
Progress of Artificial Intelligence in Drug Synthesis and Prospect of Its Application in Nitrification of Energetic Materials

 , ,
, ,
Abstract
:
1. Introduction
2. Advances of Artificial Intelligence in Drug Discovery
2.1. Artificial Intelligence High-Throughput Chemical Reaction Screening Platform
2.2. Modular Automatic Synthesis Machine Driven by Chemical Programming Language
2.3. AI Synthetic Robots with Thinking Functions to Explore New Reactions
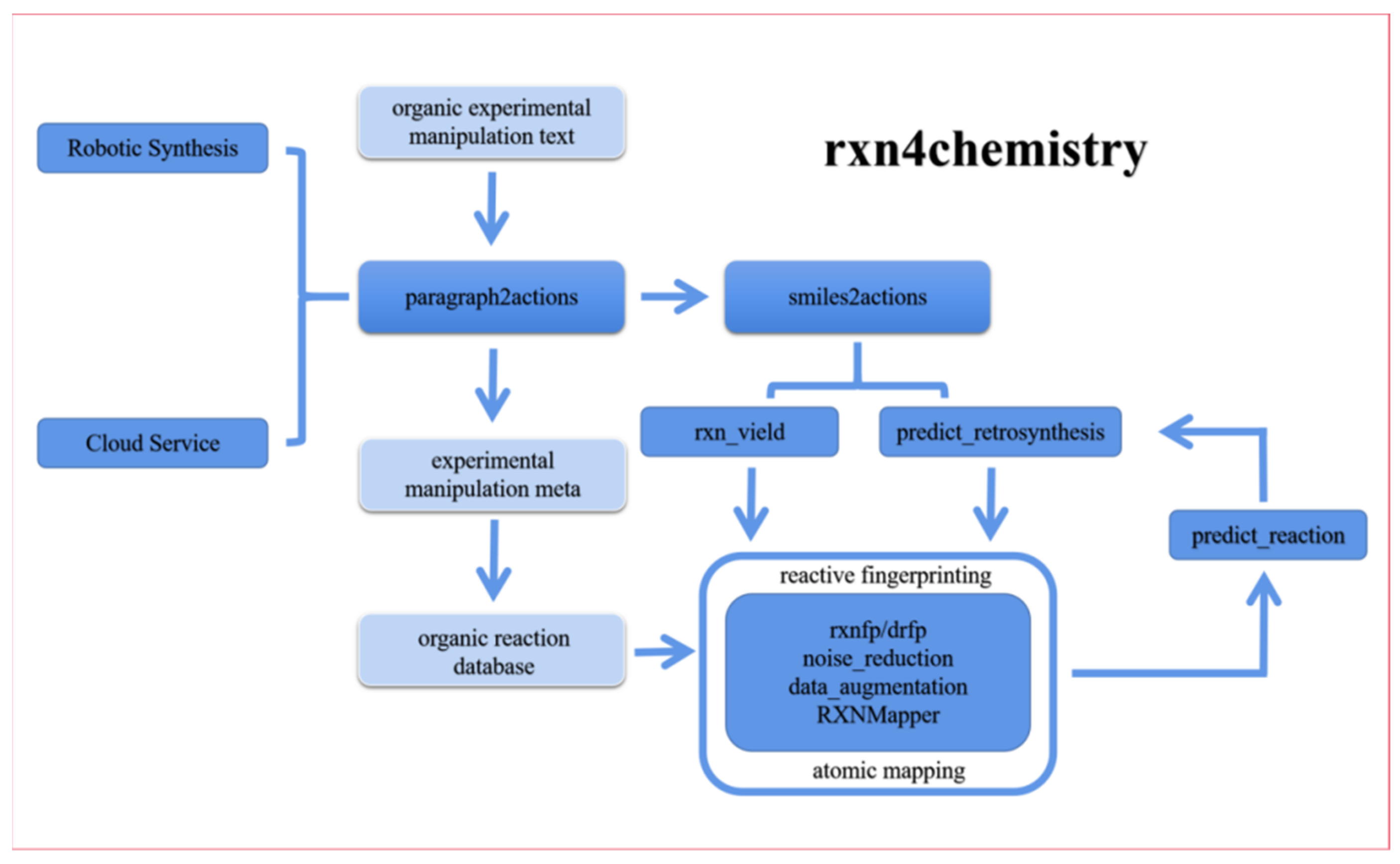
2.4. Artificial Intelligence Cloud Lab
2.5. Automated Synthesis Systems from Design to Synthesis
2.6. Artificial Intelligent Fully Autonomous Mobile Robots
3. The Research Progress of Artificial Intelligence in the Field of Energetic Materials and Its Application Prospect in the Field of Energetic Materials Nitrification
3.1. The Research Progress of Artificial Intelligence in the Field of Energetic Materials
3.1.1. Advances in Artificial Intelligence Algorithms and Methods for Performance Enhancement of Energetic Materials
3.1.2. Advances in Artificial Intelligence in Machine-Based Structure Search in Energetic Materials
3.2. The Inspiration of Artificial Intelligence in Development in the Drug Field on the Nitrification of Energetic Materials
3.3. Perspectives on the Application of Artificial Intelligence in the Field of Nitrification of Energetic Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huo, H.; Xuan, C.L.; Bi, F.Q.; Zhang, J.R.; Zhou, C.; Wang, B.T. Recent Progress insynthesis of insensitive energetic compounds. Chin. J. Explos. Propellants 2019, 42, 6–16. [Google Scholar]
- Wang, X. Current Situation of Study on Insensitive Composite Explosives in USA. Chin. J. Explos. Propellants 2007, 30, 78–80. [Google Scholar]
- Philip, P. A comparison of the structure synthesis and properties of insensitive energetic compounds. Propellants Explos. Pyrotech. 2016, 41, 452–469. [Google Scholar]
- Dioppold, A.A.; Klapotke, T.M.; Oswald, M. Asymmetrically substituted 5,5’-bistriazoles-nitrogen rich materials with various energetic functionalities. Dalton Trans. 2013, 42, 11136–11145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, N.; Izsák, D.; Klapötke, T.M.; Stierstorfer, J. The chemistry of 5-(tetrazol-1-yl)-2H-tetrazole: An extensive study of structural and energetic properties. Chem. Eur. J. 2013, 19, 8948–8957. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Wang, B.Z.; Li, H.; Li, Y.N.; Bi, F.Q.; Huo, H.; Fan, X.Z. Novel route and characterization of oxadiazolo-[1,2,3,4]tetrazine 4,6-di-N-oxide(FTDO). Chin. J. Org. Chem. 2012, 10, 1975–1980. [Google Scholar] [CrossRef] [Green Version]
- Lian-Jie, Z.; Bo-Zhou, W.; Huan, H.; Huai-Ming, H.; Su, P.-F.; Xue-Zhong, F.; Hui, L. Synthesis, crystal structure and thermal behavior of 3,4-bis(3-nitrofurazan-4-oxy)furazan. Chin. J. Energ. Mater. 2015, 23, 18–22. [Google Scholar]
- Jin, S. Risk Analysis of Benzene Nitrification Process and Discussion on Safety Facility Design. Henan Sci. Technol. 2020, 22, 110–112. [Google Scholar]
- Zhang, B.H.; He, Y.C.; Bi, J.G.; Wu, K.J.; Xu, G.H. Conceptual Design of Stirred Tank Nitration Reactors Based on an Inherently Safer Design Strategy. J. Chem. Eng. Chin. Univ. 2015, 29, 312–319. [Google Scholar]
