oncol. 2022, 16, 3761–3777. [Google Scholar] [CrossRef]
- Asciminib: The Sixth Fragment-Derived Drug Approved. Available online: http://practicalfragments.blogspot.com/2021/10/asciminib-sixth-fragment-derived-drug.html (accessed on 30 January 2022).
- Over, B.; Wetzel, S.; Grütter, C.; Nakai, Y.; Renner, S.; Rauh, D.; Waldmann, H. Natural-Product-Derived Fragments for Fragment-Based Ligand Discovery. Nat. Chem. 2013, 5, 21–28. [Google Scholar] [CrossRef]
- Pahl, A.; Waldmann, H.; Kumar, K. Exploring Natural Product Fragments for Drug and Probe Discovery. Chimia 2017, 71, 653–660. [Google Scholar] [CrossRef]
- Prescher, H.; Koch, G.; Schuhmann, T.; Ertl, P.; Bussenault, A.; Glick, M.; Dix, I.; Petersen, F.; Lizos, D.E. Construction of a 3D-Shaped, Natural Product like Fragment Library by Fragmentation and Diversification of Natural Products. Bioorg. Med. Chem. 2017, 25, 921–925. [Google Scholar] [CrossRef]
- Liu, M.; Quinn, R.J. Fragment-Based Screening with Natural Products for Novel Anti-Parasitic Disease Drug Discovery. Exp. Opin. Drug Discov. 2019, 14, 1283–1295. [Google Scholar] [CrossRef]
- Itabaiana, I., Jr.; Avelar Do Nascimento, M.; De Souza, R.O.M.A.; Dufour, A.; Wojcieszak, R. Levoglucosan: A Promising Platform Molecule? Green Chem. 2020, 22, 5859–5880. [Google Scholar] [CrossRef]
- Camp, J.E. Bio-Available Solvent Cyrene: Synthesis, Derivatization, and Applications. ChemSusChem 2018, 11, 3048–3055. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, J.; De Bruyn, M.; Constantinou, A.; Moity, L.; McElroy, C.R.; Farmer, T.J.; Duncan, T.; Raverty, W.; Hunt, A.J.; Clark, J.H. Dihydrolevoglucosenone (Cyrene) as a Bio-Based Alternative for Dipolar Aprotic Solvents. Chem. Commun. 2014, 50, 9650–9652. [Google Scholar] [CrossRef] [PubMed]
- New Solvent, Cyrene, Takes on NMP. Available online: https://cen.acs.org/business/biobased-chemicals/New-solvent-Cyrene-takes-NMP/97/i22 (accessed on 9 January 2022).
- Wu, Y.J.; Meanwell, N.A. Geminal Diheteroatomic Motifs: Some Applications of Acetals, Ketals, and Their Sulfur and Nitrogen Homologues in Medicinal Chemistry and Drug Design. J. Med. Chem. 2021, 64, 9786–9874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; White, G.B.; Ryan, M.D.; Hunt, A.J.; Katz, M.J. Dihydrolevoglucosenone (Cyrene) As a Green Alternative to N,N-Dimethylformamide (DMF) in MOF Synthesis. ACS Sustain. Chem. Eng. 2016, 4, 7186–7192. [Google Scholar] [CrossRef]
- Can Cyrene Offer a Greener Alternative to Harmful Solvents? Available online: https://www.merckgroup.com/en/research/science-space/envisioning-tomorrow/scarcity-of-resources/cyrene.html (accessed on 9 January 2022).
- Cyrene Solvent Plant Set for France. Available online: https://cen.acs.org/environment/green-chemistry/Cyrene-solvent-plant-set-France/98/i8 (accessed on 9 January 2022).
