
Red Light Absorption of [ReI(CO)3(α-diimine)Cl] Complexes through Extension of the 4,4′-Bipyrimidine Ligand’s π-System


Abstract
:1. Introduction
2. Results
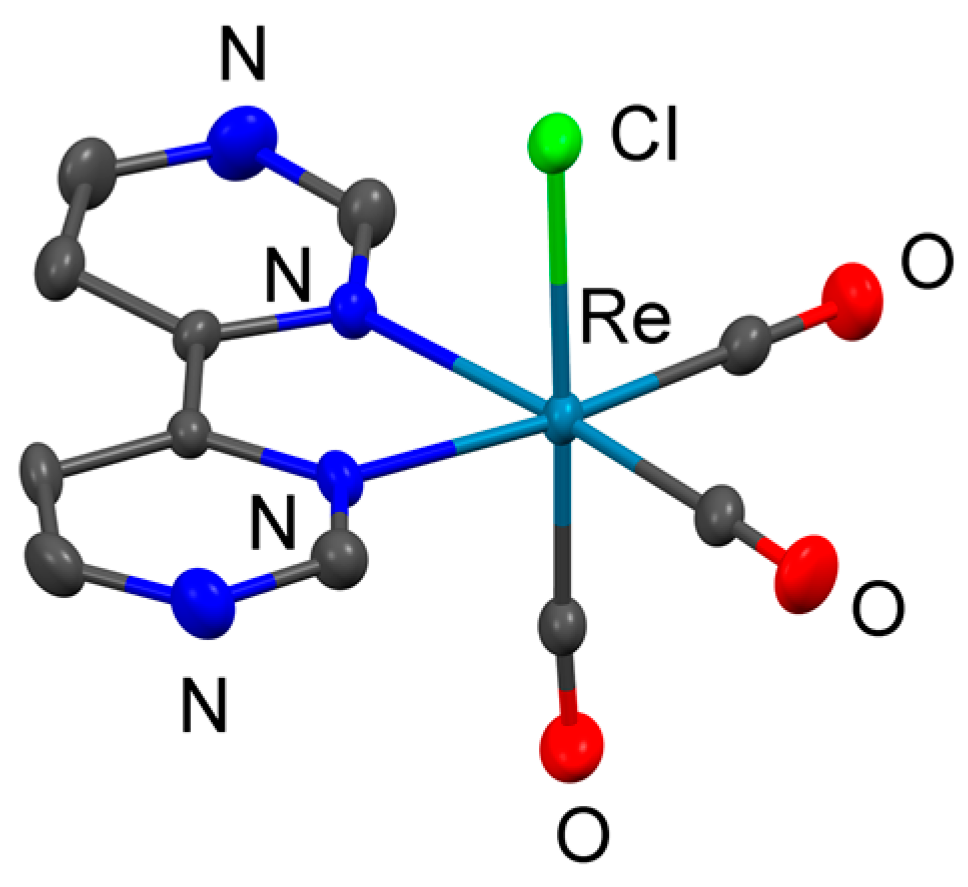
2.1. Syntheses and Structural Characterization
2.2. Crystallography
2.2.1. Details about 1
2.2.2. Details about 3
2.3. Electrochemistry
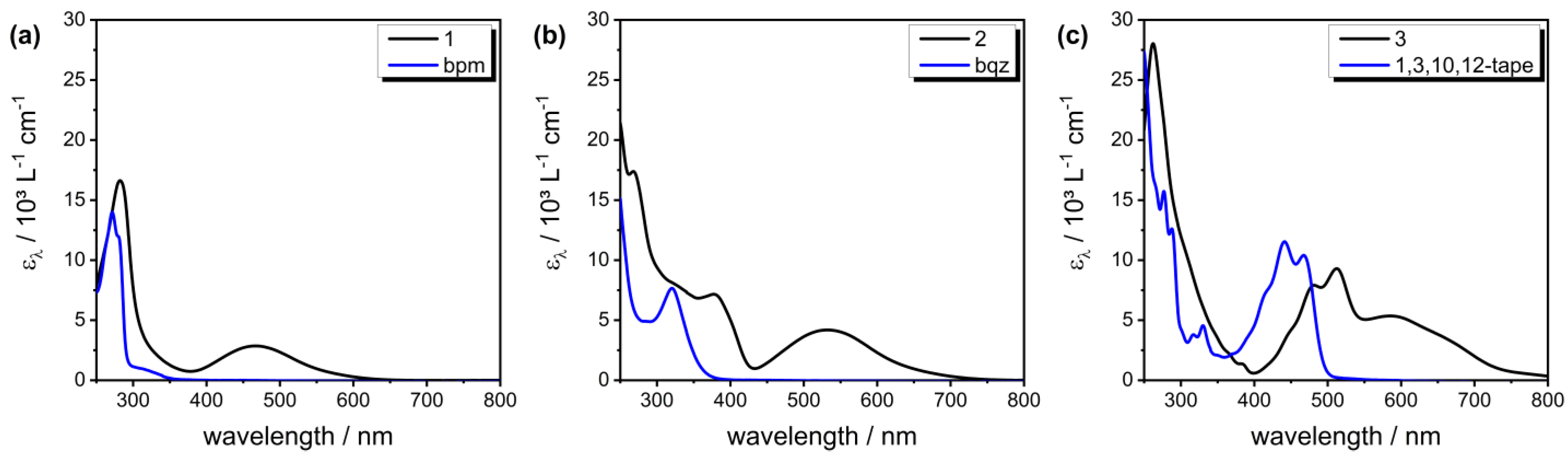
2.4. Electronic Absorption
Solvatochromism
2.5. UV–Vis NIR Spectroelectrochemistry
2.6. Steady-State Fluorescence
2.7. Transient Absorption
3. Conclusions and Outlook
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Esswein, A.J.; Nocera, D.G. Hydrogen Production by Molecular Photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Credi, A.; Venturi, M. Photochemical Conversion of Solar Energy. ChemSusChem 2008, 1, 26–58. [Google Scholar] [CrossRef] [PubMed]
- Teets, T.S.; Nocera, D.G. Photocatalytic Hydrogen Production. Chem. Commun. 2011, 47, 9268–9274. [Google Scholar] [CrossRef] [PubMed]
- Ceroni, P.; Balzani, V. Photoinduced Energy and Electron Transfer Processes. In The Exploration of Supramolecular Systems and Nanostructures by Photochemical Techniques; Ceroni, P., Ed.; Lecture Notes in Chemistry; Springer: Berlin/Heidelberg, Germany, 2012; Volume 78, pp. 21–38. ISBN 978-94-007-2041-1. [Google Scholar]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef] [PubMed]
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-Based Photosensitizers for Photodynamic Therapy: The Future of Multimodal Oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.K.-W. Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48, 2985–2995. [Google Scholar] [CrossRef]
- Lee, L.C.-C.; Leung, K.-K.; Lo, K.K.-W. Recent Development of Luminescent Rhenium(I) Tricarbonyl Polypyridine Complexes as Cellular Imaging Reagents, Anticancer Drugs, and Antibacterial Agents. Dalton Trans. 2017, 46, 16357–16380. [Google Scholar] [CrossRef]
- Takeda, H.; Ishitani, O. Development of Efficient Photocatalytic Systems for CO2 Reduction Using Mononuclear and Multinuclear Metal Complexes Based on Mechanistic Studies. Coord. Chem. Rev. 2010, 254, 346–354. [Google Scholar] [CrossRef]
- Takeda, H.; Koike, K.; Morimoto, T.; Inumaru, H.; Ishitani, O. Photochemistry and Photocatalysis of Rhenium(I) Diimine Complexes. In Advances in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 2011; Volume 63, pp. 137–186. ISBN 978-0-12-385904-4. [Google Scholar]
- Rau, S.; Görls, H. Solid State Structure of a Rhenium Bibenzimidazole Complex. J. Coord. Chem. 2004, 57, 1587–1590. [Google Scholar] [CrossRef]
