
Synthesis, Empirical and Theoretical Investigations on New Histaminium Bis(Trioxonitrate) Compound

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
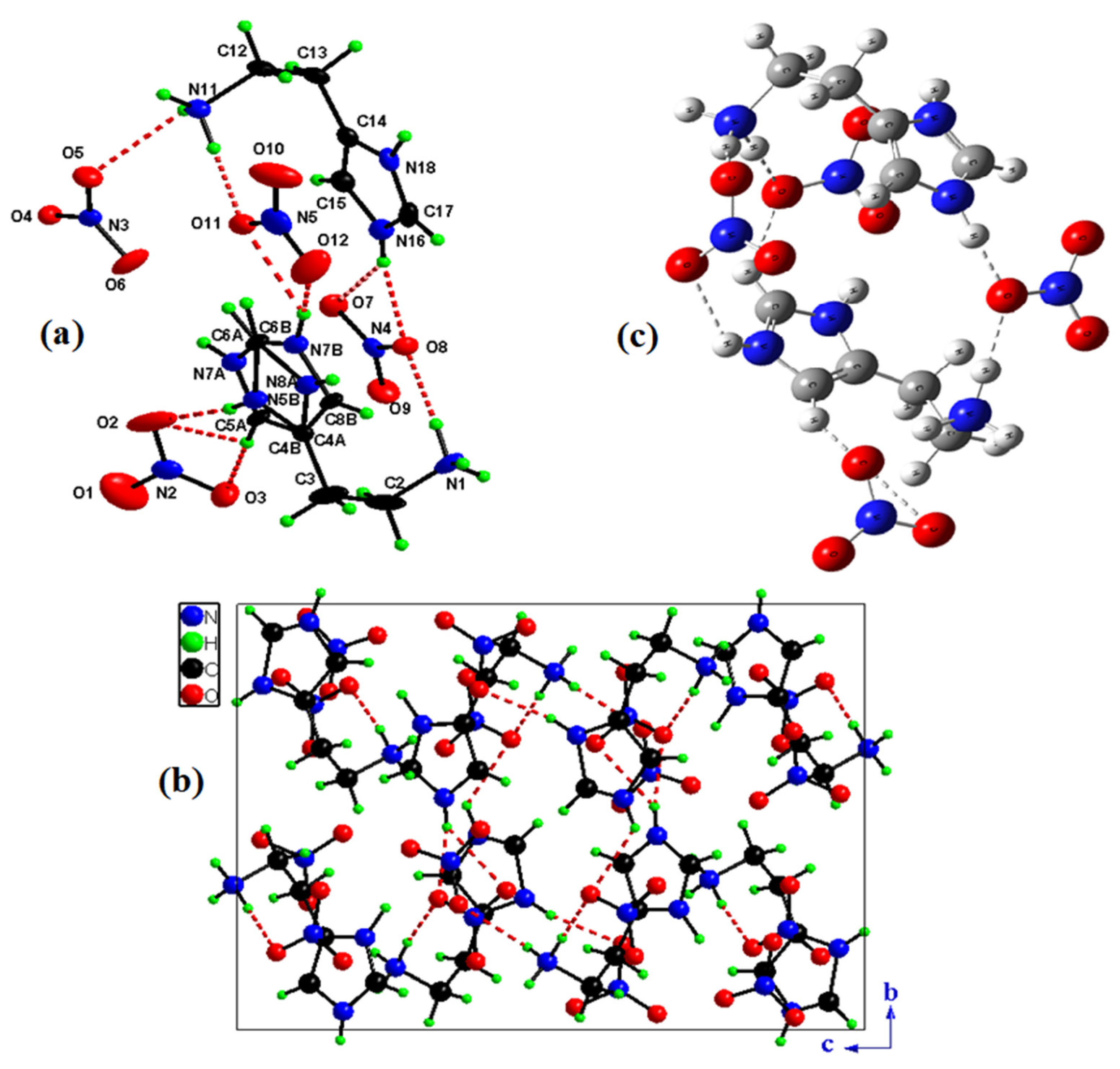
2.1. Structure Description
2.2. Hirshfeld Surface Analysis
2.3. Vibrational IR Spectral Analysis
2.3.1. Vibration Modes of Histaminium Cation
2.3.2. Vibration Modes of Nitrate Anion
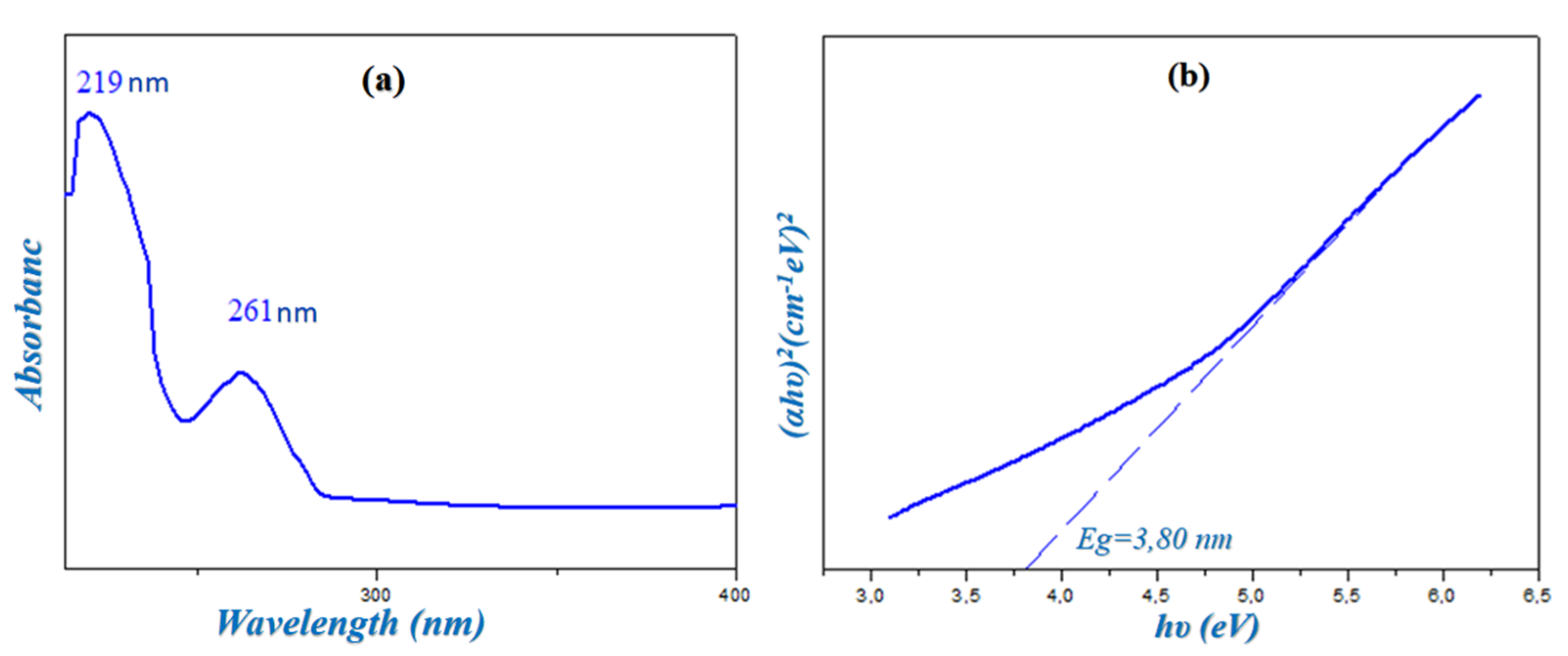
2.4. UV-Visible Spectroscopy
2.5. Spectrofluorimetry
2.6. HOMO-LUMO Analysis
2.7. AIM Topological Analysis
- − ∇2ρ(r) > 0 et H(r) > 0: weak H-bonds.
- − ∇2ρ(r) > 0 et H(r) < 0: moderate H-bonds.
- − ∇2ρ(r) < 0 et H(r) < 0: strong H-bonds.
2.8. Reduced Density Gradient (RDG)Analysis
- − λ2 < 0: H-bonding interactions.
- − λ2 close to zero: van der Waals interactions.
- − λ2 > 0: steric effect (repulsion; no interaction).