- Czubatka-Bieńkowska, A.; Sarnik, J.; Macieja, A.; Galita, G.; Witczak, Z.J.; Poplawski, T. Thio-Functionalized Carbohydrate Thiosemicarbazones and Evaluation of Their Anticancer Activity. Bioorganic Med. Chem. Lett. 2017, 27, 2713–2720. [Google Scholar] [CrossRef]
- Hughes, L.; McElroy, C.R.; Whitwood, A.C.; Hunt, A.J. Development of Pharmaceutically Relevant Bio-Based Intermediates Though Aldol Condensation and Claisen-Schmidt Reactions of Dihydrolevoglucosenone (Cyrene®). Green Chem. 2018, 20, 4423–4427. [Google Scholar] [CrossRef]
- Jung, M.E.; Kiankarimi, M. Synthesis of Methylene-Expanded 2′,3′-Dideoxyribonucleosides. J. Org. Chem. 1998, 63, 8133–8144. [Google Scholar] [CrossRef]
- Delorme, D.; Ducharme, Y.; Brideau, C.; Chan, C.C.; Chauret, N.; Desmarais, S.; Dubé, D.; Falgueyret, J.P.; Fortin, R.; Guay, J.; et al. Dioxabicyclooctanyl Naphthalenenitriles as Nonredox 5-Lipoxygenase Inhibitors: Structure-Activity Relationship Study Directed toward the Improvement of Metabolic Stability. J. Med. Chem. 1996, 39, 3951–3970. [Google Scholar] [CrossRef]
- Sharipov, B.T.; Davidova, A.N.; Ryabova, A.S.; Galimzyanova, N.F.; Valeev, F.A. Synthesis and Fungicidal Activity of Methylsulfanylmethyl Ether Derivatives of Levoglucosenone. Chem. Heterocycl. Compd. 2019, 55, 31–37. [Google Scholar] [CrossRef]
- Titarenko, Z.; Vasilevich, N.; Zernov, V.; Kirpichenok, M.; Genis, D. Oxygen-Containing Fragments in Natural Products. J. Comput.-Aided Mol. Des. 2013, 27, 125–160. [Google Scholar] [CrossRef]
- Fuller, N.; Spadola, L.; Cowen, S.; Patel, J.; Schönherr, H.; Cao, Q.; McKenzie, A.; Edfeldt, F.; Rabow, A.; Goodnow, R. An Improved Model for Fragment-Based Lead Generation at AstraZeneca. Drug Discov. Today 2016, 21, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Revillo Imbernon, J.; Jacquemard, C.; Bret, G.; Marcou, G.; Kellenberger, E. Comprehensive Analysis of Commercial Fragment Libraries. RSC Med. Chem. 2021, 13, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.J.; Dekker, T.; Klein, H.F.; Janssen, G.V.; Wijtmans, M.; O’Brien, P.; de Esch, I.J.P. Escape from Planarity in Fragment-Based Drug Discovery: A Physicochemical and 3D Property Analysis of Synthetic 3D Fragment Libraries. Drug Discov. Today Technol. 2020, 38, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.F.; Hamilton, D.J.; de Esch, I.J.P.; Wijtmans, M.; O’Brien, P. Escape from Planarity in Fragment-Based Drug Discovery: A Synthetic Strategy Analysis of Synthetic 3D Fragment Libraries. Drug Discov. Today 2022, 27, 2484–2496. [Google Scholar] [CrossRef]
- Leach, A.R.; Hann, M.M. Molecular Complexity and Fragment-Based Drug Discovery: Ten Years On. Curr. Opin. Chem. Biol. 2011, 15, 489–496. [Google Scholar] [CrossRef]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef]
