
Bio-Inspired Iron Pentadentate Complexes as Dioxygen Activators in the Oxidation of Cyclohexene and Limonene



Abstract
:1. Introduction
2. Results and Discussion
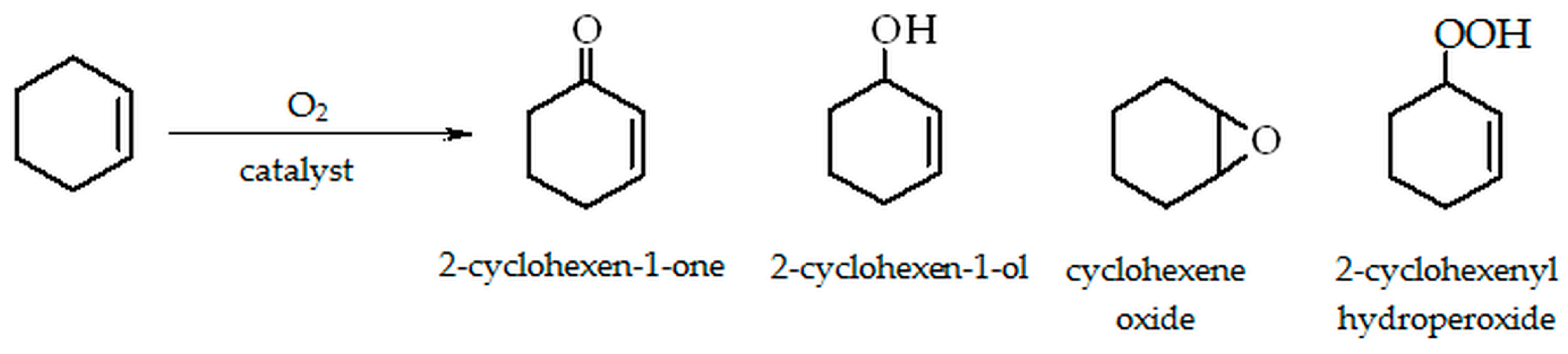
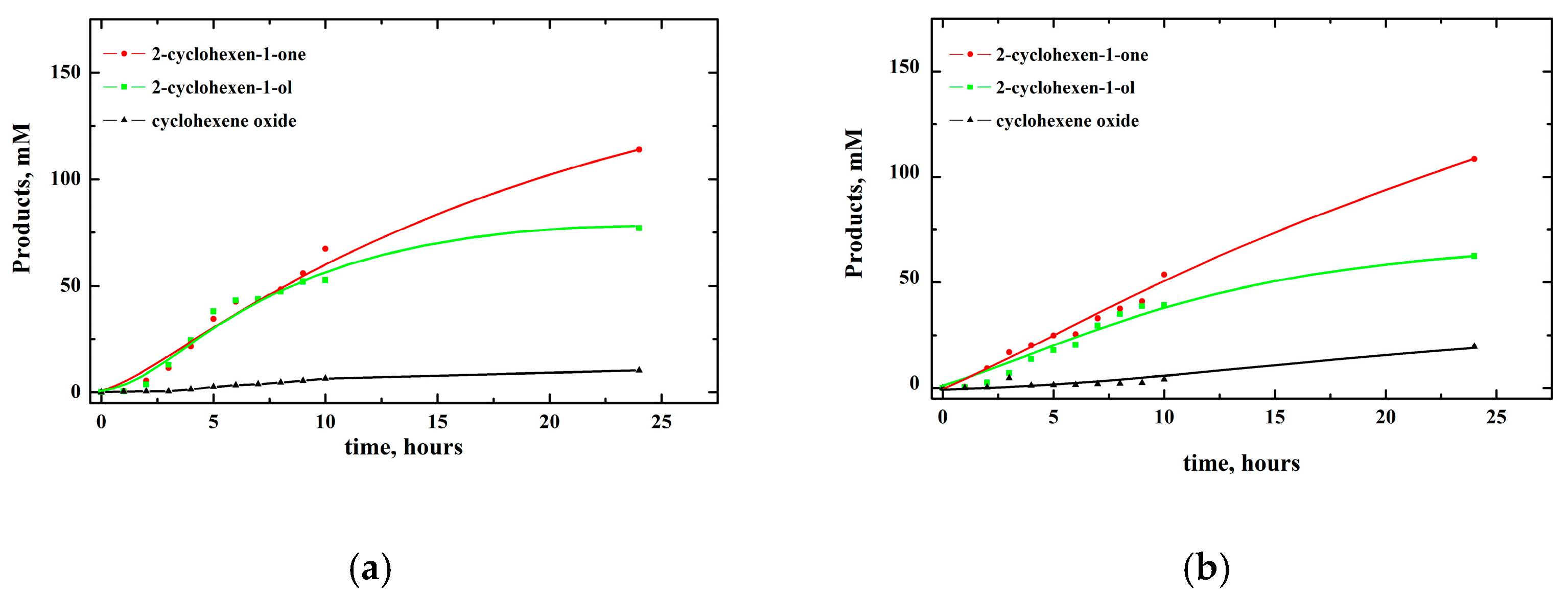
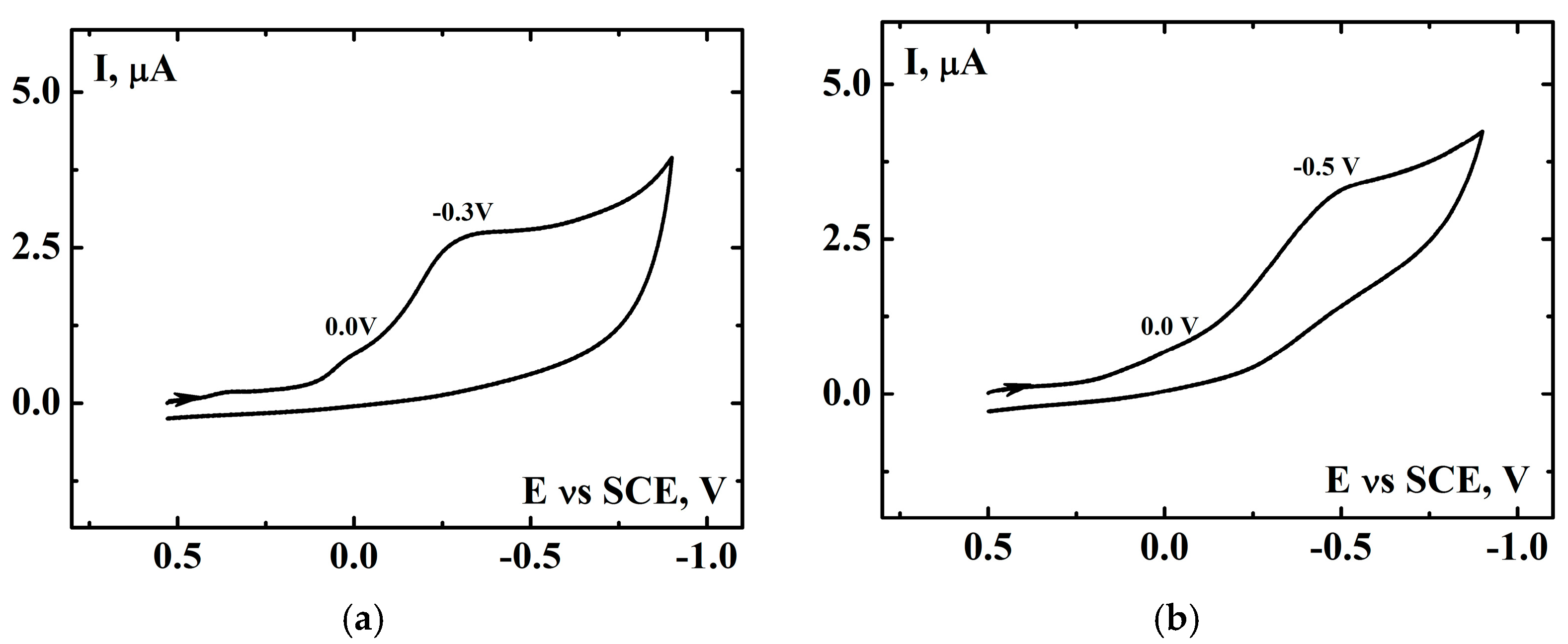
2.1. Oxidation of Cyclohexene
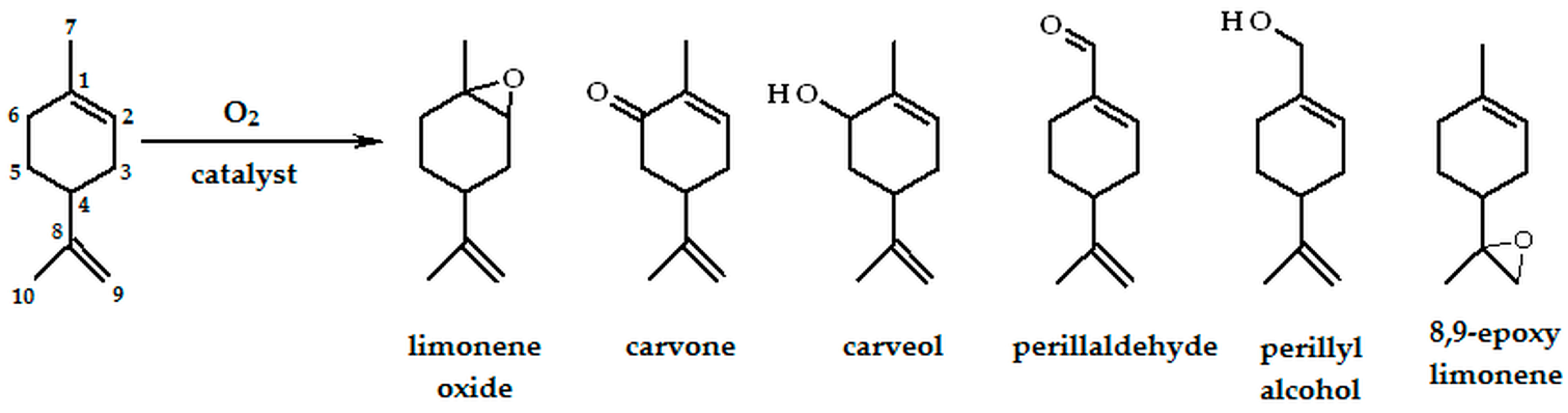
2.2. Oxidation of Limonene
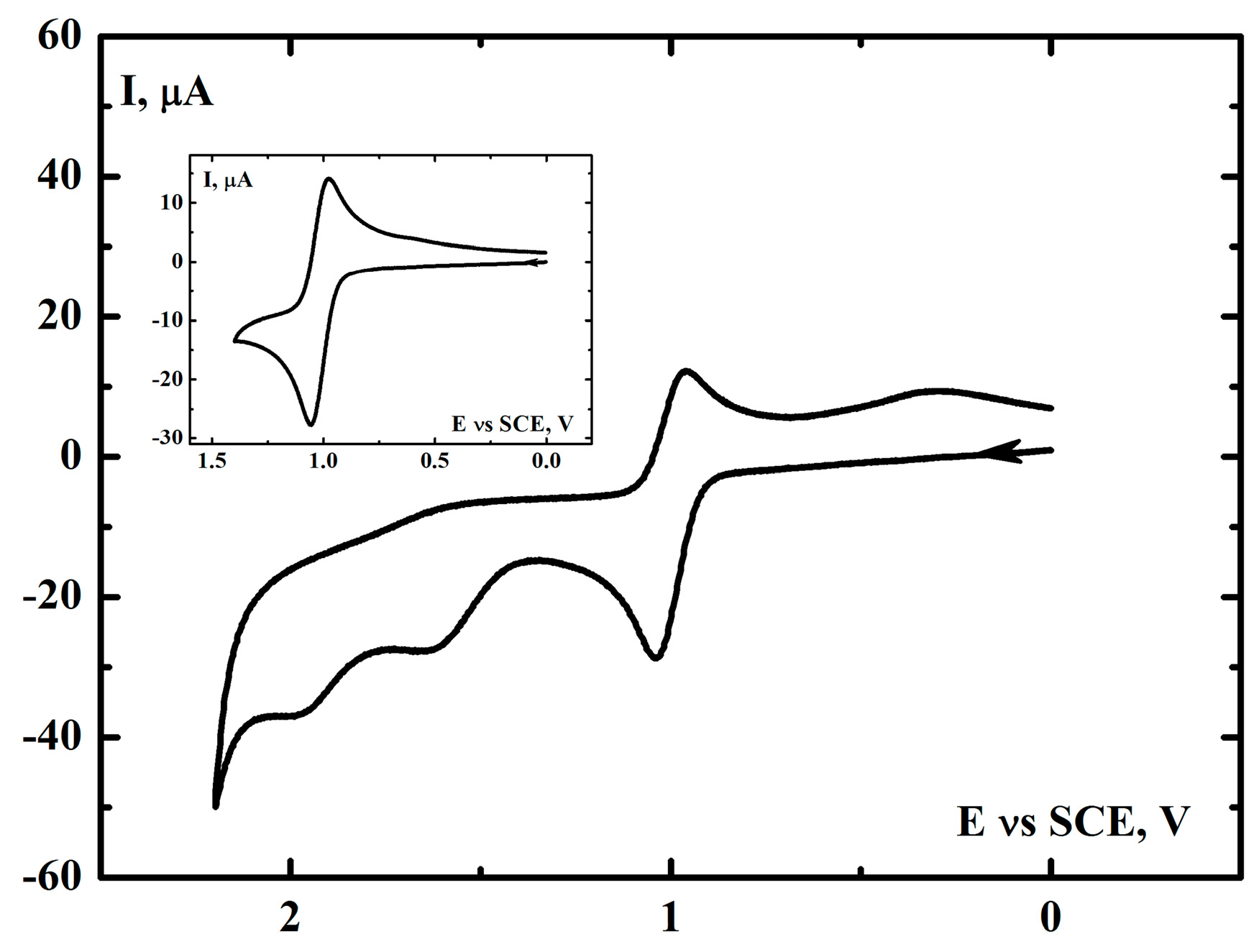
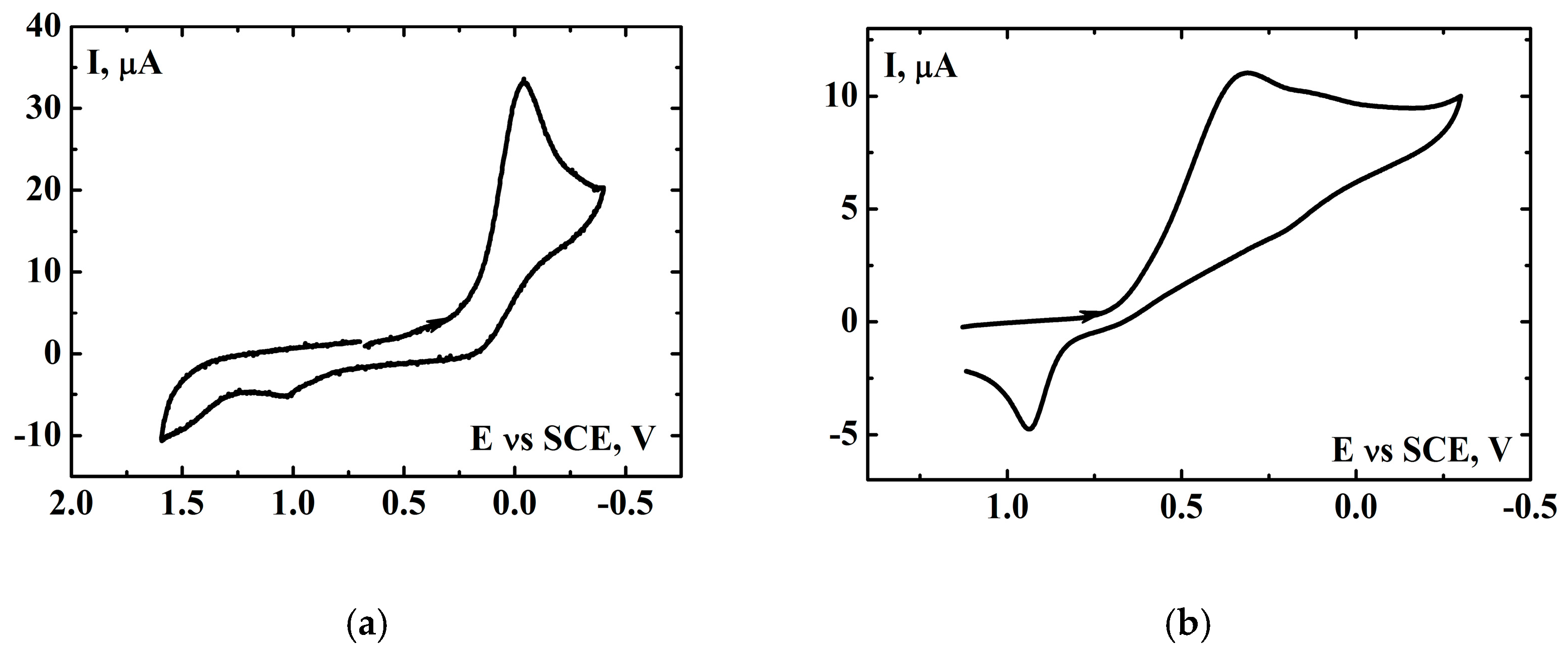
2.3. Electrochemical Investigation of the [(N4Py)FeII]2+ Complex
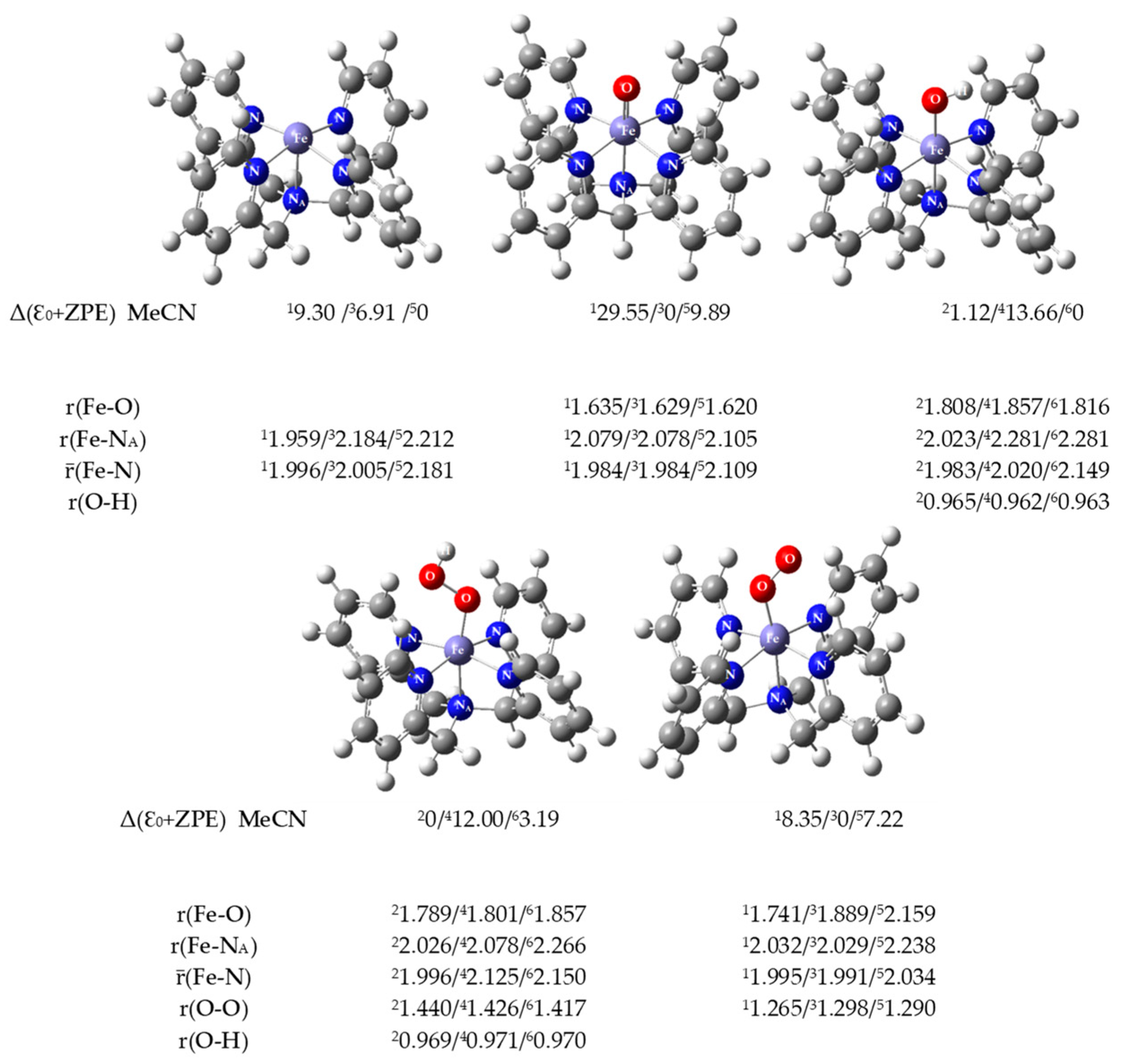
2.4. DFT Calculations
[(N4Py)FeIII-OH]2+ + C6H9

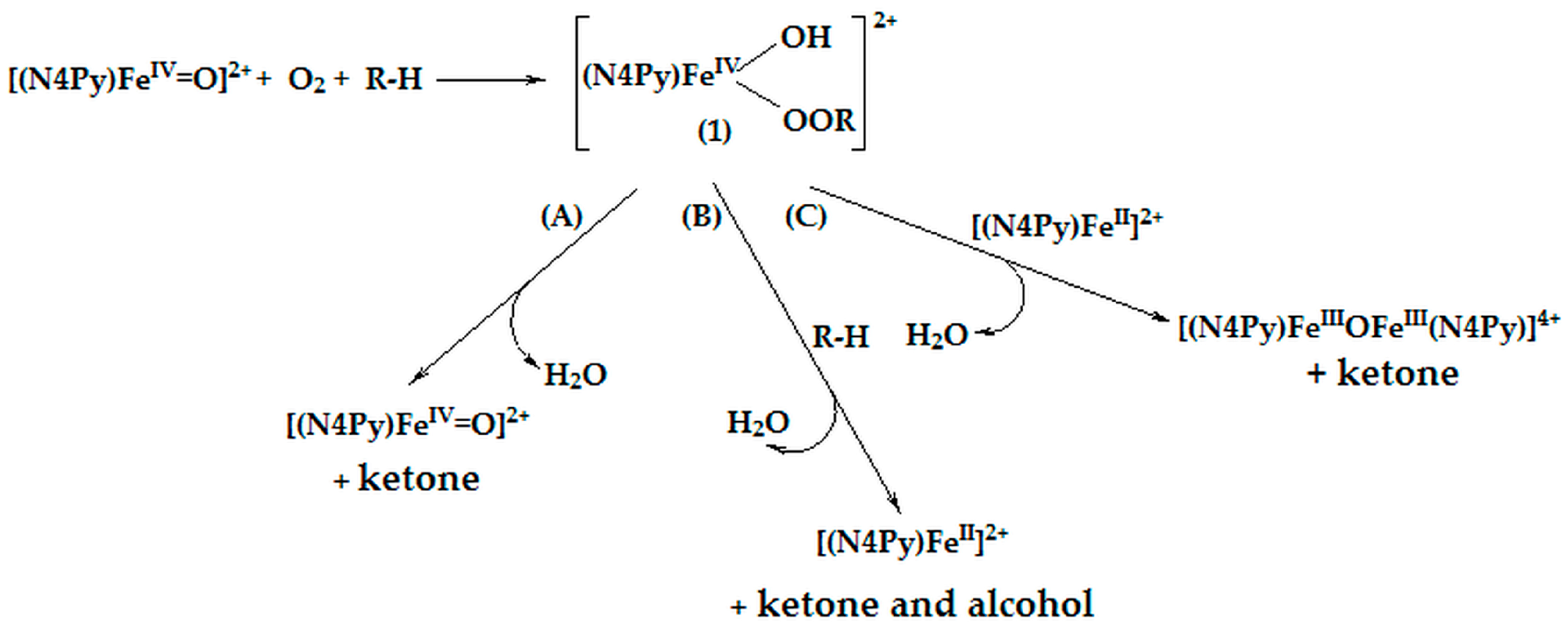
2.5. Considerations on Oxidation Mechanism
3. Materials and Methods
3.1. Equipment
3.2. Chemicals and Regents
3.3. Synthesis of [(N4Py)FeII]2+
3.4. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Simándi, L.I. Catalytic Activation of Dioxygen by Metal Complexes; Springer: Dordrecht, The Netherlands, 1992; Volume 13, pp. 1–397. [Google Scholar]
- Sawyer, D.T. Oxygen Chemistry; Oxford University Press: Oxford, UK, 1991. [Google Scholar]
- De Montellano, P.R.O. (Ed.) Cytochrome P450, 3rd ed.; Springer: New York, NY, USA, 2005; p. 690. [Google Scholar]
- Ferraro, D.J.; Gakhar, L.; Ramaswamy, S. Rieske Business: Structure–Function of Rieske Non-heme Oxygenases. Biochem. Biophys. Res. Commun. 2005, 338, 175–190. [Google Scholar] [CrossRef]
- Pati, S.G.; Bopp, C.E.; Kohler, H.-P.E.; Hofstetter, T.B. Substrate-Specific Coupling of O2 Activation to Hydroxylations of Aromatic Compounds by Rieske Non-heme Iron Dioxygenases. ACS Catal. 2022, 12, 6444–6456. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.M. Nature of Activated Bleomycin. In Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations; Meunier, B., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2000; pp. 287–303. [Google Scholar]
- Burger, R.M.; Grigoryants, V.M.; Scholes, C.P. Exchangeable Proton ENDOR as a Probe of the Redox-Active Iron Center in Activated Bleomycin and Ferric Bleomycin. Dalton Trans. 2017, 46, 13263–13272. [Google Scholar] [CrossRef]
- Wallar, B.J.; Lipscomb, J.D. Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem. Rev. 1996, 96, 2625–2658. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef] [PubMed]
- Jasniewski, A.J.; Que, L., Jr. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. [Google Scholar] [CrossRef]
- Banerjee, R.; Lipscomb, J.D. Small-Molecule Tunnels in Metalloenzymes Viewed as Extensions of the Active Site. Acc. Chem. Res. 2021, 54, 2185–2195. [Google Scholar] [CrossRef]
- de Visser, S.P.; Mukherjee, G.; Ali, H.S.; Sastri, C.V. Local Charge Distributions, Electric Dipole Moments, and Local Electric Fields Influence Reactivity Patterns and Guide Regioselectivities in α-Ketoglutarate-Dependent Non-heme Iron Dioxygenases. Acc. Chem. Res. 2022, 55, 65–74. [Google Scholar] [CrossRef]
