1. Introduction
Nonlinear optical (NLO) materials are at the front line of research in interdisciplinary science and laser-based technology due to their fundamental applications in the field of optoelectronics [
1,
2,
3,
4]. Photonic devices, laser-based technology, endoscope, and sensors are examples of well known technologies where NLO materials have possible applications [
5,
6,
7,
8,
9,
10,
11,
12]. To design and synthesize the NLO materials, much efforts are exerted to understand the origin of nonlinearity in molecules and clusters in order to correlate NLO responses to electronic structure and molecular geometry. Polarization, asymmetric charge distribution, asymmetric crystal packing, and π-conjugated electron transport routes are all required for NLO materials. Because of their high thermal stability and transparency, inorganic nonlinear optical materials have been preferred over organic ones [
13]. Some inorganic borates crystals, such as KB
5 (KB
5O
8H
2O), BBO (-BaB
2O
4), and LiB
3O
5 (LBO), have been investigated as good NLO materials, particularly in the ultraviolet range [
13].
For obtaining high-performance NLO materials, several strategies were proposed, which include bond length alternation (BLA) [
14], doping metal atoms [
15], push–pull mechanisms from donor to acceptor [
16], modification of sp
2 hybridized carbon nanomaterials [
17], designing octupolar molecules [
18], multidecker sandwich complexes [
19], and excess electron induction [
20].
The introduction of excess electrons into molecules and clusters is the most viable technique to escalate hyperpolarizability. The availability of loosely bound electrons predominantly decreases the excitation energies for the crucial transition [
21,
22,
23]. Excess electrons in molecules and crystals behave similarly to Rydberg orbitals, which are positioned outside the parent molecules and are held loosely [
24,
25,
26]. Several studies have demonstrated the substantial role of the diffuse excess electrons in compounds for developing NLO materials. Wei Chen et al. investigated the Li@calix[
4]pyrrole electride complex, which has a significant static hyperpolarizability (β
o) value of up to 7.3 × 10
3 au, where the presence of excess electrons has a significant role [
27].
Theoretically designed compounds having excess electrons that are further classified into [
28] alkalides [
29], alkaline-earthides [
30], metalides [
31], and electrides [
32]. Alkalides are complex compounds in which alkali metals bear the negative charge (Li
−, Na
−, K
−) [
33]. On the other hand, electride complexes have anionic sites occupied by the electron inside the complexes [
34]. Furthermore, the alkaline–earthides were recently introduced to excess electrons compounds, where the alkaline earth metals hold a negative charge [
35]. Interestingly, superalkali clusters are a new class of materials that can transport electrons, making them useful for the fabrication of electro-optical materials [
36].
Superalkalis clusters with lower ionization energy (IE) than alkali metal elements are well known due to their powerful reducing capabilities. The very first report about superalkalis was obtained in 1982 by Gutsev and Boldyrev for Li
3O, Li
2F, and Li
4N clusters [
37]. These clusters with unique qualities, such as tuneability in their electrical properties and the ability to function as a bridge between micro and macro materials, are of great interest to cluster science. Recent advances in cluster science show the potential applications of superalkali clusters, i.e., the reductive materials, helium and hydrogen storage, catalysis, supersalt formation, and nonlinear optics [
38,
39,
40,
41].
Superalkali clusters are excellent candidates for creating optical and NLO materials because of their excellent tunable electronic and structural properties. The decreased excitation energy may be responsible for electrons shifting from HOMO to LUMO, as they are loosely bound. Based on the intriguing characteristics of superalkali clusters, these were used to fabricate NLO materials. In this regard, two-dimensional materials doped superalkali, and they play an essential role in triggering the hyperpolarizability response. Sun et al. theoretically designed superalkali-based alkalides Li
3+(calix[
4]pyrrole)M
−, Li
3O
+(calix[
4]pyrrole)M
−, and M
3O
+(calix[
4]pyrrole)K
− (M = Li, Na, and K), where the hyperpolarizability response is recorded up to 34 718 au [
42]. Similarly, Faizan Ullah et al. reported a noticeable enhancement in the NLO response of the A1
12P
12 nanocluster by using Li
4N, Li
2F, and Li
3O superalkalis as the source of the excess electrons [
43]. Furthermore, macrocyclic oligofurans ring doped with superalkali clusters were also reported as a new kind of nonlinear optical material where a larger hyperpolarizability response is attributed to the presence of loosely bound electrons [
44].