- Pacheco, A.A.C.; Sherwood, J.; Zhenova, A.; McElroy, C.R.; Hunt, A.J.; Parker, H.L.; Farmer, T.J.; Constantinou, A.; De bruyn, M.; Whitwood, A.C.; et al. Intelligent Approach to Solvent Substitution: The Identification of a New Class of Levoglucosenone Derivatives. ChemSusChem 2016, 9, 3503–3512. [Google Scholar] [CrossRef]
- Liu, X.; Pollard, B.; Banwell, M.G.; Yu, L.-J.; Coote, M.L.; Gardiner, M.G.; van Vugt-Lussenburg, B.M.A.; van der Burg, B.; Grasset, F.L.; Campillo, E.; et al. Simple and Modestly Scalable Synthesis of Iso-Cyrene from Levoglucosenone and Its Comparison to the Bio-Derived and Polar Aprotic Solvent Cyrene®. Aust. J. Chem. 2022, 75, 331–344. [Google Scholar] [CrossRef]
- Valeev, F.A.; Kalimullina, L.K.; Salikhov, S.M.; Shitikova, O.V.; Tsypysheva, I.P.; Safarov, M.G. Synthesis of 2-Amino Derivatives of Levoglucosenone. Chem. Nat. Compd. 2004, 40, 521–525. [Google Scholar] [CrossRef]
- Brimacombe, J.S.; Hunedy, F.; Mather, A.M.; Tucker, L.C.N. Studies Related to the Synthesis of Derivatives of 2,6-Diamino-2,3,4,6-Tetradeoxy-D-Erythro-Hexose (Purpurosamine C), a Component of Gentamicin C12. Carbohydr. Res. 1979, 68, 231–238. [Google Scholar] [CrossRef]
- Kuhl, N.; Turnbull, B.W.H.; Ji, Y.; Larson, R.T.; Shevlin, M.; Prier, C.K.; Chung, C.K.; Desmond, R.; Guetschow, E.; He, C.Q.; et al. Utilizing Biocatalysis and a Sulfolane-Mediated Reductive Acetal Opening to Access Nemtabrutinib from Cyrene. Green Chem. 2023, 25, 606–613. [Google Scholar] [CrossRef]
- Chessari, G.; Grainger, R.; Holvey, R.S.; Ludlow, R.F.; Mortenson, P.N.; Rees, D.C. C-H Functionalisation Tolerant to Polar Groups Could Transform Fragment-Based Drug Discovery (FBDD). Chem. Sci. 2021, 12, 11976–11985. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Jin, C.; Willmore, L.; Feher, M.; Shaw, D.E. Fragment Hits: What Do They Look Like and How Do They Bind? J. Med. Chem. 2019, 62, 3381–3394. [Google Scholar] [CrossRef] [PubMed]
- SDS Cyrene. Available online: https://circa-group.com/wp-content/uploads/2022/02/Cyrene-SDS-feb-2022-pd-1.pdf (accessed on 1 November 2022).
- Wilson, K.L.; Kennedy, A.R.; Murray, J.; Greatrex, B.; Jamieson, C.; Watson, A.J.B. Scope and Limitations of a DMF Bio-Alternative within Sonogashira Cross-Coupling and Cacchi-Type Annulation. Beilstein J. Org. Chem. 2016, 12, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Jæger Pedersen, M.; Pedersen, C.M. Reactivity, Selectivity, and Synthesis of 4-C-Silylated Glycosyl Donors and 4-Deoxy Analogues. Angew. Chem. Int. Ed. 2021, 60, 2689–2693. [Google Scholar] [CrossRef]
- Merck Team Wins 2022 Peter J. Dunn Award for Green Chemistry and Engineering. Available online: https://communities.acs.org/t5/GCI-Nexus-Blog/Merck-team-wins-2022-Peter-J-Dunn-Award-for-Green-Chemistry-and/ba-p/87165 (accessed on 14 June 2022).