- Que, L.; Tolman, W.B. Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef]
- Company, A.; Gómez, L.; Costas, M. Chapter 6 Bioinspired Non-heme Iron Catalysts in C–H and C=C Oxidation Reactions. In Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; The Royal Society of Chemistry: London, UK, 2011; pp. 148–208. [Google Scholar]
- Oloo, W.N.; Que, L., Jr. Bioinspired Nonheme Iron Catalysts for C-H and C=C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. [Google Scholar] [CrossRef]
- Kal, S.; Xu, S.; Que, L., Jr. Bio-inspired Nonheme Iron Oxidation Catalysis: Involvement of Oxoiron(V) Oxidants in Cleaving Strong C−H Bonds. Angew. Chem. Int. Ed. 2020, 59, 7332–7349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-P.; Chandra, A.; Lee, Y.-M.; Cao, R.; Ray, K.; Nam, W. Transition Metal-Mediated O–O Bond Formation and Activation in Chemistry and Biology. Chem. Soc. Rev. 2021, 50, 4804–4811. [Google Scholar] [CrossRef] [PubMed]
- Walling, C. Fenton’s Reagent Revisited. Acc. Chem. Res. 1975, 8, 125–131. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Sobkowiak, A.; Matsushita, T. Metal [MLx; M = Fe, Cu, Co, Mn]/Hydroperoxide-Induced Activation of Dioxygen for the Oxygenation of Hydrocarbons: Oxygenated Fenton Chemistry. Acc. Chem. Res. 1996, 29, 409–416. [Google Scholar] [CrossRef]
- Barton, D.H.R. Gif Chemistry: The Present Situation. Tetrahedron 1998, 54, 5805–5817. [Google Scholar] [CrossRef]
- Walling, C. Intermediates in the Reactions of Fenton Type Reagents. Acc. Chem. Res. 1998, 31, 155–157. [Google Scholar] [CrossRef]
- MacFaul, P.A.; Wayner, D.D.M.; Ingold, K.U. A Radical Account of “Oxygenated Fenton Chemistry”. Acc. Chem. Res. 1998, 31, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Meyerstein, D. Comments on the Mechanism of the “Fenton-Like” Reaction. Acc. Chem. Res. 1999, 32, 547–550. [Google Scholar] [CrossRef]
- Stavropoulos, P.; Celenligil-Cetin, R.; Tapper, A.E. The Gif Paradox. Acc. Chem. Res. 2001, 34, 745–752. [Google Scholar] [CrossRef]
- Gozzo, F. Radical and Non-radical Chemistry of the Fenton-like Systems in the Presence of Organic Substrates. J. Mol. Catal. A Chem. 2001, 171, 1–22. [Google Scholar] [CrossRef]
- Masarwa, A.; Rachmilovich-Calis, S.; Meyerstein, N.; Meyerstein, D. Oxidation of Organic Substrates in Aerated Aqueous Solutions by the Fenton Reagent. Coord. Chem. Rev. 2005, 249, 1937–1943. [Google Scholar] [CrossRef]
- Que, L., Jr. The Road to Non-Heme Oxoferryls and Beyond. Acc. Chem. Res. 2007, 40, 493–500. [Google Scholar] [CrossRef]
- Cho, J.; Jeon, S.; Wilson, S.A.; Liu, L.V.; Kang, E.A.; Braymer, J.J.; Lim, M.H.; Hedman, B.; Hodgson, K.O.; Valentine, J.S.; et al. Structure and Reactivity of a Mononuclear Non-haem Iron(III)–peroxo Complex. Nature 2011, 478, 502–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, A.R.; Que, L. High-valent Nonheme Iron-oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar] [CrossRef]