Although a larger number of superalkali clusters were theoretically designed, very limited studies have been conducted to show the possibility of using polynuclear superalkali (undoped) clusters as NLO materials. Srivastava et al. investigated the electronic and nonlinear optical properties Li
nF (
n = 2–5) and M2X small clusters as excess electron compounds where the β
o increases up to 10
5 au for Li
2F [
45,
46]. Our group investigated the static and dynamic hyperpolarizability response of M
2OCN and M
2NCO (M = Li, Na, K) superalkali clusters as excess electrons candidates where the second hyperpolarizability γ(ω) values were calculated up to 2.1 × 10
8 au [
45].
Superalkali clusters can be mononuclear, bimetallic, and polynuclear based on their rational design and elemental composition. We are interested to investigate carbon-based polynuclear clusters for electronic and NLO properties. These clusters are more stable than conventional mononuclear superalkali clusters and might possess better electronic and NLO properties. The previous development in the family of superalkalis and their tunable electronic properties prompted us to further investigate polynuclear clusters for optical and NLO properties. Polynuclear carbon-based clusters C3O[C(CN)2]2M3 (where M = Li, Na, and K) are investigated using DFT.
3. Computational Details
All density functional theory (DFT) calculations are performed in the gas phase with Gaussian 09 software, whereas visualization is achieved using the GaussView 5.0 program [
56,
57]. Geometries of all polynuclear C
3O[C(CN)
2]
2M
3 (where M = Li, Na, and K) clusters are optimized at CAM-B3LYP/6-311++G(d,p) functionality [
58]. The quantum mechanics-based Coulomb attenuating method (CAM-B3LYP) is a hybrid exchange-correlation functional that combines B3LYP’s hybrid features with the CAM functional’s long-range corrected parameter. It was found that this long-range corrected density functional substantially reduces the overestimation seen with conventional techniques and typically provides results that are comparable to those of coupled cluster calculations. Previous research has demonstrated that this method is well recognized for examining molecules and clusters, as well as for determining NLO properties [
59,
60]. Besides, the choice of a suitable basis set is crucial for obtaining reliable results. Thus, the CAM-B3LYP method with 6-311+G(d,p) split valence basis set is a reliable level of theory for geometry optimization and accuracy in results for electronic properties [
61,
62,
63,
64,
65].
To determine whether the presented structures are true minima on the potential energy surface, frequency calculations are carried out. For thermodynamic stability, we calculated binding energy per atom for these clusters. Electronic stability and superalkali nature are validated through computed ionization energies (IE) and electron affinities (EA). To further explore the electronic properties, we performed frontier molecular orbital (FMO) analysis. Natural bonding orbitals (NBO) study is carried out to explore the charge distribution on atoms within superalkali clusters [
66]. The binding energy per atom (E
B) is given by the following relations:
where E
T is the total electronic energy of studied (X) superalkali clusters, E
A(X) is the total energy of individual atoms within clusters, and n is the total number of atoms. The vertical ionization energy, electron affinity, and electrical conductivity (σ) can be represented by the equation:
where VIE and VEA are vertical ionization energies and electron affinities of studied clusters. In Equation (4), σ, E
G, k, and T represent the electrical conductivity, energy gap, Boltzmann constant, and temperature, respectively. To further explore the electronic properties of studied clusters, we also performed total density of state (TDOS) analysis at the same method by using the GaussSum software [
67]. The following equation can be used to explain the molecules under the static electric field.
where F is an external applied electric field, F
i is the component of field along i direction, E
0 is the total energy of the superalkali clusters without a static electric field, and µ
i, α
ij, β
ijk, and γ
ijkl are dipole moment, polarizability, hyperpolarizability, and second-order hyperpolarizability, respectively. The mean dipole moment (µ
o), change in dipole moment (Δµ), static polarizability (α
o), and static first hyperpolarizability (β
o) are calculated to illustrate the NLO response and associated responsible factors.
where β
x = β
xxx + β
xyy + β
xzz, β
y = β
yyy + β
yzz + β
yxx and β
z = β
zzz + β
zxx + β
zyy.
To obtain absorption behaviors and excited state parameters of studied clusters, we performed TD-DFT simulations. We considered 30 states for getting excited states parameters. The Gaussian band shape and the absorption spectra were obtained by using the following relation,
:
where the
i subscript represents the electronic excitation of interest. The other symbols in the equation have the following meanings:
, shows the excitation energy (in wavenumbers) corresponding to the required electronic excitation in TD-DFT
is the value of at the maximum of the band shape
Sigma (σ) is a wavenumber representation of the standard deviation that is related to the simulated band’s width.