- Fan, H.J. Fire and Explosion Danger of Industry Nitrification Process and Accident Prevention Inquiry. Guangzhou Chem. Ind. 2013, 41, 174–176. [Google Scholar]
- Jiang, J. Security Hidden Troubles and Countermeasures of Several Reaction Processes in Chemical Industry. Liaoning Chem. Ind. 2011, 40, 490–492. [Google Scholar]
- Pan, X.T.; Wang, J.P.; Che, Z.H. Safety Management of Nitrification Process. Guangzhou Chem. Ind. 2016, 44, 177–178. [Google Scholar]
- Liu, S.Y.; Lv, X.F. Safety production technology in nitrification process. Hebei Chem. Ind. 2010, 33, 69–70. [Google Scholar]
- Zeng, L.Y.; Mao, M.Z.; Wang, W.; Wang, L.; Zhang, J.G.; Ning, B.K. Application of Microreactors in Nitration. Chem. Reag. 2018, 40, 1054–1058. [Google Scholar]
- Du, S. Study on Hazard and Safety Control of Typical Nitrification Process. Jiangxi Chem. Ind. 2021, 4, 11–13. [Google Scholar]
- Meng, M.Y.; Ma, Y.; Liu, D.F. Review and progress on nitrification of aromatic compounds. Dyest. Color. 2015, 52, 36–40. [Google Scholar]
- Li, B.M.; Zhao, Y.Q. Study on cationic modification technology of cotton spinning fiber. Text. Dye. Finish. J. 2011, 33, 19–21. [Google Scholar]
- Wei, W.; Xu, W.C.; Yan, D.M. Progress in Nitrification in Micro-reactors. Dyest. Color. 2019, 56, 55–60. [Google Scholar]
- Hu, C. Denitrification Based on Sieve Plate Tower Biofilm Reactor; Shanghai Normal University: Shanghai, China, 2020. [Google Scholar]
- Wu, C.L. The Application of Panel Foaming Equipment in CPU External Wall Insulation. Polyurethane Ind. 2010, 25, 32–35. [Google Scholar]
- Wu, J. Study on the Aromatic Nitrationm a Microreactor; Nanjing University of Science and Technology: Nanjing, China, 2012. [Google Scholar]
- Xu, W.L.; Wang, Y.Q.; Zhang, X.X. Process design for production of nitrochlorobenzene by continuous adiabatic nitration of chlorobenzene. Chem. Eng. 2012, 40, 71–74. [Google Scholar]
- Jiang, S.K.; Han, B.; Zhao, X.; Yu, W.H.; Luo, G.S.; Deng, J.; Liu, G.Q.; Wang, J.Q.; Wang, J.B. Preparation of mononitrotoluene by continuous adiabatic nitration of Progress excess toluene in microreactor. Prog. Chem. 2022, 41, 2910–2914. [Google Scholar]
- Wang, D. Research status and technical progress of dangerous nitrification process. Shandong Chem. Ind. 2021, 50, 86–90. [Google Scholar]
- Yu, H.W.; Zhao, J.Y.; Zhou, P.C.; Yu, Z.Q. Research Progress on Continuous Flow Nitrification Reaction Technology. Zhejiang Chem. Ind. 2020, 51, 26–31. [Google Scholar]
- Zhao, X.F. Lebanon big bang again alarm-the whole process of nitrification industry rupture safety management. China Pet. Chem. Ind. 2020, 9, 10–13. [Google Scholar]
- Peplow, M. Organic synthesis: The robo-chemist. Nature 2014, 512, 20–22. [Google Scholar] [CrossRef] [Green Version]
- Ley, S.V.; Fitzpatrick, D.E.; Myers, R.M.; Battilocchio, C.; Richard, J.I. Machine-Assisted Organic Synthesis. Angew. Chem. Int. Ed. 2015, 54, 10122–10137. [Google Scholar] [CrossRef] [Green Version]
- Markus, M. Machine Learning for Chemical Reactions. Chem. Rev. 2021, 121, 10218–10239. [Google Scholar]
- Clark, A.M.; Gedeck, P.; Cheung, P.P.; Bunin, B.A. Using Machine Learning to Parse Chemical Mixture Descriptions. ACS Omega 2021, 6, 22400–22409. [Google Scholar] [CrossRef]
- Gu, B.; Yu, B.; Dong, X.G.; Li, X.F.; Zhong, R.M.; Yang, M.F. Intelligent Program Synthesis Techniques: Literature Review. J. Softw. 2021, 32, 1373–1384. [Google Scholar]