2.9. Analysis of the Molecular Electrostatic Potential Surface (MEPS)
2.10. Mulliken Population Analysis
3. Experimental Section
3.1. Chemical Preparation
3.2. Characterization Techniques
3.2.1. Single-Crystal X-ray Diffraction
3.2.2. Physical Measurements
3.2.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shipway, A.; Katz, E.; Willner, I. Nanoparticle Arrays on Surfaces for Electronic, Optical, and Sensor Applications. ChemPhysChem 2000, 1, 18–52. [Google Scholar] [CrossRef] [PubMed]
- Trindade, T.; Brien, P.; Pickett, N. Nanocrystalline Semiconductors: Synthesis, Properties, and Perspectives. Chem. Mater. 2001, 13, 3843–3858. [Google Scholar] [CrossRef]
- Daniel, M.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Boissiere, C.; Cassaignon, S.; Chaneac, C.; Durupthy, O.; Faustini, M.; Grosso, D.; Laberty-Robert, C.; Nicole, L.; Portehault, D.; et al. Molecular Engineering of Functional Inorganic and Hybrid Materials. Chem. Mater. 2014, 26, 221–238. [Google Scholar] [CrossRef]
- Pillai, S.K.; Kleyi, P.; Beer, M.; Mudaly, P. Layered double hydroxides: An advanced encapsulation and delivery system for cosmetic ingredients-an overview. Appl. Clay Sci. 2020, 199, 105868. [Google Scholar] [CrossRef]
- Bronshtein, A.; Aharonson, N.; Turniansky, A.; Altstein, M. Sol−Gel-Based Immunoaffinity Chromatography: Application to Nitroaromatic Compounds. Chem. Mater. 2000, 12, 2050–2058. [Google Scholar] [CrossRef]
- Sanchez, C.; Arribart, H.; Giraud-Guille, M.M. Biomimetism and bioinspiration as tools for the design of innovative materials and systems. Nat. Mater. 2005, 4, 277–288. [Google Scholar] [CrossRef]
- Shin, H.; Jo, S.; Mikos, A.G. Biomimetic materials for tissue engineering. Biomaterials 2003, 24, 4353–4364. [Google Scholar] [CrossRef]
- Popall, M.; Andrei, M.; Kappel, J.; Kron, J.; Olma, K.; Olsowski, B. ORMOCERs as inorganic–organic electrolytes for new solid state lithium batteries and supercapacitors. Electrochim. Acta 1998, 43, 1155–1161. [Google Scholar] [CrossRef]
- Faustini, M.; Nicole, L.; Ruiz-Hitzky, E.; Sanchez, C. History of Organic–Inorganic Hybrid Materials: Prehistory, Art, Science, and Advanced Applications. Adv. Funct. Mater. 2018, 28, 1704158. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Issaoui, N.; Brandán, S.A.; Roisnel, T.; Marouani, H. Synthesis and characterization of p-xylylenediaminium bis(nitrate). Effects of the coordination modes of nitrate groups on their structural and vibrational properties. J. Mol. Struct. 2018, 1151, 152–168. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Marouani, H.; Rzaigui, M. 4-Methylbenzylammonium nitrate. Acta Cryst. 2013, 69, o1453. [Google Scholar] [CrossRef] [PubMed]
- Gatfaoui, S.; Mezni, A.; Roisnel, T.; Marouani, H. Synthesis, characterization, Hirshfeld surface analysis and antioxidant activity of a novel organic–inorganic hybrid material 1-methylpiperazine-1,4-diium bis(nitrate). J. Mol. Struct. 2017, 1139, 52–59. [Google Scholar] [CrossRef]
- Guesmi, A.; Gatfaoui, S.; Roisnel, T.; Marouani, H. m-Xylylenediaminium sulfate: Crystal structure and Hirshfeld surface analysis. Acta Cryst. 2016, 72, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Sumner, S.; Roche, F.; Taylor, S. Factors Controlling Histamine Production in Swiss Cheese Inoculated with Lactobacillus buchneri. JDS 1990, 73, 3050–3058. [Google Scholar] [CrossRef] [PubMed]