- Gottschaldt, M.; Koth, D.; Müller, D.; Klette, I.; Rau, S.; Görls, H.; Schäfer, B.; Baum, R.P.; Yano, S. Synthesis and Structure of Novel Sugar-Substituted Bipyridine Complexes of Rhenium and 99m-Technetium. Chem. Eur. J. 2007, 13, 10273–10280. [Google Scholar] [CrossRef]
- Braumüller, M.; Schulz, M.; Staniszewska, M.; Sorsche, D.; Wunderlin, M.; Popp, J.; Guthmuller, J.; Dietzek, B.; Rau, S. Synthesis and Characterization of Ruthenium and Rhenium Dyes with Phosphonate Anchoring Groups. Dalton Trans. 2016, 45, 9216–9228. [Google Scholar] [CrossRef] [PubMed]
- Probst, B.; Kolano, C.; Hamm, P.; Alberto, R. An Efficient Homogeneous Intermolecular Rhenium-Based Photocatalytic System for the Production of H2. Inorg. Chem. 2009, 48, 1836–1843. [Google Scholar] [CrossRef]
- Probst, B.; Rodenberg, A.; Guttentag, M.; Hamm, P.; Alberto, R. A Highly Stable Rhenium−Cobalt System for Photocatalytic H2 Production: Unraveling the Performance-Limiting Steps. Inorg. Chem. 2010, 49, 6453–6460. [Google Scholar] [CrossRef]
- Probst, B.; Guttentag, M.; Rodenberg, A.; Hamm, P.; Alberto, R. Photocatalytic H2 Production from Water with Rhenium and Cobalt Complexes. Inorg. Chem. 2011, 50, 3404–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttentag, M.; Rodenberg, A.; Kopelent, R.; Probst, B.; Buchwalder, C.; Brandstätter, M.; Hamm, P.; Alberto, R. Photocatalytic H2 Production with a Rhenium/Cobalt System in Water under Acidic Conditions. Eur. J. Inorg. Chem. 2012, 2012, 59–64. [Google Scholar] [CrossRef]
- Abram, U.; Alberto, R. Technetium and Rhenium: Coordination Chemistry and Nuclear Medical Applications. J. Braz. Chem. Soc. 2006, 17, 1486–1500. [Google Scholar] [CrossRef]
- Alberto, R.; Meola, G.; Valdés, D.H. Technetium and Rhenium Complexes with Aromatic Hydrocarbons as Ligands. In Advances in Bioorganometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 215–241. ISBN 978-0-12-814197-7. [Google Scholar]
- Alberto, R.; Braband, H.; Nadeem, Q. Bioorganometallic Technetium and Rhenium Chemistry: Fundamentals for Applications. Chimia 2020, 74, 953. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.L.; Scott, S.M.; Flood, A.H.; Gordon, K.C. The Effect of Reduction on Rhenium(I) Complexes with Binaphthyridine and Biquinoline Ligands: A Spectroscopic and Computational Study. J. Phys. Chem. A 2005, 109, 3745–3753. [Google Scholar] [CrossRef]