- Almeida, A.F.; Moreira, R.; Rodrigues, T. Synthetic organic chemistry driven by artificial intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Jethava, K.P.; Fine, J.; Chen, Y.Q.; Hossain, A.; Chopra, G. Accelerated Reactivity Mechanism and Interpretable Machine Learning Model of N-Sulfonylimines toward Fast Multicomponent Reactions. Org. Lett. 2020, 22, 8480–8486. [Google Scholar] [CrossRef]
- Kletz, T.A. What you don’t have can’t leak. Chem. Ind. 1978, 42, 287–292. [Google Scholar]
- Kletz, T.A. Inherently safer design-its scope and future. Process Saf. Environ. Prot. 2003, 81, 401–405. [Google Scholar] [CrossRef]
- Li, M.; Lin, Z.J.; Liao, W.B.; Chen, T.B.; Li, J.Q.; Chen, J.; Xiao, M.F. Application of Artificial Intelligence in Synthetic Biology: A Review. J. Integr. Technol. 2021, 10, 43–46. [Google Scholar]
- Wang, Y.; Wang, H.C.; Yan, M.H.; Hu, G.H.; Wang, X.W. Design of biomolecular sequences by artificial intelligence. Synth. Biol. J. 2021, 2, 1–14. [Google Scholar]
- Reymond, J.L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Engkvist, O.; Norrby, P.-O.; Selmi, N.; Lam, Y.-h.; Peng, Z.; Sherer, E.C.; Amberg, W.; Erhard, T.; Smyth, L.A. Computational prediction of chemical reactions: Current status and outlook. Drug Discov. Today 2018, 23, 1203–1218. [Google Scholar] [CrossRef]
- Maryasin, B.; Marquetand, P.; Maulide, N. Machine Learning for Organic Synthesis: Are Robots Replacing Chemists? Angew. Chem. Int. Ed. 2018, 57, 6978–6980. [Google Scholar] [CrossRef]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Wilbraham, L.; Mehr, S.H.; Leroy, C. Digitizing Chemistry Using the Chemical Processing Unit: From Synthesis to Discovery. Acc. Chem. Res. 2021, 54, 253–262. [Google Scholar] [CrossRef]
- Malig, T.C.; Yunker, L.P.E.; Steiner, S.; Hein, J.E. Online High-Performance Liquid Chromatography Analysis of Buchwald−Hartwig Aminations from within an Inert Environment. ACS Catal. 2020, 10, 13236–13244. [Google Scholar] [CrossRef]
- Salley, D.S.; Keenan, G.A.; Long, D.L.; Bell, N.L.; Cronin, L. A Modular Programmable Inorganic Cluster Discovery Robot for the Discovery and Synthesis of Polyoxometalates. ACS Cent. Sci. 2020, 6, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zhao, H.; Han, T. Research and enlightenment of artificial intelligence strategies in major countries. High Technol. Commun. 2017, 27, 755–762. [Google Scholar]
- Li, J.; Li, J.; Liu, R.; Tu, Y.; Li, Y.; Cheng, J.; He, T.; Zhu, X. Autonomous discovery of optically active chiral inorganic perovskite nanocrystals through an intelligent cloud lab. Nat. Commun. 2020, 11, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Buitrago Santanilla, A.; Regalado, E.L.; Pereira, T.; Shevlin, M.; Bateman, K.; Campeau, L.-C.; Schneeweis, J.; Berritt, S.; Shi, Z.-C.; Nantermet, P.; et al. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 2015, 347, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Perera, D.; Tucker, J.W.; Brahmbhatt, S. A platform for automated nanomolescale reaction screening and micromole-scale synthesis in flow. Science 2018, 359, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Gesmundo, N.J.; Sauvagnat, B.; Curran, P.J.; Richards, M.P.; Andrews, C.L.; Dandliker, P.J.; Cernak, T. Nanoscale synthesis and affinity ranking. Nature 2018, 557, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Ahneman, D.T.; Estrada, J.G.; Lin, S.S.; Dreher, S.D.; Doyle, A.G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Science 2018, 555, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ballmer, S.G.; Gillis, E.P.; Fujii, S.; Schmidt, M.J.; Palazzolo, A.M.; Lehmann, J.W.; Morehouse, G.F.; Burke, M.D. Synthesis of many different types of organic small molecules using one automated process. Science 2015, 347, 1221–1226. [Google Scholar] [CrossRef] [Green Version]
- Mehr, S.H.; Craven, M.; Leonov, A.I.; Keenan, G.; Cronin, L. A universal system for digitization and automatic execution of the chemical synthesis literature. Science 2020, 370, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Guidi, M.; Seeberger, P.H.; Gilmore, K. Automated radial synthesis of organic molecules. Nature 2020, 579, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Granda, J.M.; Donina, L.; Dragone, V.; Long, D.L.; Cronin, L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 2018, 559, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Angelone, D.; Hammer, A.J.; Rohrbach, S.; Krambeck, S.; Granda, J.M.; Wolf, J.; Zalesskiy, S.; Chisholm, G.; Cronin, L. Convergence of multiple synthetic paradigms in a universally programmable chemical synthesis machine. Nat. Chem. 2021, 13, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Bédard, A.-C.; Adamo, A.; Aroh, K.C.; Russell, M.G.; Bedermann, A.A.; Torosian, J.; Yue, B.; Jensen, K.F.; Jamison, T. Reconfigurable system for automated optimization of diverse chemical reactions. Science 2018, 361, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Steiner, S.; Wolf, J.; Glatzel, S.; Andreou, A.; Granda, J.M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P.J.; Angelone, D.; et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 2019, 363, 144–152. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, S. AI-driven robotic laboratories show promise. Engineering 2021, 7, 1351–1353. [Google Scholar] [CrossRef]
- Coley, C.W.; Thomas, D.A., III; Lummiss, J.A.; Jaworski, J.N.; Breen, C.P.; Schultz, V.; Hart, T.; Fishman, J.S.; Rogers, L.; Gao, H.; et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, 1–9. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, Z.; Mahjour, B.; Wang, D.; Zhang, R.; Shim, E.; McGrath, A.; Shen, Y.; Brugger, N.; Turnbull, R.; et al. Reinforcing the supply chain of COVID-19 therapeutics with expert-coded retrosynthetic software. CernakChemRxiv 2020. [Google Scholar] [CrossRef]
- Burger, B.; Maffettone, P.M.; Gusev, V.V.; Aitchison, C.M.; Bai, Y.; Wang, X.; Li, X.; Alston, B.M.; Li, B.; Clowes, R.; et al. A mobile robotic chemist. Nature 2020, 583, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Nariman-Zadeh, N.; Darvizeh, A.; Jamali, A.; Moeini, A. Evolutionary design of generalized polynomial neural networks for modelling and prediction of explosive forming process. J. Mater. Process. Technol. 2005, 164, 1561–1571. [Google Scholar] [CrossRef]
