
The Contribution of Theoretical Prediction Studies to the Antioxidant Activity Assessment of the Bioactive Secoiridoids Encountered in Olive Tree Products and By-Products




Abstract
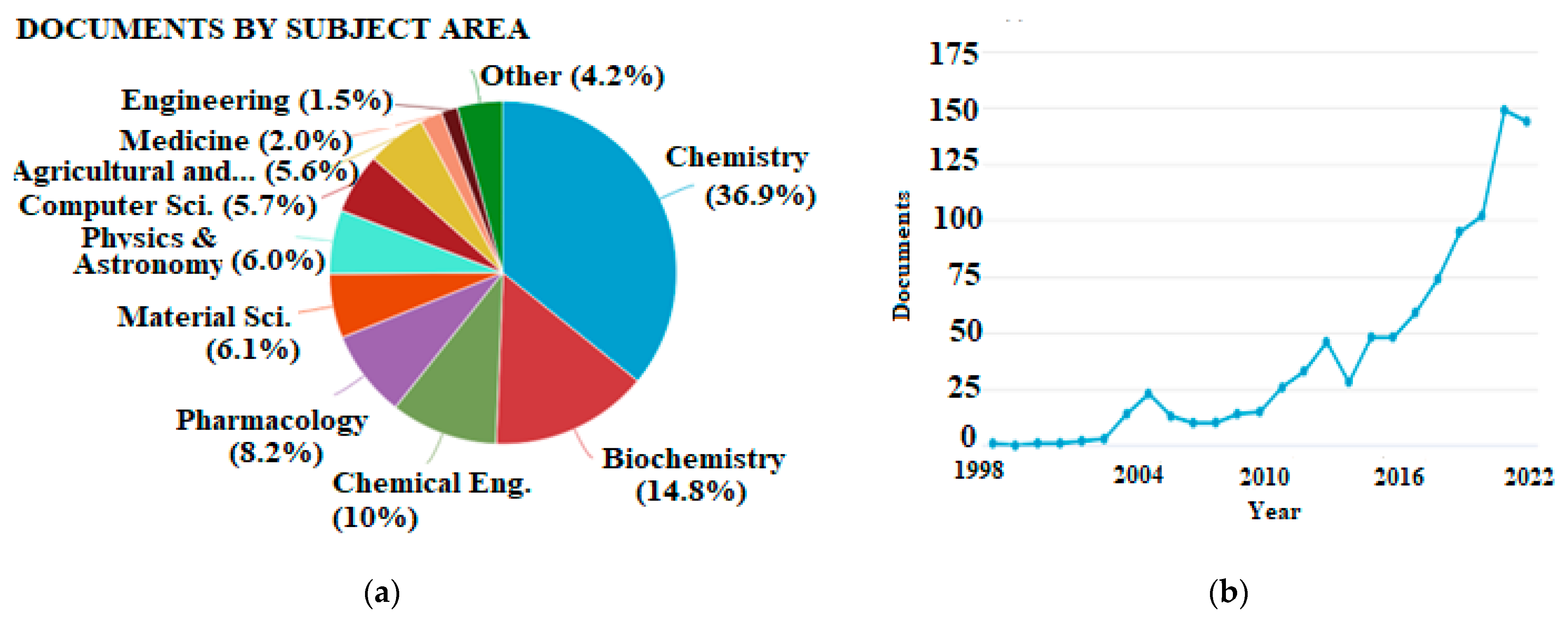
1. Introduction
- (a)
- Hydrogen atom transfer (HAT) /Proton coupled electron transfer (PCET) to free radicals:AH + ROO● → ROOH + A●
- (b)
- Single electron transfer followed by proton transfer (SET-PT) or not (SET):AH + ROO● → [ROO−---AH●+ ] → ROOH + A●
- (c)
- Sequential proton loss followed by electron transfer (SPLET):AH + ROO● → A− + ROO● + H+ → ROO− + A● + H+ → ROOH + A●
- (d)
- Sequential proton loss−hydrogen atom transfer (SPLHAT):AH → A− + H+ → A− + ROO● →ROOH + A●−
- (e)
- Radical adduct formation (RAF):AH + ROO● → [AH-OOR]●
2. Theoretical Prediction Strategies
- Consideration of tunneling effects for reactions involving small particles;
- Contribution of different mechanisms of reaction, and reaction sites, to the whole activity of the examined compound;
- Inclusion of reactions that do not fulfill the Bell–Evans–Polanyi principle;
- Incorporation of the pH influence on the reactivity of antioxidants bearing ionizable groups such as acids, considering the molar fraction of different forms present when the pH value is different;
- Consideration of single electron transfer reactions situated in the inverted region of the Marcus parabola.