- Zhao, F.; Zhang, L.D.; Hao, Y.; Chen, N.; Bai, R.; Wang, Y.J.; Zhang, C.C.; Li, G.S.; Hao, L.J.; Shi, C.; et al. Identification of 3-Substituted-6-(1-(1H-[1,2,3]Triazolo [4,5-b]Pyrazin-1-Yl)Ethyl)Quinoline Derivatives as Highly Potent and Selective Mesenchymal-Epithelial Transition Factor (c-Met) Inhibitors via Metabolite Profiling-Based Structural Optimization. Eur. J. Med. Chem. 2017, 134, 147–158. [Google Scholar] [CrossRef]
- Valeev, F.A.; Gorobets, E.V.; Tsypysheva, I.P.; Singizova, G.S.; Kalimullina, L.K.; Safarov, M.G.; Shitikova, O.V.; Miftakhov, M.S. Stereochemical Aspects of the Beckman Rearrangement of Oximes of Levoglucosenone and Its Dihydro Derivative. Enantioselective Synthesis of (+)-γ-Pelargonolactone. Chem. Nat. Compd. 2003, 39, 563–568. [Google Scholar] [CrossRef]
- Okada, M.; Banno, A.; Sumitomo, H. Chemical Synthesis of 4-Deoxy-(1→6)-α-d-Xylo-Hexopyranan and 3,4-Dideoxy-(1→6)-α-d-Erythro-Hexopyranan. Carbohydr. Res. 1992, 226, 345–352. [Google Scholar] [CrossRef]
- Klepp, J.; Sumby, C.J.; Greatrex, B.W. Synthesis of a Chiral Auxiliary Family from Levoglucosenone and Evaluation in the Diels-Alder Reaction. Synlett 2018, 29, 1441–1446. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “Rule of Three” for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The “Rule of Three” for Fragment-Based Drug Discovery: Where Are We Now? Nat. Rev. Drug Discov. 2013, 12, 644. [Google Scholar] [CrossRef] [PubMed]
- Bevan, C.D.; Lloyd, R.S. A High-Throughput Screening Method for the Determination of Aqueous Drug Solubility Using Laser Nephelometry in Microtiter Plates. Anal. Chem. 2000, 72, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Hoelke, B.; Gieringer, S.; Arlt, M.; Saal, C. Comparison of Nephelometric, UV-Spectroscopic, and HPLC Methods for High-Throughput Determination of Aqueous Drug Solubility in Microtiter Plates. Anal. Chem. 2009, 81, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Downes, T.D.; Jones, S.P.; Klein, H.F.; Wheldon, M.C.; Atobe, M.; Bond, P.S.; Firth, J.D.; Chan, N.S.; Waddelove, L.; Hubbard, R.E.; et al. Design and Synthesis of 56 Shape-Diverse 3D Fragments. Chem. Eur. J. 2020, 26, 8969–8975. [Google Scholar] [CrossRef]
- Firth, N.C.; Brown, N.; Blagg, J. Plane of Best Fit: A Novel Method to Characterize the Three-Dimensionality of Molecules. J. Chem. Inf. Model. 2012, 52, 2516–2525. [Google Scholar] [CrossRef]
- ZINC. Available online: https://zinc15.docking.org/patterns/home/ (accessed on 29 June 2022).
- Han, Y.; Xing, K.; Zhang, J.; Tong, T.; Shi, Y.; Cao, H.; Yu, H.; Zhang, Y.; Liu, D.; Zhao, L. Application of Sulfoximines in Medicinal Chemistry from 2013 to 2020. Eur. J. Med. Chem. 2021, 209, 112885. [Google Scholar] [CrossRef]