- Li, H.; Zhang, L.; Petrutik, N.; Wang, K.C.; Ma, Q.; Shem-Tov, D.; Zhao, F.Q.; Gozin, M. Molecular and Crystal Features of Thermostable Energetic Materials: Guidelines for Architecture of “Bridged” Compounds. ACS Cent. Sci. 2020, 6, 54–75. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Li, H.; Zong, H.H.; Huang, Y.L.; Gozin, M.; Sun, C.Q.; Zhang, L. Strategies for Achieving Balance between Detonation Performance and Crystal Stability of High-Energy-Density Materials. iScience 2020, 23, 100944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.B.; Nguyen, P.C.H.; Sen, O.; Udaykumar, H.S. Stephen Baek1,3 Artificial intelligence approaches for materials-by-design of energetic materials: State-of-the-art, challenges, and future directions. ArXiv 2022, arXiv:2211.08179. [Google Scholar]
- Cao, Y.; Lai, W.; Yu, T.; Liu, Y.; Wang, B. Can N-oxidation alleviate the energy-safety contradiction of energetic materials. FirePhysChem 2021, 1, 27–32. [Google Scholar] [CrossRef]
- Yao, Y.; Lin, Q.; Zhou, X.; Lu, M. Recent research on the synthesis pentazolate anion cyclo-N5−. FirePhysChem 2021, 1, 33–45. [Google Scholar] [CrossRef]
- Liu, Z. Review and prospect of thermal analysis technology applied to study thermal properties of energetic materials. FirePhysChem 2021, 1, 129–138. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.L.; Wang, B.Z.; Qiu, L.L.; Xu, R.Q.; Sheremetev, A.B. Recent synthetic efforts towards high energy density materials: How to design high-performance energetic structures? FirePhysChem 2022, 2, 83–139. [Google Scholar] [CrossRef]
- Wespiser, C.; Mathieu, D. Application of Machine Learning to the Design of Energetic Materials: Preliminary Experience and Comparison with Alternative Techniques. Propellants Explos. Pyrotech. 2022. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Butrico, M.S.; Fuge, M.D.; Chung, P.W. Applying machine learning techniques to predict the properties of energetic materials. Sci. Rep. 2018, 8, 9059–9071. [Google Scholar] [CrossRef] [Green Version]
- Barnes, B.C.; Elton, D.C.; Boukouvalas, Z.; Taylor, D.E.; Mattson, W.D.; Fuge, M.D.; Chung, P.W. Machine Learning of Energetic Material Properties. arXiv 2018. [Google Scholar] [CrossRef]
- Tian, X.L.; Song, S.W.; Chen, F.; Qi, X.J.; Wang, Y.; Zhang, Q.H. Machine learning-guided property prediction of energetic materials: Recent advances, challenges, and perspectives. Energetic Mater. Front. 2022, 3, 177–186. [Google Scholar] [CrossRef]
- Casey, A.D.; Son, S.F.; Bilionis, I.; Barnes, B.C. Prediction of Energetic Material Properties from Electronic Structure Using 3D Convolutional Neural Networks. J. Chem. Inf. Model. 2020, 60, 4457–4473. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.; Rai, N.; Sen, O.; Perry, W.L. Toward a machine-guided approach to energetic material discovery. J. Appl. Phys. 2022, 131, 234902–234916. [Google Scholar] [CrossRef]
- Zang, X.W.; Zhou, X.; Bian, H.T.; Jin, W.P.; Pan, X.H.; Jiang, J.C.; Koroleva, M.Y.; Shen, R.Q. Prediction and Construction of Energetic Materials Based on Machine Learning Methods. Molecules 2023, 28, 322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, Q. Application of Artificial Intelligence in Microfluidic Systems. Chin. J. Anal. Chem. 2020, 48, 439–448. [Google Scholar]
- Jacob, P.M.; Yamin, P.; Perez-Storey, C.; Hopgood, M.; Lapkin, A.A. Towards automation of chemical process route selection based on data mining. Green Chem. 2017, 19, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Tai, X.Y.; Zhang, H.; Niu, Z.; Christie, S.D.; Xuan, J. The future of sustainable chemistry and process: Convergence of artificial intelligence, data and hardware. Energy AI 2020, 2, 100036. [Google Scholar] [CrossRef]