The second static hyperpolarizability
(γ
o) and the projection of hyperpolarizability on the dipole moment vector
(β
vec) are also calculated for our studied superalkali clusters at the same level of theory. Static second hyperpolarizability
(γ
o) and vector part of hyperpolarizability
(β
vec) are expressed as:
Moreover, the molecular parameters relevant to electro-optical Pockel’s effect (EOPE) and second harmonic generation (SHG) are calculated at externally applied frequencies (532 and 1064 nm).
4. Conclusions
In summary, we presented the geometric, electronic, and nonlinear optical properties of polynuclear carbon-based clusters at CAM-B3LYP/6-311++G(d,p) level. These clusters are thermodynamically stable, and their binding energies per atom range from −160.07 to −162.07 kcal mol−1. The electronic stability and superalkali nature are characterized through calculated ionization potential (IP) and FMO analyses. Small ionization potential further suggests their superalkali nature. NBO charge analysis reveals excellent charge separation within clusters. The performed DOS analysis shows size-dependent electronic and conductive properties, where C3 is a potential candidate. The significant first and second hyperpolarizabilities, up to 5.78 × 103 and 5.55 × 106 au, respectively, are calculated for the clusters. The βo response shows dependence on the size of alkali metals. Furthermore, the absorption study shows their transparency in the deep-UV region for NLO applications and absorption at longer wavelengths. The significant scattering hyperpolarizability (βHRS) value is (1.62 × 104), calculated for the C3 cluster, where octupolar contribution to βHRS is 92%. The dynamic first hyperpolarizability β(ω) is more pronounced for the EOPE effect at 532 nm, whereas SHG is more prominent for second hyperpolarizability γ(ω).

 and
and 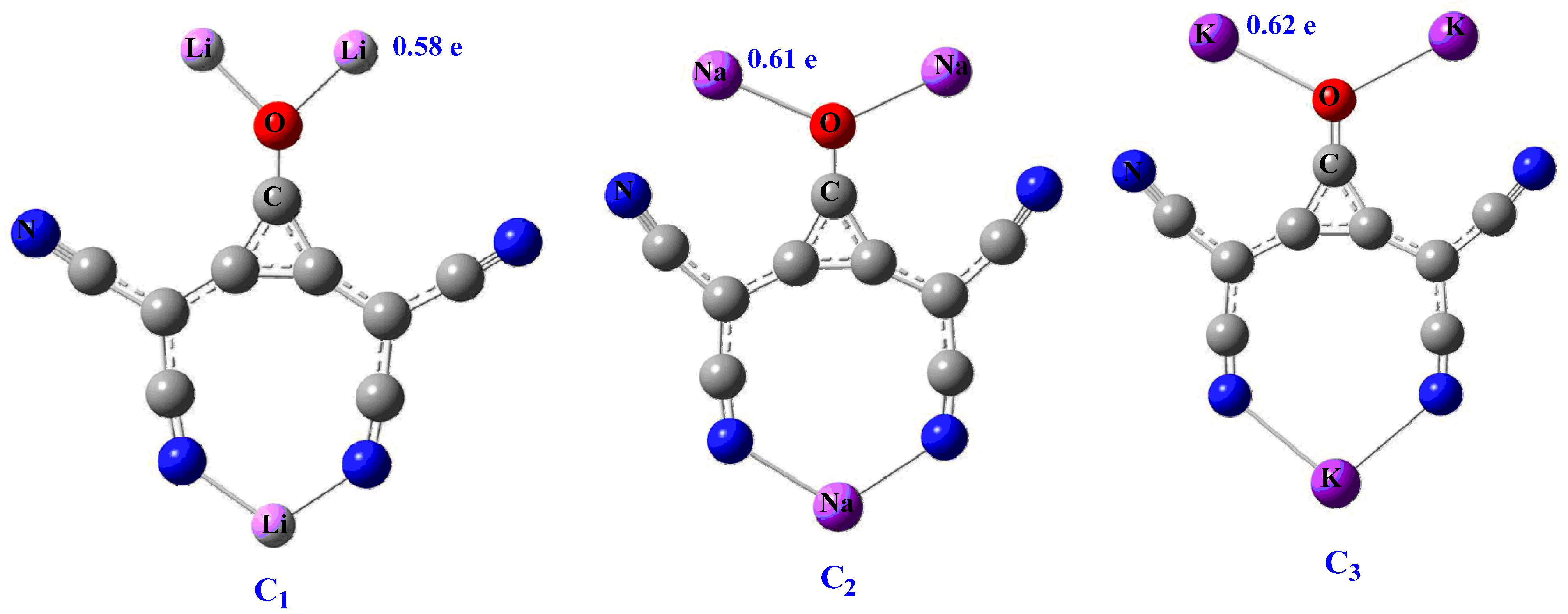





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}