- Yi, X.; Zhao, J.; Wu, W.; Huang, D.; Ji, S.; Sun, J. Rhenium(I) Tricarbonyl Polypyridine Complexes Showing Strong Absorption of Visible Light and Long-Lived Triplet Excited States as a Triplet Photosensitizer for Triplet–Triplet Annihilation Upconversion. Dalton Trans. 2012, 41, 8931–8940. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Schaugaard, R.N.; Noffke, B.W.; Liu, Y.; Li, D.; Liu, L.; Raghavachari, K.; Li, L. Well-Defined Nanographene–Rhenium Complex as an Efficient Electrocatalyst and Photocatalyst for Selective CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 3934–3937. [Google Scholar] [CrossRef]
- Hino, J.K.; Della Ciana, L.; Dressick, W.J.; Sullivan, B.P. Substituent Constant Correlations as Predictors of Spectroscopic, Electrochemical, and Photophysical Properties in Ring-Substituted 2,2′-Bipyridine Complexes of Rhenium(I). Inorg. Chem. 1992, 31, 1072–1080. [Google Scholar] [CrossRef]
- Wrighton, M.; Morse, D.L. Nature of the Lowest Excited State in Tricarbonylchloro-1,10-Phenanthrolinerhenium(I) and Related Complexes. J. Am. Chem. Soc. 1974, 96, 998–1003. [Google Scholar] [CrossRef]
- Komreddy, V.; Ensz, K.; Nguyen, H.; Paul Rillema, D. Synthesis and Characterization of Rhenium(I) 4,4′-Dicarboxy-2,2′-Bipyridine Tricarbonyl Complexes for Solar Energy Conversion. Inorg. Chim. Acta 2020, 511, 119815. [Google Scholar] [CrossRef]
- Artem’ev, A.V.; Petyuk, M.Y.; Berezin, A.S.; Gushchin, A.L.; Sokolov, M.N.; Bagryanskaya, I.Y. Synthesis and Study of Re(I) Tricarbonyl Complexes Based on Octachloro-1,10-Phenanthroline: Towards Deep Red-to-NIR Emitters. Polyhedron 2021, 209, 115484. [Google Scholar] [CrossRef]
- Ernst, S.; Kaim, W. Coordination Characteristics of Four Isomeric .Alpha.-Diimine Ligands. .Pi. Molecular Orbital Perturbation Calculations for the Bidiazines and Their Correlation with the Properties of Group 6 Metal Carbonyl Complexes. J. Am. Chem. Soc. 1986, 108, 3578–3586. [Google Scholar] [CrossRef]
- Kaim, W.; Kramer, H.E.A.; Vogler, C.; Rieker, J. Synthesis, Electrochemistry and Emission Spectroscopy in Fluid Solution of Four Isomeric (α-Diimine)Re(CO)3Hal Complexes. J. Organomet. Chem. 1989, 367, 107–115. [Google Scholar] [CrossRef]
- Knör, G.; Leirer, M.; Keyes, T.E.; Vos, J.G. Non-Luminescent 1,2-Diiminetricarbonylrhenium(I) Chloride Complexes–Synthesis, Electrochemical and Spectroscopic Properties of Re(DIAN)(CO)3Cl with DIAN =p-Substituted Bis(Arylimino)Acenaphthene. Eur. J. Inorg. Chem. 2000, 2000, 749–751. [Google Scholar] [CrossRef]
- Schnierle, M.; Winkler, M.; Filippou, V.; Slageren, J.; Ringenberg, M.R. (Spectro)Electrochemistry of 3-(Pyrid-2-yl)-s-Tetrazine- and 1,2- (Dihydro)Pyridazine Tricarbonylrhenium(I)Chloride. Euro. J. Inorg. Chem. 2022, 2022, e202100998. [Google Scholar] [CrossRef]