- Veidis, M.V.; Palenik, G.J. Crystal Structure of Histamine Diphosphate Monohydrate. J. Chem. Soc. A Inorg. Phys. Theor. 1969, 91, 2659–2666. [Google Scholar] [CrossRef]
- Iarosh, O.O.; Kanevs’ka, S.A. The characteristics of the blood histamine indices and of the pathomorphological changes in the gastric mucosa of patients with multiple sclerosis. Likars’ka Sprav. 1992, 1, 75–76. [Google Scholar]
- Brown, I.D. On the geometry of O-H···O hydrogen bonds. Acta Cryst. 1976, 32, 24–31. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystal. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Tomilin, F.N.; Pozdnyakova, A.A.; Vasilyeva, N.Y.; Malyar, Y.N.; Kuznetsova, S.A.; Avramov, P.V. Theoretical DFT interpretation of infrared spectra of biologically active arabinogalactan sulphated derivatives. Chem. Pap. 2020, 74, 4103–4113. [Google Scholar] [CrossRef]
- Frost, R.L.; Erickson, K.L.; Kloprogge, T.J. Vibrational spectroscopic study of the nitrate containing hydrotalcitembobomkulite. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 61, 2919–2925. [Google Scholar] [CrossRef] [PubMed]
- Goebbert, D.; Garand, E.; Wende, T.; Bergmann, R.; Meijer, G.; Asmis, K.; Neumark, D. Infrared Spectroscopy of the Microhydrated Nitrate Ions NO3−(H2O)1–6. J. Phys. Chem. 2009, 113, 7584–7592. [Google Scholar] [CrossRef] [PubMed]
- Héléne, M.; Cance, H.; Potier, A.; Potier, J. Etude par spectroscopie de vibration de l’autoprotolyse de l’acide nitrique absolu. Can. J. Chem. 1985, 63, 1492–1501. [Google Scholar]
- Torreggiani, A.; Tamba, M.; Bonora, S.; Fini, G. Raman and IR study on copper binding of histamine. Biopolymers 2003, 72, 290–298. [Google Scholar] [CrossRef]
- Tauc, J. Optical properties and electronic structure of amorphous Ge and Si. Mater. Res. Bull. 1968, 3, 37–46. [Google Scholar] [CrossRef]
- Sidir, I.; Sidir, Y.G.; Kumalar, M.; Taşal, E. Ab initio Hartree–Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 7-acetoxy-6-(2,3-dibromopropyl)-4,8-dimethylcoumarin molecule. J. Mol. Struct. 2010, 964, 134–151. [Google Scholar] [CrossRef]
- Okulik, N.; Jubert, A.H. Theoretical Analysis of the Reactive Sites of Non–steroidal Anti–inflammatory Drugs. Int. Elect. J. Mol. Des. 2005, 4, 17–30. [Google Scholar]
- Jomaa, I.; Noureddine, O.; Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. Experimental, computational, and in silico analysis of (C8H14N2)2[CdCl6] compound. J. Mol. Struct. 2020, 1213, 128186. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Issaoui, N.; Mezni, A.; Bardak, F.; Roisnel, T.; Atak, A.; Marouani, H. Synthesis, structural and spectroscopic features, and investigation of bioactive nature of a novel organic-inorganic hybrid material 1H-1,2,4-triazole-4-ium trioxonitrate. J. Mol. Struct. 2017, 1150, 242–257. [Google Scholar] [CrossRef]
- Salihovic, M.; Huseinovic, S.; Spirtovic-Halilovic, S.; Osmanovic, A.; Dedic, A.; Asimovic, Z.; Zavrsnik, D. DFT Study and Biological Activity of Some Methylxanthines. Bull. Chem. Technol. Bos. Herzeg. 2014, 42, 31–36. [Google Scholar]
- Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium trioxonitrate. J. Mol. Struct. 2020, 1225, 129132. [Google Scholar] [CrossRef]
- Akman, F.; Issaoui, N.; Kazachenko, A. Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n=1, …, 5): X-ray, DFT, NBO, AIM, and RDG analyses. J. Mol. Model. 2020, 26, 161. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Sagaama, A.; Issaoui, N.; Al-Dossary, O.; Kazachenko, A.; Wojcik, M. Non covalent interactions and molecular docking studie on morphine compound. J. King Saud Univ. Sci. 2021, 33, 101606. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. A proton transfer compound template phenylethylamine: Synthesis, a collective experimental and theoretical investigations. J. Mol. Struct. 2019, 1191, 183–196. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. Adv. Quant. Chem. 1978, 11, 115–193. [Google Scholar]
- Fliszar, S. Charge Distributions and Chemical Effects; Springer: Berlin/Heidelberg, Germany; New York, NY, USA; Tokyo, Japan, 1983. [Google Scholar]
- Thul, P.; Gupta, V.P.; Ram, V.J.; Tandon, P. Structural and spectroscopic studies on 2-pyranones. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2010, 75, 251–260. [Google Scholar] [CrossRef]
- Mahendra, K.; Fernandes, J.; Udayashankar, N.K. A novel approach to the synthesis of semiorganic ammonium hydrogen oxalate oxalic acid dihydrate single crystal and its characterization. J. Therm. Anal. Calorim. 2021, 146, 93–102. [Google Scholar] [CrossRef]
- Maksic, Z.B. Theoretical Treatment of Large Molecules and Their Interactions: Part 4 Theoretical Models of Chemical Bonding; Springer: Berlin/Heidelberg, Germany, 1991; Volume 139. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView, version 5; Semichem. Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Chiodo, S.; Russo, N.; Sicilia, E. LANL2DZ basis sets recontracted in the framework of density functional theory. J. Chem. Phys. 2006, 125, 104107. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, O.; Gatfaoui, S.; Brandan, S.A.; Marouani, H.; Issaoui, N. Structural, docking and spectroscopic studies of a new piperazine derivative, 1-Phenylpiperazine-1,4-diium bis(hydrogen sulfate). J. Mol. Struct. 2020, 1202, 127351. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2020, 132, 154104. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC Number | 2,236,747 |
|---|---|
| Temperature | 150 K |
| Empirical formula | (C5H11N3)(NO3)2 |
| Formula weight (g·mol−1) | 474.37 |
| Crystal size (mm) | 0.58 × 0.52 × 0.50 |
| Crystal system | monoclinic |
| Space group | P21/c |
| a (Å) | 10.4807 (16) |
| b (Å) | 11,8747 (15) |
| c (Å) | 16,194 (2) |
| β (°) | 95,095 (6) |
| Z | 8 |
| V (Å3) | 2007.4 (5) |
| F (000) | 992 |
| Mo Kα (mm−1) | 0.14 |
| Reflections collected | 15,384 |
| Independent reflections | 4589 |
| Reflections with I > 2σ(I) | 3901 |
| Rint | 0.049 |
| Absorption correction: | multi-scan Tmin = 0.838, Tmax = 0.931 |
| Refined parameters | 310 |
| R[F2 ˃ 2σ(F2)] | 0.073 |
| wR(F2) | 0.186 |
| Goodness-of-fit on F2 | 1.097 |
| Parameters | X-ray | Calculated |
|---|---|---|
| Bond length (Å) | ||
| Organic | ||
| N1–C2 | 1.479(5) | 1.5144 |
| C2–C3 | 1.522(4) | 1.5402 |
| C3–C4B | 1.469(4) | 1.5068 |
| C4B–N5B | 1.350(6) | 1.4099 |