- Gao, W.H.; Coley, C.W. The Synthesizability of Molecules Proposed by Generative Models. J. Chem. Inf. Model. 2020, 60, 5714–5723. [Google Scholar] [CrossRef] [Green Version]
- Gromski, P.S.; Granda, J.M.; Cronin, L. Universal Chemical Synthesis and Discovery with “The Chemputer”. Trends Chem. 2020, 2, 4–12. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Institution | Name Of The Platform | Features |
|---|---|---|---|
| 2015 | Merck | High throughput chemical reaction screening platform | Screening of substrates and reaction conditions for Buchwald-Hartwig coupling reactions at the nanomolar level is possible, with 1536 reactions completed per day. |
| 2018 | Pfizer | High throughput chemical reaction screening platform | Screening of over 1500 nano-molar scale Suzuki-Miyaura coupling reactions in 1 day; synthesis of hundreds of micro-molar scale products. |
| 2018 | University of Glasgow, USA | Chemputer | (1) standardization of language and modularity of equipment, resulting in the preparation of three high-quality medicinal compounds without human intervention, with yields and purity comparable to those of artificial synthesis. (2) The ability to accurately predict the outcome of chemical reactions and to "think" independently after completing experiments, allowing for the independent exploration of new chemical reactions and molecules. (3) Automated synthesis "from literature to compound". |
| 2019 | MIT | - | The synthetic route design software ASKCOS was developed to automate the synthesis of 15 drug molecules based on the designed routes. |
| 2020 | IBM | RoboRXN | The three functions of artificial intelligence, cloud technology and experimental robotics have been integrated to develop the synthesis design system RXN for cloud-controlled automated synthesis based on designed synthesis routes. |
| 2020 | University of Liverpool | Mobile chemist | Freedom of movement within the laboratory to perform various tasks in experiments independently. The analysis of 10 dimensional variables in more than 98 million alternative experiments and the autonomous adjustment of the catalyst composition led to the discovery of a photocatalyst system with six times higher activity than the initial catalyst. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, B.; Zhang, J.; Xiao, C.; Liu, Y.; Yang, X.; Wang, W.; Li, Y.; Liu, N. Progress of Artificial Intelligence in Drug Synthesis and Prospect of Its Application in Nitrification of Energetic Materials. Molecules 2023, 28, 1900. https://doi.org/10.3390/molecules28041900
Tan B, Zhang J, Xiao C, Liu Y, Yang X, Wang W, Li Y, Liu N. Progress of Artificial Intelligence in Drug Synthesis and Prospect of Its Application in Nitrification of Energetic Materials. Molecules. 2023; 28(4):1900. https://doi.org/10.3390/molecules28041900
Chicago/Turabian StyleTan, Bojun, Jing Zhang, Chuan Xiao, Yingzhe Liu, Xiong Yang, Wei Wang, Yanan Li, and Ning Liu. 2023. "Progress of Artificial Intelligence in Drug Synthesis and Prospect of Its Application in Nitrification of Energetic Materials" Molecules 28, no. 4: 1900. https://doi.org/10.3390/molecules28041900