- Chakrabortty, S.; Agrawalla, B.K.; Stumper, A.; Vegi, N.M.; Fischer, S.; Reichardt, C.; Kögler, M.; Dietzek, B.; Feuring-Buske, M.; Buske, C.; et al. Mitochondria Targeted Protein-Ruthenium Photosensitizer for Efficient Photodynamic Applications. J. Am. Chem. Soc. 2017, 139, 2512–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftus, L.M.; Al-Afyouni, K.F.; Turro, C. New RuII Scaffold for Photoinduced Ligand Release with Red Light in the Photodynamic Therapy (PDT) Window. Chem. Eur. J. 2018, 24, 11550–11553. [Google Scholar] [CrossRef]
- Chen, Q.; Dan, H.; Tang, F.; Wang, J.; Li, X.; Cheng, J.; Zhao, H.; Zeng, X. Photodynamic Therapy Guidelines for the Management of Oral Leucoplakia. Int. J. Oral Sci. 2019, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Ioachim, E.; Medlycott, E.A.; Hanan, G.S. Synthesis and Properties of Re(I) Tricarbonyl Complexes of 6,6′-Disubstituted-4,4′-Bipyrimidines with High Energy Excited States Suitable for Incorporation into Polynuclear Arrays. Inorg. Chim. Acta 2006, 359, 2599–2607. [Google Scholar] [CrossRef]
- Mosberger, M.; Probst, B.; Spingler, B.; Alberto, R. Influence of Hetero-Biaryl Ligands on the Photo-Electrochemical Properties of [ReINCS(N∩N)(CO)3]-Type Photosensitizers. Eur. J. Inorg. Chem. 2019, 2019, 3518–3525. [Google Scholar] [CrossRef]
- Ernst, S.; Kaim, W. D6-Metal Complexes of 4,4?-Bipyrimidine, an Ambident Ligand with High?-Acceptor Ability. Angew. Chem. Int. Ed. Engl. 1985, 24, 430–431. [Google Scholar] [CrossRef]
- Ademi, L.; Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schaffner, S. Spontaneous Resolution of a Diastereomeric Ruthenium(II) Complex with an Atropisomeric 4,4′-Biquinazoline Ligand. Dalton Trans. 2003, 24, 4565–4567. [Google Scholar] [CrossRef]
- Ioachim, E.; Medlycott, E.A.; Polson, M.I.J.; Hanan, G.S. Synthesis of a Novel Series of 6,6′-Disubstituted 4,4′-Bipyrimidines by Radical Anion Coupling: New π-Accepting Ligands for Coordination Chemistry. Eur. J. Org. Chem. 2005, 2005, 3775–3780. [Google Scholar] [CrossRef]
- Ucar, S.; Dastan, A. One-Pot Homo- and Cross-Coupling of Diazanaphthalenes via C-H Substitution: Synthesis of Bis- and Tris-Diazanaphthalenes. J. Heterocycl. Chem. 2020, 57, 4013–4022. [Google Scholar] [CrossRef]
- Schmelz, O.; Mews, A.; Basché, T.; Herrmann, A.; Müllen, K. Supramolecular Complexes from CdSe Nanocrystals and Organic Fluorophors. Langmuir 2001, 17, 2861–2865. [Google Scholar] [CrossRef] [Green Version]
- Brietzke, T.; Mickler, W.; Kelling, A.; Holdt, H.-J. Mono- and Dinuclear Ruthenium(Ii) 1,6,7,12-Tetraazaperylene Complexes. Dalton Trans. 2012, 41, 2788–2797. [Google Scholar] [CrossRef]