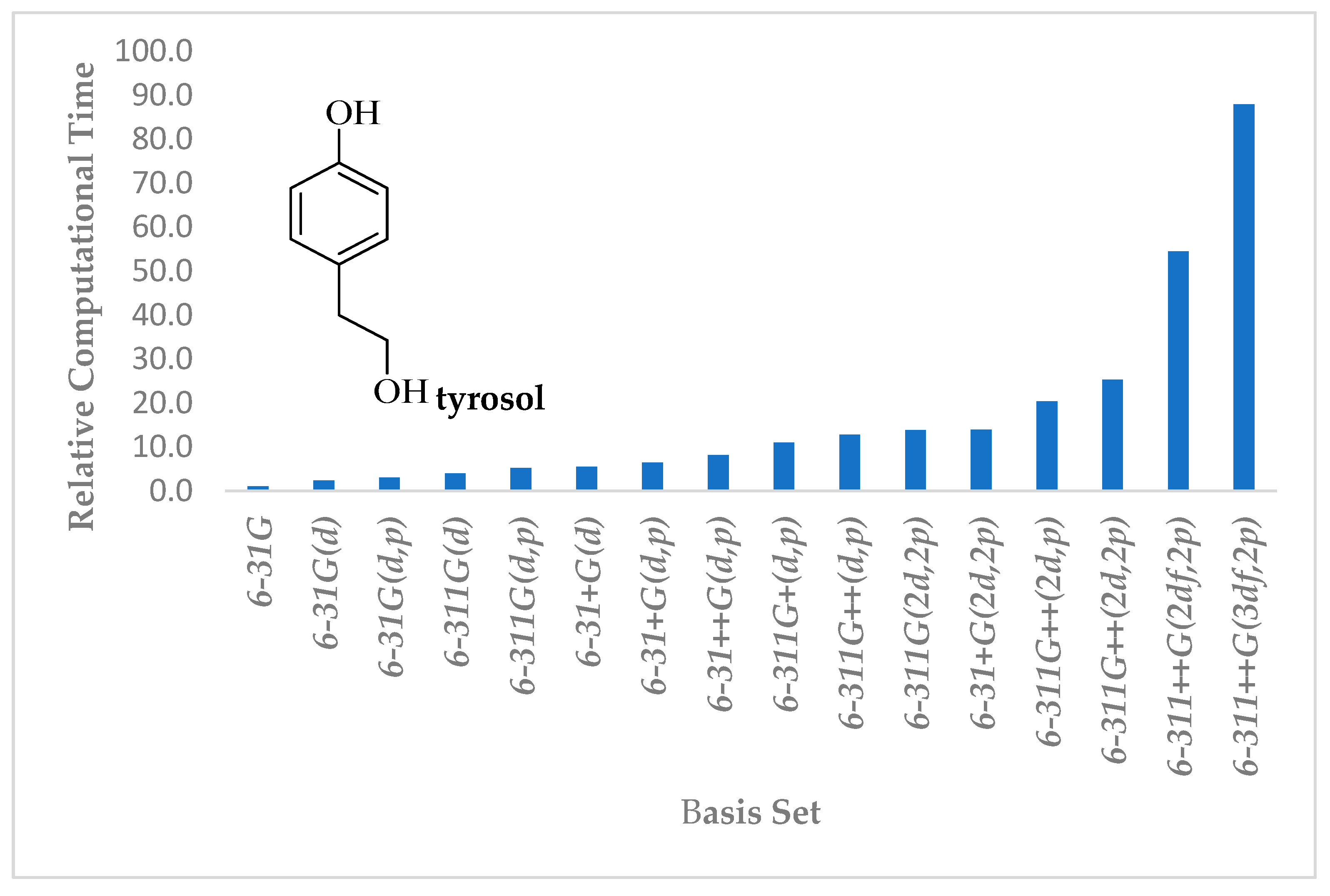
3. Computational Methods