- Hong, S.; Lee, Y.-M.; Ray, K.; Nam, W. Dioxygen Activation Chemistry by Synthetic Mononuclear Nonheme Iron, Copper and Chromium Complexes. Coord. Chem. Rev. 2017, 334, 25–42. [Google Scholar] [CrossRef]
- Ehudin, M.A.; Gee, L.B.; Sabuncu, S.; Braun, A.; Moënne-Loccoz, P.; Hedman, B.; Hodgson, K.O.; Solomon, E.I.; Karlin, K.D. Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands. J. Am. Chem. Soc. 2019, 141, 5942–5960. [Google Scholar] [CrossRef]
- Roelfes, G.; Lubben, M.; Hage, R.; Que, J.L.; Feringa, B.L. Catalytic Oxidation with a Non-Heme Iron Complex that Generates a Low-Spin FeIIIOOH Intermediate. Chem. Eur. J. 2000, 6, 2152–2159. [Google Scholar] [CrossRef]
- Shan, X.; Que, L., Jr. High-valent Nonheme Iron-oxo Species in Biomimetic Oxidations. J. Inorg. Biochem. 2006, 100, 421–433. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Hong, S.; Morimoto, Y.; Shin, W.; Fukuzumi, S.; Nam, W. Dioxygen Activation by a Non-Heme Iron(II) Complex: Formation of an Iron(IV)−Oxo Complex via C−H Activation by a Putative Iron(III)−Superoxo Species. J. Am. Chem. Soc. 2010, 132, 10668–10670. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Selective Oxygenation of Cyclohexene by Dioxygen via an Iron(V)-Oxo Complex-Autocatalyzed Reaction. Inorg. Chem. 2017, 56, 5096–5104. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Amphoteric Reactivity of Metal–oxygen Complexes in Oxidation Reactions. Coord. Chem. Rev. 2018, 365, 41–59. [Google Scholar] [CrossRef]
- Singh, R.; Ganguly, G.; Malinkin, S.O.; Demeshko, S.; Meyer, F.; Nordlander, E.; Paine, T.K. A Mononuclear Nonheme Iron(IV)-Oxo Complex of a Substituted N4Py Ligand: Effect of Ligand Field on Oxygen Atom Transfer and C–H Bond Cleavage Reactivity. Inorg. Chem. 2019, 58, 1862–1876. [Google Scholar] [CrossRef] [PubMed]
- Masferrer-Rius, E.; Borrell, M.; Lutz, M.; Costas, M.; Klein Gebbink, R.J.M. Aromatic C−H Hydroxylation Reactions with Hydrogen Peroxide Catalyzed by Bulky Manganese Complexes. Adv. Synth. Catal. 2021, 363, 3783–3795. [Google Scholar] [CrossRef]
- Das, P.; Que, L., Jr. Iron Catalyzed Competitive Olefin Oxidation and ipso-Hydroxylation of Benzoic Acids: Further Evidence for an FeV=O Oxidant. Inorg. Chem. 2010, 49, 9479–9485. [Google Scholar] [CrossRef] [PubMed]
- Ezhov, R.; Ravari, A.K.; Pushkar, Y. Characterization of the FeV=O Complex in the Pathway of Water Oxidation. Angew. Chem. Int. Ed. 2020, 59, 13502–13505. [Google Scholar] [CrossRef]
- Schröder, K.; Junge, K.; Bitterlich, B.; Beller, M. Fe-Catalyzed Oxidation Reactions of Olefins, Alkanes, and Alcohols: Involvement of Oxo- and Peroxo Complexes. In Iron Catalysis; Topics in Organometallic, Chemistry; Plietker, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 33, pp. 83–109. [Google Scholar]