| C4B–C8B | 1.377(7) | 1.3862 |
| N5B–C6B | 1.500(6) | 1.3496 |
| C6B–N7B | 1.102(6) | 1.3452 |
| N7B–C8B | 1.438(7) | 1.3982 |
| C3–C4A | 1.469(4) | 1.5068 |
| C4A–N8A | 1.356(6) | 1.4099 |
| C4A–C5A | 1.403(7) | 1.3862 |
| C5A–N7A | 1.346(7) | 1.3982 |
| N7A–C6A | 1.136(6) | 1.3452 |
| C6A–N8A | 1.520(6) | 1.3496 |
| C12–N11 | 1.492(3) | 1.5181 |
| C12–C13 | 1.522(4) | 1.5497 |
| C13–C14 | 1.489(4) | 1.5042 |
| C14–C15 | 1.359(3) | 1.5042 |
| C14–N18 | 1.382(3) | 1.3846 |
| C15–N16 | 1.381(4) | 1.3975 |
| N16–C17 | 1.326(4) | 1.3447 |
| C17–N18 | 1.317(4) | 1.3540 |
| Inorganic | ||
| N2–O1 | 1.218 (4) | 1.2716 |
| N2–O3 | 1.232 (3) | 1.3078 |
| N2–O2 | 1.238 (3) | 1.3576 |
| N3–O6 | 1.241 (3) | 1.2880 |
| N3–O5 | 1.246 (3) | 1.3065 |
| N3–O4 | 1.267 (3) | 1.3327 |
| N4–O9 | 1.240 (3) | 1.2847 |
| N4–O7 | 1.247 (3) | 1.2883 |
| N4–O8 | 1.269 (3) | 1.3598 |
| N5–O10 | 1.227 (4) | 1.2961 |
| N5–O12 | 1.254 (4) | 1.2992 |
| N5–O11 | 1.272 (3) | 1.3299 |
| Bond angle (°) | ||
| Organic | ||
| N1–C2–C3 | 112.7(3) | 113.5539 |
| C4B–C3–C2 | 113.16(2) | 116.4708 |
| N5B–C4B–C8B | 110.7(4) | 105.9464 |
| N5B–C4B–C3 | 132.7(4) | 121.0151 |
| C8B–C4B–C3 | 116.5(4) | 133.0338 |
| C4B–N5B–C6B | 101.1(4) | 109.6545 |
| N7B–C6B–N5B | 112.1(4) | 107.7707 |
| C6B–N7B–C8B | 113.6(5) | 109.8616 |
| C4B–C8B–N7B | 102.5(5) | 106.7661 |
| C4A–C3–C2 | 113.6(2) | 116.4708 |
| N8A–C4A–C5A | 107.9(4) | 105.9464 |
| N8A–C4A–C3 | 142.3(4) | 121.0151 |
| C5A–C4A–C3 | 109.7(3) | 133.0338 |
| N7A–C5A–C4A | 107.7(5) | 106.7661 |
| C6A–N7A–C5A | 111.3(5) | 109.8616 |
| N7A–C6A–N8A | 113.3(4) | 107.7707 |
| C4A–N8A–C6A | 99.8(4) | 109.6545 |
| Inorganic | ||
| O1–N2–O3 | 121.1 (3) | 123.5990 |
| O1–N2–O2 | 119.4 (3) | 116.8351 |
| O3–N2–O2 | 119.5 (3) | 119.5647 |
| O6–N3–O5 | 121.9 (2) | 121.8126 |
| O6–N3–O4 | 118.4 (2) | 118.7139 |
| O5–N3–O4 | 119.7 (2) | 119.4460 |
| O9–N4–O7 | 121.0 (2) | 123.9298 |
| O9–N4–O8 | 120.0 (2) | 118.2186 |
| O7–N4–O8 | 119.0 (2) | 117.8391 |
| O10–N5–O12 | 120.8 (3) | 122.0878 |
| O10–N5–O11 | 120.4 (3) | 119.2345 |
| O12–N5–O11 | 118.8 (3) | 118.6568 |
| D–H…A | D–H (Å) | H…A (Å) | D…A (Å) | D–H…A (°) |
|---|---|---|---|---|
| N1–H1A···O4 i | 0.86(4) | 2.20(4) | 2.951(3) | 146(3) |
| N1–H1A···O6 i | 0.86(4) | 2.38(4) | 3.101(4) | 141(3) |
| N1–H1B···O12 ii | 0.92(4) | 2.19(4) | 2.978(4) | 144(3) |
| N1–H1B···O2 i | 0.92(4) | 2.25(4) | 2.885(4) | 126(3) |
| N1–H1C···O8 | 0.94(4) | 1.95(4) | 2.881(3) | 170(3) |
| N5B–H5B···O2 | 0.88 | 2.13 | 3.001(7) | 169 |
| N7B–H7B···O12 | 0.88 | 1.79 | 2.646(6) | 164.2 |
| N7B–H7B···O11 | 0.88 | 2.30 | 2.897(5) | 124.9 |
| N11–H11A···O11 | 0.97(3) | 1.87(3) | 2.835(3) | 178(3) |
| N11–H11B···O5 | 0.87(3) | 2.34(3) | 2.809(3) | 114(3) |
| N11–H11B···O7 iii | 0.87(3) | 2.39(4) | 3.212(3) | 156(3) |
| N11–H11B···O9 iii | 0.87(3) | 2.53(3) | 3.255(3) | 142(3) |
| N11–H11C···O3 iv | 0.87(4) | 2.12(4) | 2.902(3) | 149(3) |
| N16–H16···O8 | 0.82(4) | 2.04(4) | 2.843(3) | 168(3) |
| N16–H16···O7 | 0.82(4) | 2.51(4) | 3.157(3) | 136(3) |
| N18–H18···O4 v | 1.04(4) | 1.80(4) | 2.821(3) | 167(3) |
| N18–H18···O5 v | 1.04(4) | 2.23(4) | 2.993(3) | 129(3) |
| C2–H2A···O10 vi | 0.99 | 2.47 | 3.214(4) | 131.8 |
| C3–H3B···O12 ii | 0.99 | 2.53 | 3.298(5) | 134.4 |