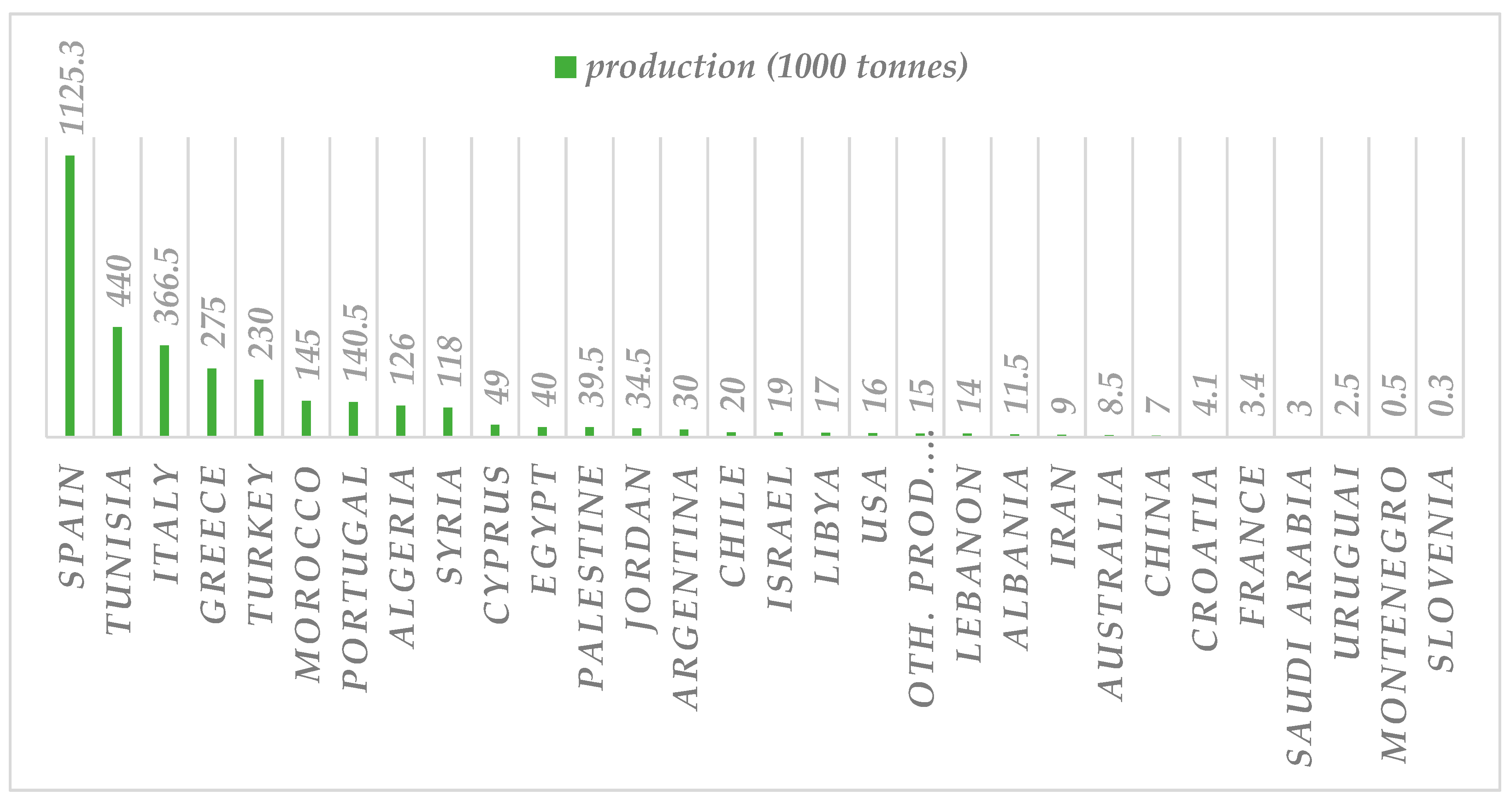
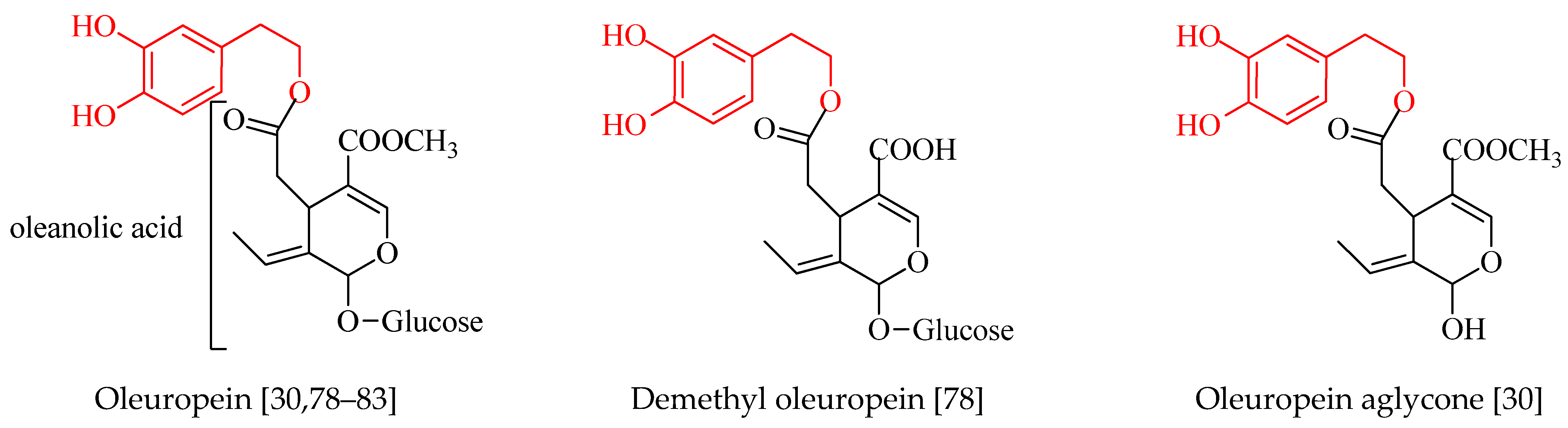