- Canta, M.; Rodríguez, M.; Costas, M. Recent Advances in the Selective Oxidation of Alkyl C-H Bonds Catalyzed by Iron Coordination Complexes. Top. Curr. Chem. 2016, 372, 27–54. [Google Scholar]
- Shul’pin, G.B. New Trends in Oxidative Functionalization of Carbon–Hydrogen Bonds: A Review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef] [Green Version]
- Yadav, G.D.; Mewada, R.K.; Wagh, D.P.; Manyar, H.G. Advances and Future Trends in Selective Oxidation Catalysis: A Critical Review. Catal. Sci. Technol. 2022, 12, 7245–7269. [Google Scholar] [CrossRef]
- Denekamp, I.M.; Antens, M.; Slot, T.K.; Rothenberg, G. Selective Catalytic Oxidation of Cyclohexene with Molecular Oxygen: Radical Versus Nonradical Pathways. ChemCatChem 2018, 10, 1035–1041. [Google Scholar] [CrossRef]
- Cao, H.; Zhu, B.; Yang, Y.; Xu, L.; Yu, L.; Xu, Q. Recent Advances on Controllable and Selective Catalytic Oxidation of Cyclohexene. Chin. J. Catal. 2018, 39, 899–907. [Google Scholar] [CrossRef]
- Leising, R.A.; Takeuchi, K.J. Aerobic Oxidation of Cyclohexene with a Phosphine-ruthenium(II)-aquo Catalyst. Inorg. Chem. 1987, 26, 4391–4393. [Google Scholar] [CrossRef]
- Kojima, T.; Matsuda, Y. Catalytic Hydrocarbon Oxygenation by a Dinuclear Ruthenium(II) Complex with Molecular Oxygen. Chem. Lett. 1999, 28, 81–82. [Google Scholar] [CrossRef]
- Seok, W.K.; Dobson, J.C.; Meyer, T.J. Mechanisms of Oxidation of Phenol and Cyclohexene by an Oxo Complex of Ruthenium(IV). Inorg. Chem. 1988, 27, 3–5. [Google Scholar] [CrossRef]
- Stultz, L.K.; Huynh, M.H.V.; Binstead, R.A.; Curry, M.; Meyer, T.J. Allylic Oxidation of Cyclohexene and Indene by cis-[RuIV(bpy)2(py)(O)]2+. J. Am. Chem. Soc. 2000, 122, 5984–5996. [Google Scholar] [CrossRef]
- Wang, R.-M.; Hao, C.-J.; Wang, Y.-P.; Li, S.-B. Amino Acid Schiff Base Complex Catalyst for Effective Oxidation of Olefins with Molecular Oxygen. J. Mol. Catal. A Chem. 1999, 147, 173–178. [Google Scholar] [CrossRef]
- Matsushita, T.; Sawyer, D.T.; Sobkowiak, A. MnIIILx/t-BuOOH-induced Activation of Dioxygen for the Oxygenation of Cyclohexene. J. Mol. Catal. A Chem. 1999, 137, 127–133. [Google Scholar] [CrossRef]
- Rydel-Ciszek, K.; Pacześniak, T.; Miłaczewska, A.; Chmielarz, P.; Sobkowiak, A. ‘Oxygen-Consuming Complexes’-Catalytic Effects of Iron-Salen Complexes with Dioxygen. Catalysts 2021, 11, 1462. [Google Scholar] [CrossRef]
- Sobkowiak, A.; Naróg, D.; Sawyer, D.T. Iron(III, II)-induced Activation of Dioxygen for the Oxygenation of Cyclohexene and Related Unsaturated Hydrocarbons. J. Mol. Catal. A Chem. 2000, 159, 247–256. [Google Scholar] [CrossRef]
- Malik, D.D.; Chandra, A.; Seo, M.S.; Lee, Y.-M.; Farquhar, E.R.; Mebs, S.; Dau, H.; Ray, K.; Nam, W. Formation of Cobalt–Oxygen Intermediates by Dioxygen Activation at a Mononuclear Nonheme Cobalt(II) Center. Dalton Trans. 2021, 50, 11889–11898. [Google Scholar] [CrossRef]