- Mäder, P.; Kattner, L. Sulfoximines as Rising Stars in Modern Drug Discovery? Current Status and Perspective on an Emerging Functional Group in Medicinal Chemistry. J. Med. Chem. 2020, 63, 14243–14275. [Google Scholar] [CrossRef]
- Bonneau, G.; Peru, A.A.M.; Flourat, A.L.; Allais, F. Organic Solvent- and Catalyst-Free Baeyer-Villiger Oxidation of Levoglucosenone and Dihydrolevoglucosenone (Cyrene®): A Sustainable Route to (S)-γ-Hydroxymethyl-α,β-Butenolide and (S)-γ-Hydroxymethyl-γ-Butyrolactone. Green Chem. 2018, 20, 2455–2458. [Google Scholar] [CrossRef]
- Lederman, N.G. Students’ and Teachers’ Conceptions of the Nature of Science: A Review of the Research. J. Res. Sci. Teach. 1992, 29, 331–359. [Google Scholar] [CrossRef]
- Abd-El-khalick, F.; Lederman, N.G. Improving Science Teachers’ Conceptions of Nature of Science: A Critical Review of the Literature. Int. J. Sci. Educ. 2000, 22, 665–701. [Google Scholar] [CrossRef]
- Wijtmans, M.; Edink, E.; van Linden, O.P.; Zheng, Y.; Blaazer, A.R.; Siderius, M.; van Muijlwijk-Koezen, J.E. From Recipe to Research: Introducing Undergraduate Students to the Nature of Science Using a Hybrid Practical Course Centred on Drug Discovery for Neglected Diseases. Drug Discov. Today 2021, 26, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Sivén, M.; Teppo, J.; Lapatto-Reiniluoto, O.; Teräsalmi, E.; Salminen, O.; Sikanen, T. Generation Green—A Holistic Approach to Implementation of Green Principles and Practices in Educational Programmes in Pharmaceutical and Medical Sciences at the University of Helsinki. Sustain. Chem. Pharm. 2020, 16, 100262. [Google Scholar] [CrossRef]
- International Master in Sustainable Drug Discovery. Available online: https://sustainabledrugdiscovery.eu/ (accessed on 20 February 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Template | L | X | Y | Z | R1 | R2 | |
| Ethers | 5a | I | –OCH2– | CH | CH | - | - | - |
| 5b | I | –OCH2– | CH | CH | - | 2-Me | - | |
| 5c | I | –OCH2– | CH | CH | - | 4-Me | - | |
| 5d | I | –OCH2– | CH | CH | - | 3-CN | - | |
| 5e | I | –OCH2– | CH | CH | - | 4-CN | - | |
| 5f | I | –OCH2– | CH | CH | - | 3-OMe | - | |
| 5g | I | –OCH2– | CH | CH | - | 4-COOMe | - | |
| 5h | I | –OCH2– | N | CH | - | - | - | |
| 5i | I | –OCH2– | CH | N | - | - | - | |
| Esters | 6a | I | –O(CO)– | CH | CH | - | - | - |
| 6b | I | –O(CO)– | CH | CH | - | 2-F | - | |
| 6c | I | –O(CO)– | CH | CH | - | 4-Cl | - | |
| 6d | I | –O(CO)– | CH | CH | - | 3-CN | - | |
| 6e | I | –O(CO)– | CH | CH | - | 2-OMe | - | |
| 6f | I | –O(CO)– | CH | CH | - | 4-NMe2 | - | |
| 6g | II | –O(CO)– | CH | O | CH | - | - | |
| 6h | II | –O(CO)– | O | CH | CH | - | - | |
| 6i | II | –O(CO)– | S | CH | CH | - | - | |
| 6j | II | –O(CO)– | NMe | CH | CH | - | - | |
| 6k | II | –O(CO)– | O | CH | N | - | - | |
| 6l | II | –O(CO)– | N | O | CH | - | - | |
| 6m | II | –O(CO)– | N | CH | S | - | - | |