- Hirono, A.; Sakai, H.; Kochi, S.; Sato, T.; Hasobe, T. Electrochemical Properties and Excited-State Dynamics of Azaperylene Derivatives. J. Phys. Chem. B 2020, 124, 9921–9930. [Google Scholar] [CrossRef]
- Grunwald, N.; Schilde, U.; Kelling, A.; Holdt, H.-J. CCDC 631826: Experimental Crystal Structure Determination Cambridge Crystallographic Data Centre: Cambridge, England, 2016.
- Fitchett, C.M.; Richardson, C.; Steel, P.J. Solid State Conformations of Symmetrical Aromatic Biheterocycles: An X-Ray Crystallographic Investigation. Org. Biomol. Chem. 2005, 3, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.; Vogler, C.; Kaim, W. The δ in 18 + δ Electron Complexes: Importance of the Metal/Ligand Interface for the Substitutional Reactivity of “Re(0)” Complexes (α-Diimine-)ReI(CO)3(X). Organometallics 1996, 15, 236–244. [Google Scholar] [CrossRef]
- Kaim, W.; Ernst, S. ESR and ENDOR Study of Three Isomeric Bidiazine Anion Radicals and of Their Group 6 Metal Carbonyl Complexes. Coordinative Effects on the Spin Distribution. J. Phys. Chem. 1986, 90, 5010–5014. [Google Scholar] [CrossRef]
- Ladwig, M.; Kaim, W. Spectroscopic and Electrochemical Properties of the Isomeric Bidiazine Complexes [(C5Me5)ClRh(Bdz)]+ and (C5Me5)Rh(Bdz) and Their Relevance to the Catalysis of the 2 H+ → H2 Reaction by 2,2′-Bipyridine Analogues. J. Organomet. Chem. 1991, 419, 233–243. [Google Scholar] [CrossRef]
- Zoric, M.R.; Askins, E.J.; Qiao, X.; Glusac, K.D. Strong Electronic Coupling of Graphene Nanoribbons onto Basal Plane of a Glassy Carbon Electrode. ACS Appl. Electron. Mater. 2021, 3, 854–860. [Google Scholar] [CrossRef]
- Xiong, T.; Włodarczyk, R.; Saalfrank, P. Vibrationally Resolved Absorption and Fluorescence Spectra of Perylene and N-Substituted Derivatives from Autocorrelation Function Approaches. Chem. Phys. 2018, 515, 728–736. [Google Scholar] [CrossRef]
- Jacobi, R.; Hernández-Castillo, D.; Sinambela, N.; Bösking, J.; Pannwitz, A.; González, L. Computation of Förster Resonance Energy Transfer in Lipid Bilayer Membranes. J. Phys. Chem. A 2022, 126, 8070–8081. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Luminescence and Redox Reactions of the Metal-to-Ligand Charge-Transfer Excited State of Tricarbonylchloro-(Polypyridyl)Rhenium(I) Complexes. J. Chem. Soc. Faraday Trans. 2 1986, 82, 2401–2415. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Saldías, M.; Guzmán, N.; Palominos, F.; Sandoval-Altamirano, C.; Günther, G.; Pizarro, N.; Vega, A. Electronic and Photophysical Properties of ReI(CO)3Br Complexes Modulated by Pyrazolyl–Pyridazine Ligands. ACS Omega 2019, 4, 4679–4690. [Google Scholar] [CrossRef] [Green Version]