- Malko, M.W.; Wróblewska, A. The Importance of R-(+)-Limonene as the Raw Material for Organic Syntheses and for Organic Industry. Chemik 2016, 70, 193–202. [Google Scholar]
- Denicourt-Nowicki, A.; Rauchdi, M.; Ait Ali, M.; Roucoux, A. Catalytic Oxidation Processes for the Upgrading of Terpenes: State-of-the-Art and Future Trends. Catalysts 2019, 9, 893. [Google Scholar] [CrossRef] [Green Version]
- Gusevskaya, E.; Gonsalves, J. Palladium(II) Catalyzed Oxidation of Naturally Occurring Terpenes with Dioxygen. J. Mol. Catal. A Chem. 1997, 121, 131–137. [Google Scholar] [CrossRef]
- El Firdoussi, L.; Baqqa, A.; Allaoud, S.; Ait Allal, B.; Karim, A.; Castanet, Y.; Mortreux, A. Selective Palladium-Catalysed Functionalization of Limonene: Synthetic and Mechanistic Aspects. J. Mol. Catal. A Chem. 1998, 135, 11–22. [Google Scholar] [CrossRef]
- Silva, A.D.; Patitucci, M.L.; Bizzo, H.R.; D’Elia, E.; Antunes, O.A.C. Wacker PdCl2–CuCl2 Catalytic Oxidation Process: Oxidation of Limonene. Catal. Commun. 2002, 3, 435–440. [Google Scholar] [CrossRef]
- Gonçalves, J.A.; Gusevskaya, E.V. Palladium Catalyzed Oxidation of Monoterpenes: Multistep Electron Transfer Catalytic Systems Pd(OAc)2/Benzoquinone/M(OAc)2 (M=Cu, Co or Mn) for the Allylic Oxidation of Limonene with Dioxygen. Appl. Catal. A Gen. 2004, 258, 93–98. [Google Scholar] [CrossRef]
- Gomes, M.d.F.T.; Antunes, O.A.C. Autoxidation of Limonene, α-Pinene and β-Pinene by Dioxygen Catalyzed by Co(OAc)2/Bromide. J. Mol. Catal. A Chem. 1997, 121, 145–155. [Google Scholar] [CrossRef]
- da Silva, M.J.; Robles-Dutenhefner, P.; Menini, L.; Gusevskaya, E.V. Cobalt Catalyzed Autoxidation of Monoterpenes in Acetic Acid and Acetonitrile Solutions. J. Mol. Catal. A Chem. 2003, 201, 71–77. [Google Scholar] [CrossRef]
- Mastrorilli, P.; Nobile, C.F.; Suranna, G.P.; Lopez, L. Aerobic Oxidations of Unsaturated Substrates under Mukaiyama’s Conditions: The Role of the Metal and of the Sacrificial Aldehyde. Tetrahedron 1995, 51, 7943–7950. [Google Scholar] [CrossRef]
- Corain, B.; Tessari, A.; Zecca, M. Epoxidation of Cyclohexene Catalyzed by Copper(II) Complexes under Mukaiyama’s Conditions. J. Mol. Catal. A Chem. 1995, 96, L9–L10. [Google Scholar] [CrossRef]
- Wentzel, B.B.; Alsters, P.L.; Feiters, M.C.; Nolte, R.J.M. Mechanistic Studies on the Mukaiyama Epoxidation. J. Org. Chem. 2004, 69, 3453–3464. [Google Scholar] [CrossRef] [Green Version]
- Naróg, D.; Szczepanik, A.; Sobkowiak, A. Iron(II, III)-Catalyzed Oxidation of Limonene by Dioxygen. Catal. Lett. 2008, 120, 320–325. [Google Scholar] [CrossRef]
- Szczepanik, A.; Sobkowiak, A. Manganese(II)-Induced Oxidation of Limonene by Dioxygen. Catal. Lett. 2008, 126, 261–267. [Google Scholar] [CrossRef]