| 6n | II | –O(CO)– | NH | N | CH | - | - | |
| 6o | II | –O(CO)– | NMe | N | CH | - | - | |
| 6p | I | –O(CO)– | CH | N | - | - | - | |
| 6q | I | –O(CO)– | N | CH | - | 3-F | - | |
| 6r | III | –O(CO)– | - | - | - |  | - | |
| O-Carbamates | 7ab | I | –O(CO)NH– | CH | CH | - | 2-Me | 3-Me |
| 7bb | I | –O(CO)NH– | CH | CH | - | 4-F | - | |
| 7c | I | –O(CO)NH– | CH | CH | - | 4-Cl | - | |
| 7d | I | –O(CO)NHCH2– | CH | CH | - | 3-F | - | |
| Amines | 8a | I | –NHCH2– | CH | CH | - | 4-Br | - |
| 8b | III | –NHCH2– | - | - | - | cPr | - | |
| 8c | I | –NHCH2– | CH | CH | - | 3-Cl | 4-Cl | |
| 8d | I | –NHCH2– | CH | CH | - | 2-OMe | 4-OMe | |
| 8e | I | –NHCH2– | CH | CH | - | 3,4-methylenedioxy | ||
| 8fa,b | II | –NHCH2– | N | NMe | CH | - | - | |
| 8g | I | –NHCH2– | CH | CH | - | 4-OMe | - | |
| 8hb | I | –NHCH2– | CH | CH | - | 2-Cl | 6-Cl | |
| 8ic | I | –NHCH2– | CH | CH | - | 2-Cl | 6-Cl | |
| 8j | I | –NHCH2– | N | CH | - | - | - | |
| 8k | I | –NHCH2– | CH | N | - | - | - | |
| 8l | I | –NHCH2– | CH | CH | - | 4-Cl | - | |
| 8m | I | –NHCH2– | CH | CH | - | 3-F | - | |
| 8na | I | –NHCH2– | CH | CH | - | 2-Me | - | |
| 8o | I | –NHCH2– | CH | CH | - | 4-Me | - | |
| 8pa | I | –NHCH2– | CH | CH | - | 4-SO2Me | - | |
| 8qb | I | –NHCH2– | CH | CH | - | 4-CN | - | |
| 8rc | I | –NHCH2– | CH | CH | - | 4-CN | - | |
| 8sa | I | –NHCH2– | CH | CH | - | - | - | |
| Amides | 9ab | I | –NH(CO)– | CH | CH | - | - | - |
| 9bc | I | –NH(CO)– | CH | CH | - | - | - | |
| 9c | III | –NH(CO)– | - | - | - | Et | - | |
| 9d | III | –NH(CO)– | - | - | - |  | - | |
| 9e | I | –NH(CO)– | CH | CH | - | 2-OMe | - | |
| 9fb | I | –NH(CO)– | CH | CH | - | 3-OMe | - | |
| 9gc | I | –NH(CO)– | CH | CH | - | 3-OMe | - | |
| 9hb | I | –NH(CO)– | CH | CH | - | 4-OMe | - | |
| 9ic | I | –NH(CO)– | CH | CH | - | 4-OMe | - | |
| 9j | II | –NH(CO)– | NH | CH | CH | - | - | |
| 9k | III | –NH(CO)– | - | - | - | | - | |
| 9l | I | –NH(CO)CH2– | CH | N | - | - | - | |
| 9m | I | –NH(CO)(CH2)2– | CH | CH | - | - | - | |
| 9nb | I | –NH(CO)– | CH | CH | - | 3-NMe2 | - | |
| 9o b | II | –NH(CO)– | S | CH | CH | - | - | |
| 9p | II | –NH(CO)– | S | CH | CH | - | - | |
| 9q | I | –NH(CO)– | CH | N | - | - | - | |
| 9r | I | –NH(CO)– | CH | CH | - | 3-F | - | |
| 9s | I | –NH(CO)– | CH | CH | - | 4-Cl | - | |
| 9t | I | –NH(CO)– | CH | CH | - | 3-CN | - | |
| 9u | I | –NH(CO)– | CH | CH | - | 4-CN | - | |
| 9v | I | –NH(CO)– | CH | CH | - | 2-F | - | |
| 9w | I | –NH(CO)– | CH | CH | - | 3-Cl | - | |
| 9x | II | –NH(CO)– | O | CH | N | - | - | |
| N-Carbamates | 10a | I | –NH(CO)OCH2– | CH | CH | - | 3-F | - |
| 10b | I | –NH(CO)O– | CH | CH | - | 3-OMe | - | |
| 10c | I | –NH(CO)O– | CH | CH | - | 3-F | - | |
| Ureas | 11a | I | –NH(CO)NH– | CH | CH | - | 4-F | - |
| 11b | I | –NH(CO)NH– | CH | CH | - | - | - | |