- Manuta, D.M.; Lees, A.J. Solvent and Substituent Effects on the Lowest Energy Excited States of M(CO)4(Diimine) (M = Cr, Mo, W) Complexes. Inorg. Chem. 1983, 22, 3825–3828. [Google Scholar] [CrossRef]
- Hori, H.; Ishihara, J.; Koike, K.; Takeuchi, K.; Ibusuki, T.; Ishitani, O. Photocatalytic Reduction of Carbon Dioxide Using [Fac-Re(Bpy)(CO)3(4-Xpy)]+ (Xpy=pyridine Derivatives). J. Photochem. Photobiol. A Chem. 1999, 120, 119–124. [Google Scholar] [CrossRef]
- Rodríguez, L.; Ferrer, M.; Rossell, O.; Duarte, F.J.S.; Gil Santos, A.; Lima, J.C. Solvent Effects on the Absorption and Emission of [Re(R2Bpy)(CO)3X] Complexes and Their Sensitivity to CO2 in Solution. J. Photochem. Photobiol. A Chem. 2009, 204, 174–182. [Google Scholar] [CrossRef]
- Manuta, D.M.; Lees, A.J. Solvatochromism of the Metal to Ligand Charge-Transfer Transitions of Zero-Valent Tungsten Carbonyl Complexes. Inorg. Chem. 1986, 25, 3212–3218. [Google Scholar] [CrossRef]
- Krejčík, M.; Záliš, S.; Ladwig, M.; Matheis, W.; Kaim, W. A Study of the Reduced States of Four Isomeric Bidiazines (Diaza-2,2′-Bipyridines) by UV–VIS/NIR-Spectroelectrochemistry. J. Chem. Soc. Perkin Trans. 2 1992, 11, 2007–2010. [Google Scholar] [CrossRef]
- Ferguson, J. Absorption and Emission Spectra of the Perylene Dimer. J. Chem. Phys. 1966, 44, 2677–2683. [Google Scholar] [CrossRef]
- Portenkirchner, E.; Kianfar, E.; Sariciftci, N.S.; Knör, G. Two-Electron Carbon Dioxide Reduction Catalyzed by Rhenium(I) Bis(Imino)Acenaphthene Carbonyl Complexes. ChemSusChem 2014, 7, 1347–1351. [Google Scholar] [CrossRef] [Green Version]
- Wähler, K.; Ludewig, A.; Szabo, P.; Harms, K.; Meggers, E. Rhenium Complexes with Red-Light-Induced Anticancer Activity. Eur. J. Inorg. Chem. 2014, 2014, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A Found Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, F. Enoläther, II: Synthese und Reaktionen von 1.4-Bis-äthoxymethylen-butandion-(2.3). Chem. Ber. 1965, 98, 2260–2265. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Willette, R.E. 220. Quinazolines. Part VI. 2,2′-And 4,4′-Biquinazolinyls. J. Chem. Soc. 1965, 0, 1258–1262. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | /V vs. FC+/Fc (ΔE/mV) | /V vs. FC+/Fc | /V vs. FC+/Fc |
|---|---|---|---|
| bpm b | −1.863 (70) | −2.554 | - |
| bqz | −1.609 (72) | −2.043 | - |
| 1,3,10,12-tape | −1.131 (64) | −1.771 | - |
| [Re(CO)3(bpm)Cl] (1) c | −1.095 (70) | −1.695 | - |
| [Re(CO)3(bqz)Cl] (2) | −0.738 (66) | −1.283 | - |
| [Re(CO)3(1,3,10,12-tape)Cl] (3) | −0.523 (66) | −1.164 | −2.702 |
| Compound | CHCl3 | CH3CN | ||