- Roelfes, G.; Lubben, M.; Leppard, S.W.; Schudde, E.P.; Hermant, R.M.; Hage, R.; Wilkinson, E.C.; Que, L.; Feringa, B.L. Functional Models for Iron-bleomycin. J. Mol. Catal. A Chem. 1997, 117, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Klinker, E.J.; Kaizer, J.; Brennessel, W.W.; Woodrum, N.L.; Cramer, C.J.; Que, L., Jr. Structures of Nonheme Oxoiron(IV) Complexes from X-ray Crystallography, NMR Spectroscopy, and DFT Calculations. Angew. Chem. Int. Ed. 2005, 44, 3690–3694. [Google Scholar] [CrossRef]
- Draksharapu, A.; Li, Q.; Logtenberg, H.; van den Berg, T.A.; Meetsma, A.; Killeen, J.S.; Feringa, B.L.; Hage, R.; Roelfes, G.; Browne, W.R. Ligand Exchange and Spin State Equilibria of FeII(N4Py) and Related Complexes in Aqueous Media. Inorg. Chem. 2012, 51, 900–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizer, J.; Klinker, E.J.; Oh, N.Y.; Rohde, J.-U.; Song, W.J.; Stubna, A.; Kim, J.; Münck, E.; Nam, W.; Que, L. Nonheme FeIVO Complexes That Can Oxidize the C−H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc. 2004, 126, 472–473. [Google Scholar] [CrossRef]
- Draksharapu, A.; Li, Q.; Roelfes, G.; Browne, W.R. Photo-induced Oxidation of [FeII(N4Py)CH3CN] and Related Complexes. Dalton Trans. 2012, 41, 13180–13190. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, G.; Lee, C.W.Z.; Nag, S.S.; Alili, A.; Cantú Reinhard, F.G.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Dramatic Rate-enhancement of Oxygen Atom Transfer by an Iron(IV)-oxo Species by Equatorial Ligand Field Perturbations. Dalton Trans. 2018, 47, 14945–14957. [Google Scholar] [CrossRef] [Green Version]
- Rohde, J.-U.; Torelli, S.; Shan, X.; Lim, M.H.; Klinker, E.J.; Kaizer, J.; Chen, K.; Nam, W.; Que, L. Structural Insights into Nonheme Alkylperoxoiron(III) and Oxoiron(IV) Intermediates by X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2004, 126, 16750–16761. [Google Scholar] [CrossRef]
- Collins, M.J.; Ray, K.; Que, L. Electrochemical Generation of a Nonheme Oxoiron(IV) Complex. Inorg. Chem. 2006, 45, 8009–8011. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lee, Y.-M.; Shin, W.; Fukuzumi, S.; Nam, W. Dioxygen Activation by Mononuclear Nonheme Iron(II) Complexes Generates Iron−Oxygen Intermediates in the Presence of an NADH Analogue and Proton. J. Am. Chem. Soc. 2009, 131, 13910–13911. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, M.; Bühlmann, P.; Que, L., Jr. Redox Potential and C−H Bond Cleaving Properties of a Nonheme FeIV=O Complex in Aqueous Solution. J. Am. Chem. Soc. 2010, 132, 7638–7644. [Google Scholar] [CrossRef] [Green Version]
- Greef, R.; Peat, R.; Peter, L.M.; Pletcher, D.; Robinson, J. Instrumental Methods in Electrochemistry, Southampton Electrochemistry Group; Halsted Press: Chichester, NY, USA, 1985. [Google Scholar]