| 11c | I | –NH(CO)NH– | CH | CH | - | 2-F | 4-F | |
| 11d | I | –NH(CO)NH– | CH | CH | - | 2-Me | 3-Me | |
| 11e | I | –NH(CO)NH– | CH | CH | - | 4-OMe | - | |
| 11f | I | –NH(CO)NH– | CH | CH | - | 3-Me | 4-Me | |
| 11g | I | –NH(CO)NHCH2– | CH | CH | - | 3-Me | - | |
| 11h | I | –NH(CO)NH– | CH | CH | - | 3-Me | - | |
| 11i | III | –NH(CO)NEt2 | - | - | - | - | - | |
| 11j | I | –NH(CO)NHCH2– | CH | CH | - | 4-Cl | - | |
| 11k | I | –NH(CO)NH– | CH | CH | - | 3-OMe | - | |
| 11l | I | –NH(CO)NH– | CH | CH | - | 2-OMe | - | |
| 11m | I | –NH(CO)NHCH2– | CH | CH | - | 3-F | - | |
| 11n | I | –NH(CO)NH– | CH | CH | - | 3-F | - | |
| 11o | I | –NH(CO)NH– | CH | CH | - | 2-F | - | |
| 11p | I | –NH(CO)NHCH2– | CH | CH | - | 2-F | - | |
| Sulfonamides | 12ab | I | –NHSO2– | CH | CH | - | - | - |
| 12bc | I | –NHSO2– | CH | CH | - | - | - | |
| 12cb | I | –NHSO2– | CH | CH | - | 4-Me | - | |
| 12dc | I | –NHSO2– | CH | CH | - | 4-Me | - | |
| 12e | I | –NHSO2– | CH | CH | - | 3-F | - | |
| 12f | III | –NHSO2– | - | - | - | cPr | - | |
| 12g | III | –NHSO2– | - | - | - | nPr | - | |
| Property | Ro3 Limits | Fsp3/3D Commercial Libraries | 3D Synthetic Libraries | Current Work |
|---|---|---|---|---|
| cLogP a | 3 | 1.56 | 1.13 | 1.52 |
| HBA | 3 | 3.77 | 3.23 | 3.92 |
| HBD | 3 | 1.18 | 1.08 | 0.90 |
| nRot | 3 | 4.5 | 2.0 | 2.6 |
| TPSA | 60 | 58.1 | 53.5 | 53.0 |
| MW | 300 | 262 | 232 | 254 |
| HAC | 20 | 18.3 | 16.2 | 17.9 |
| #Aromatic rings | - | 0.88 | 0.65 | 0.94 |
| #Rings | - | 2.01 | 2.28 | 2.98 |
| #Saturated rings | - | 1.07 | 1.32 | 2.03 |
| #Stereocenters | - | 1.07 | 1.67 | 3.00 |
| % Carbon atoms b | - | 71.6 | 71.0 | 71.8 |
| % heteroatoms b | - | 28.4 | 29.0 | 28.2 |
| % Nitrogen atoms b | - | 13.8 | 10.2 | 6.7 |
| % Oxygen atoms b | - | 11.4 | 14.7 | 19.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dekker, T.; Harteveld, J.W.; Wágner, G.; de Vries, M.C.M.; Custers, H.; van de Stolpe, A.C.; de Esch, I.J.P.; Wijtmans, M. Green Drug Discovery: Novel Fragment Space from the Biomass-Derived Molecule Dihydrolevoglucosenone (CyreneTM). Molecules 2023, 28, 1777. https://doi.org/10.3390/molecules28041777
Dekker T, Harteveld JW, Wágner G, de Vries MCM, Custers H, van de Stolpe AC, de Esch IJP, Wijtmans M. Green Drug Discovery: Novel Fragment Space from the Biomass-Derived Molecule Dihydrolevoglucosenone (CyreneTM). Molecules. 2023; 28(4):1777. https://doi.org/10.3390/molecules28041777
Chicago/Turabian StyleDekker, Tom, Jaap W. Harteveld, Gábor Wágner, Max C. M. de Vries, Hans Custers, Andrea C. van de Stolpe, Iwan J. P. de Esch, and Maikel Wijtmans. 2023. "Green Drug Discovery: Novel Fragment Space from the Biomass-Derived Molecule Dihydrolevoglucosenone (CyreneTM)" Molecules 28, no. 4: 1777. https://doi.org/10.3390/molecules28041777