|---|---|---|---|---|
| /nm (ελ/M−1 cm−1) | /nm (ελ/M−1 cm−1) | /nm (ελ/M−1 cm−1) | /nm (ελ/M−1 cm−1) | |
| bpm | - | 271 (13,995) | - | - |
| bqz | - | 320 (7660) | - | - |
| 1,3,10,12-tape | - | 414 (sh) 440 (11,525) 467 (10,400) | - | - |
| 1 | 467 (2910) | 283 (16,610) | 427 (2700) | 279 (15,325) |
| 2 | 532 (4160) | 378 (7155) | 491 (3320) | 378 (5730) |
| 3 a | 586 (5450) | 447 (sh) 480 (7900) 512 (9290) | ~550 b | 444 (sh) 476 506 |
| [Re(CO)3(2,2′-bpy)Cl] c | 384 | - | 370 (3420) | - |
| [Re(CO)3(1,10-phen)Cl] c | 374 | - | 364 (3350) | - |
| [Re(CO)3(3,3′-bipyridazine)Cl] d | 463 (3649) | 363 | - | - |
| [Re(dpb)(CO)3Cl] e | 464 (2380) | - | - | - |
| [Re(dmpb)(CO)3Cl] e | 460 (790) | - | - | - |
| [Re(dnb)(CO)3Cl] e | 431 (2790) | - | - | - |
| Compound | L | L●− | L2− |
|---|---|---|---|
| /nm | /nm | /nm | |
| bpm b | 272, 318 (br) | 350 (sh), 362, 445 (sh), 468 (sh), 496, 530, 603, 660, 840, 950, 1100 | 363 |
| 1,3,10,12-tape | 278, 319 (sh), 331, 439, 458 (sh) | 292, 311 (sh), 380, 406, 470 (sh), 502, 626, 670, 683 (sh), 740 (sh), 755 | 271, 296, 342 (sh), 407, 490 (sh), 513 |
| 1 | 278, 424 (br) | 328, 345, 435 (sh), 456, 486, 610, 680, 755 | 355 |
| 2 | 271, 328 (sh), 378, 388, 489 | 268, 293, 348 (sh), 392, 468, 526 (sh), 718, 768 (sh) | 271, 305 (sh), 365 (sh), 518 |
| 3 | 268, 388, 482 (sh), 509, 555 (sh), 620 (br) | 298, 336, 354, 400, 472, 530 (sh), 560 (sh), 633, 703, 722, (sh), 780 (sh), 803 | 294, 345, 407, 484, 506 (sh), 563, 612 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meitinger, N.; Mandal, S.; Sorsche, D.; Pannwitz, A.; Rau, S. Red Light Absorption of [ReI(CO)3(α-diimine)Cl] Complexes through Extension of the 4,4′-Bipyrimidine Ligand’s π-System. Molecules 2023, 28, 1905. https://doi.org/10.3390/molecules28041905
Meitinger N, Mandal S, Sorsche D, Pannwitz A, Rau S. Red Light Absorption of [ReI(CO)3(α-diimine)Cl] Complexes through Extension of the 4,4′-Bipyrimidine Ligand’s π-System. Molecules. 2023; 28(4):1905. https://doi.org/10.3390/molecules28041905
Chicago/Turabian StyleMeitinger, Nicolas, Subrata Mandal, Dieter Sorsche, Andrea Pannwitz, and Sven Rau. 2023. "Red Light Absorption of [ReI(CO)3(α-diimine)Cl] Complexes through Extension of the 4,4′-Bipyrimidine Ligand’s π-System" Molecules 28, no. 4: 1905. https://doi.org/10.3390/molecules28041905
APA StyleMeitinger, N., Mandal, S., Sorsche, D., Pannwitz, A., & Rau, S. (2023). Red Light Absorption of [ReI(CO)3(α-diimine)Cl] Complexes through Extension of the 4,4′-Bipyrimidine Ligand’s π-System. Molecules, 28(4), 1905. https://doi.org/10.3390/molecules28041905