- Ghosh, A.; Steene, E. High-valent Transition Metal Centers and Noninnocent Ligands in Metalloporphyrins and Related Molecules: A Broad Overview Based on Quantum Chemical Calculations. J. Biol. Inorg. Chem. 2001, 6, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Pacześniak, T.; Błoniarz, P.; Rydel, K.; Sobkowiak, A. Electrochemical Catalytic Processes with Hydrogen Peroxide Showing Oxidative and Reductive Properties (Acting as Oxidant or Reductant). Electroanalysis 2007, 19, 945–951. [Google Scholar] [CrossRef]
- Awasthi, A.; Leach, I.F.; Engbers, S.; Kumar, R.; Eerlapally, R.; Gupta, S.; Klein, J.E.M.N.; Draksharapu, A. Formation and Reactivity of a Fleeting NiIII Bisphenoxyl Diradical Species. Angew. Chem. Int. Ed. 2022, 61, e202211345. [Google Scholar] [CrossRef] [PubMed]
- Pacześniak, T.; Sobkowiak, A. The Influence of Solvent on the Reaction Between Iron(II), (III) and Hydrogen Peroxide. J. Mol. Catal. A Chem. 2003, 194, 1–11. [Google Scholar] [CrossRef]
- Oh, N.Y.; Suh, Y.; Park, M.J.; Seo, M.S.; Kim, J.; Nam, W. Mechanistic Insight into Alcohol Oxidation by High-Valent Iron–Oxo Complexes of Heme and Nonheme Ligands. Angew. Chem. Int. Ed. 2005, 44, 4235–4239. [Google Scholar] [CrossRef]
- Rana, S.; Dey, A.; Maiti, D. Mechanistic Elucidation of C–H Oxidation by Electron Rich Non-heme Iron(IV)–oxo at Room Temperature. Chem. Commun. 2015, 51, 14469–14472. [Google Scholar] [CrossRef] [Green Version]
- Terencio, T.; Andris, E.; Gamba, I.; Srnec, M.; Costas, M.; Roithová, J. Chemoselectivity in the Oxidation of Cycloalkenes with a Non-Heme Iron(IV)-Oxo-Chloride Complex: Epoxidation vs. Hydroxylation Selectivity. J. Am. Soc. Mass Spectrom. 2019, 30, 1923–1933. [Google Scholar] [CrossRef] [Green Version]
- Karasevich, E.K.; Kulikova, V.S.; Shilov, A.E.; Shteinman, A.A. Biomimetic Alkane Oxidation Involving Metal Complexes. Russ. Chem. Rev. 1998, 67, 335. [Google Scholar] [CrossRef]
- Taktak, S.; Kryatov, S.V.; Haas, T.E.; Rybak-Akimova, E.V. Diiron(III) Oxo-Bridged Complexes with BPMEN and Additional Monodentate or Bidentate Ligands: Synthesis and Reactivity in Olefin Epoxidation with H2O2. J. Mol. Catal. A Chem. 2006, 259, 24–34. [Google Scholar] [CrossRef]
- Saltzman, H.; Sharefkin, J.G. Iodosobenzene. Org. Synth. 1963, 43, 60. [Google Scholar]
- Saltzman, H.; Sharefkin, J.G. Iodosobenzene diacetate. Org. Synth. 1963, 43, 62. [Google Scholar]
- Blicke, F.F.; Maxwell, C.E. Naphthylaminoalkanes. J. Am. Chem. Soc. 1939, 61, 1780–1782. [Google Scholar] [CrossRef]
- Niemers, E.; Hiltmann, R. Pyridylalkyl-substituierte Amine. Synthesis 1976, 1976, 593–595. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Sobkowiak, A.; Roberts, J.L. Electrochemistry for Chemists, 2nd ed.; Wiley: New York, NY, USA, 1995. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01. 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst, mM | Substrate, M | O2, atm | Ketone, mM | Alcohol, mM | Epoxide, mM | TON |
|---|---|---|---|---|---|---|