| C5A–H5A···O2 | 0.95 | 2.14 | 2.716(8) | 117.9 |
| C5A–H5A···O3 | 0.95 | 1.90 | 2.849(7) | 177.9 |
| C12–H12B···O9 iv | 0.99 | 2.55 | 3.276(3) | 130.2 |
| ρ(r) | V(r) | G(r) | H(r) | (Eint) kJ·mol−1 | ||
|---|---|---|---|---|---|---|
| C8–H10…O49 | 0.6060935637 | 0.2429405290 | −0.3508914227 | 0.4791213726 | 0.1282299500 | 0.4552689789 |
| C8–H10…O50 | 0.5767933635 | 0.2356048186 | −0.3274099481 | 0.4582109973 | 0.1308010491 | 0.4816467982 |
| C12–H13…O41 | 0.2484511638 | 0.1052256607 | −0.2249624021 | 0.2440132769 | 0.1905087482 | 0.9567061030 |
| C16–H17…O53 | 0.1559634154 | 0.7150801709 | −0.1170034581 | 0.1478867504 | 0.3088329231 | 0.6862805495 |
| C27–H28…O45 | 0.1130722919 | 0.5092741191 | −0.7359125289 | 0.1004548913 | 0.2686363845 | 0.4567065471 |
| C31–H32…O44 | 0.1108296139 | 0.4708767916 | −0.7156373759 | 0.9464146774 | 0.2307773015 | 0.4925806261 |
| C35–H36…O48 | 0.1354238925 | 0.6309719703 | −0.9453516802 | 0.1261390803 | 0.3160391227 | 0.5748029379 |
| C24–H25…O52 | 0.5940881497 | 0.2260591172 | −0.2941928340 | 0.4296703135 | 0.1354774795 | 0.5076018638 |
| N1–H2…O41 | 0.6472117739 | 0.1796462357 | −0.6363728811 | 0.5427442351 | −0.9362864596 | 0.3174672431 |
| N14–H15…O46 | 0.1742905363 | 0.7616909236 | −0.1319345448 | 0.1611786379 | 0.2924409302 | 0.1015512073 |
| N20–H22…O45 | 0.6370579352 | 0.1861499875 | −0.6422615363 | 0.5538182526 | −0.8844328377 | 0.3685782364 |
| N20–H21…O35 | 0.5414942347 | 0.1750245594 | −0.5291885920 | 0.4833749953 | −0.4581359667 | 0.3247555529 |
| N18–H19…O54 | 0.2685397276 | 0.1003081517 | −0.2228771104 | 0.2368237449 | 0.1394663448 | 0.1784953431 |
| N37–H38…O52 | 0.1437474207 | 0.6870277662 | −01021190421 | 0.1369379918 | 0.3481894974 | 0.1017548931 |
| N1–H4…O49 | 0.3973762041 | 0.1368061188 | −0.3618440937 | 0.3519296954 | −0.9914398322 | 0.2094545303 |
| N33–H34…O49 | 0.5365923450 | 0.1691408063 | −0.5113382011 | 0.4670951085 | −0.4424309259 | 0.3126712014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jmai, M.; Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Kazachenko, A.S.; Al-Dossary, O.; Marouani, H.; Kazachenko, A.S. Synthesis, Empirical and Theoretical Investigations on New Histaminium Bis(Trioxonitrate) Compound. Molecules 2023, 28, 1931. https://doi.org/10.3390/molecules28041931
Jmai M, Gatfaoui S, Issaoui N, Roisnel T, Kazachenko AS, Al-Dossary O, Marouani H, Kazachenko AS. Synthesis, Empirical and Theoretical Investigations on New Histaminium Bis(Trioxonitrate) Compound. Molecules. 2023; 28(4):1931. https://doi.org/10.3390/molecules28041931
Chicago/Turabian StyleJmai, Mahdi, Sofian Gatfaoui, Noureddine Issaoui, Thierry Roisnel, Aleksandr S. Kazachenko, Omar Al-Dossary, Houda Marouani, and Anna S. Kazachenko. 2023. "Synthesis, Empirical and Theoretical Investigations on New Histaminium Bis(Trioxonitrate) Compound" Molecules 28, no. 4: 1931. https://doi.org/10.3390/molecules28041931
APA StyleJmai, M., Gatfaoui, S., Issaoui, N., Roisnel, T., Kazachenko, A. S., Al-Dossary, O., Marouani, H., & Kazachenko, A. S. (2023). Synthesis, Empirical and Theoretical Investigations on New Histaminium Bis(Trioxonitrate) Compound. Molecules, 28(4), 1931. https://doi.org/10.3390/molecules28041931