| 0.5 | 1 | 1 | 135 | 71 | 13 | 438 |
| 0.5 | 1 | 0.2 | 92 | 47 | 11 | 300 |
| 1 | 1 | 1 | 114 | 77 | 10 | 201 |
| 1 | 1 | 0.2 | 108 | 62 | 20 | 190 |
| 1 a | 1 | 0.2 | 144 | 74 | 23 | 241 |
| 2.5 | 1 | 1 | 115 | 28 | 12 | 62 |
| 2.5 | 1 | 0.2 | 104 | 55 | 8 | 67 |
| 5 | 1 | 1 | 51 | 32 | 3 | 17 |
| 5 | 1 | 0.2 | 126 | 50 | 7 | 37 |
| 1 | 0.5 | 1 | 55 | 18 | 9 | 82 |
| 1 | 0.5 | 0.2 | 39 | 10 | 8 | 57 |
| 1 b | 1 | 1 | 114 | 77 | 10 | 201 |
| 1 b | 1 | 0.2 | 108 | 62 | 20 | 190 |
| 1 | 1.5 | 1 | 174 | 107 | 15 | 296 |
| 1 | 1.5 | 0.2 | 177 | 100 | 16 | 293 |
| 1 | 2 | 1 | 248 | 119 | 42 | 409 |
| 1 | 2 | 0.2 | 293 | 129 | 40 | 462 |
| Catalyst, mM | Substrate, M | Oxidant | Limonene Oxide, mM | Carvone, mM | Carveol, mM | Perill Aldehyde, mM | Perillyl Alcohol, mM | TON |
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 1 | 48 | 33 | 24 | 0 | 0 | 210 |
| 0.5 | 1 | 0.2 | 43 | 32 | 22 | 1 | 3 | 202 |
| 1 | 1 | 1 | 89 | 52 | 31 | 6 | 3 | 181 |
| 1 | 1 | 0.2 | 63 | 42 | 27 | 3 | 3 | 138 |
| 1 a | 1 | 0.2 | 93 | 55 | 32 | 3 | 7 | 190 |
| 2.5 | 1 | 1 | 56 | 40 | 28 | 1 | 2 | 51 |
| 2.5 | 1 | 0.2 | 74 | 47 | 29 | 2 | 5 | 63 |
| 5 | 1 | 1 | 49 | 36 | 26 | 1 | 1 | 23 |
| 5 | 1 | 0.2 | 59 | 43 | 29 | 1 | 2 | 27 |
| 7.5 | 1 | 1 | 51 | 37 | 28 | 0 | 0 | 15 |
| 7.5 | 1 | 0.2 | 60 | 42 | 31 | 2 | 4 | 19 |
| 10 | 1 | 1 | 57 | 41 | 33 | 5 | 1 | 14 |
| 10 | 1 | 0.2 | 57 | 41 | 28 | 2 | 3 | 13 |
| 1 | 0.5 | 1 | 46 | 22 | 10 | 2 | 3 | 83 |
| 1 | 0.5 | 0.2 | 51 | 22 | 13 | 2 | 3 | 91 |
| 1 b | 1 | 1 | 89 | 52 | 31 | 6 | 3 | 181 |
| 1 b | 1 | 0.2 | 63 | 42 | 27 | 3 | 3 | 138 |
| 1 | 1.5 | 1 | 75 | 59 | 35 | 2 | 5 | 176 |
| 1 | 1.5 | 0.2 | 58 | 46 | 30 | 0 | 0 | 134 |
| 1 | 2 | 1 | 91 | 70 | 42 | 3 | 4 | 210 |
| 1 | 2 | 0.2 | 64 | 58 | 35 | 1 | 2 | 160 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rydel-Ciszek, K.; Pacześniak, T.; Chmielarz, P.; Sobkowiak, A. Bio-Inspired Iron Pentadentate Complexes as Dioxygen Activators in the Oxidation of Cyclohexene and Limonene. Molecules 2023, 28, 2240. https://doi.org/10.3390/molecules28052240
Rydel-Ciszek K, Pacześniak T, Chmielarz P, Sobkowiak A. Bio-Inspired Iron Pentadentate Complexes as Dioxygen Activators in the Oxidation of Cyclohexene and Limonene. Molecules. 2023; 28(5):2240. https://doi.org/10.3390/molecules28052240
Chicago/Turabian StyleRydel-Ciszek, Katarzyna, Tomasz Pacześniak, Paweł Chmielarz, and Andrzej Sobkowiak. 2023. "Bio-Inspired Iron Pentadentate Complexes as Dioxygen Activators in the Oxidation of Cyclohexene and Limonene" Molecules 28, no. 5: 2240. https://doi.org/10.3390/molecules28052240
APA StyleRydel-Ciszek, K., Pacześniak, T., Chmielarz, P., & Sobkowiak, A. (2023). Bio-Inspired Iron Pentadentate Complexes as Dioxygen Activators in the Oxidation of Cyclohexene and Limonene. Molecules, 28(5), 2240. https://doi.org/10.3390/molecules28052240