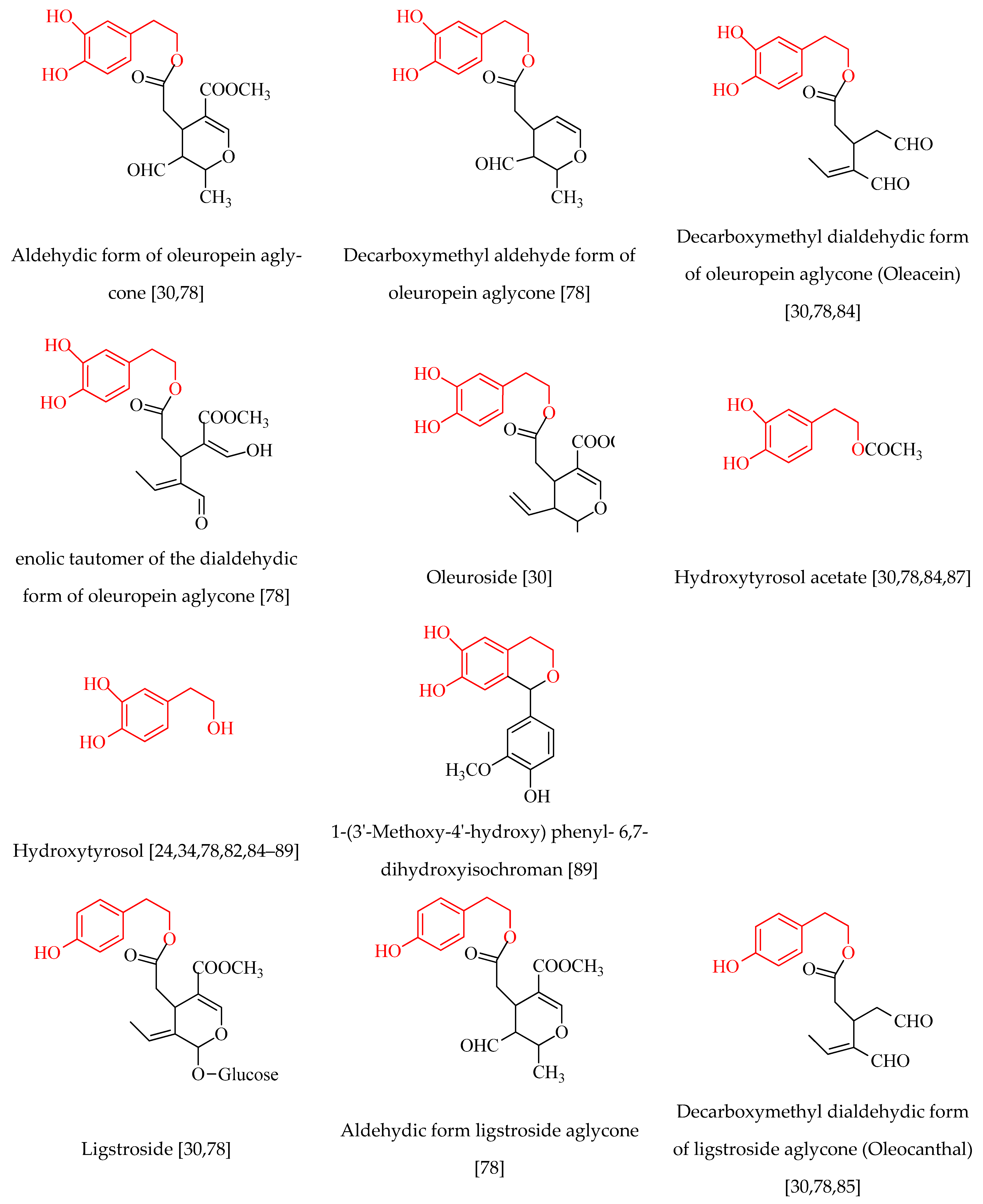
4. Phenolic Compounds of Olive Tree Products and By-Products
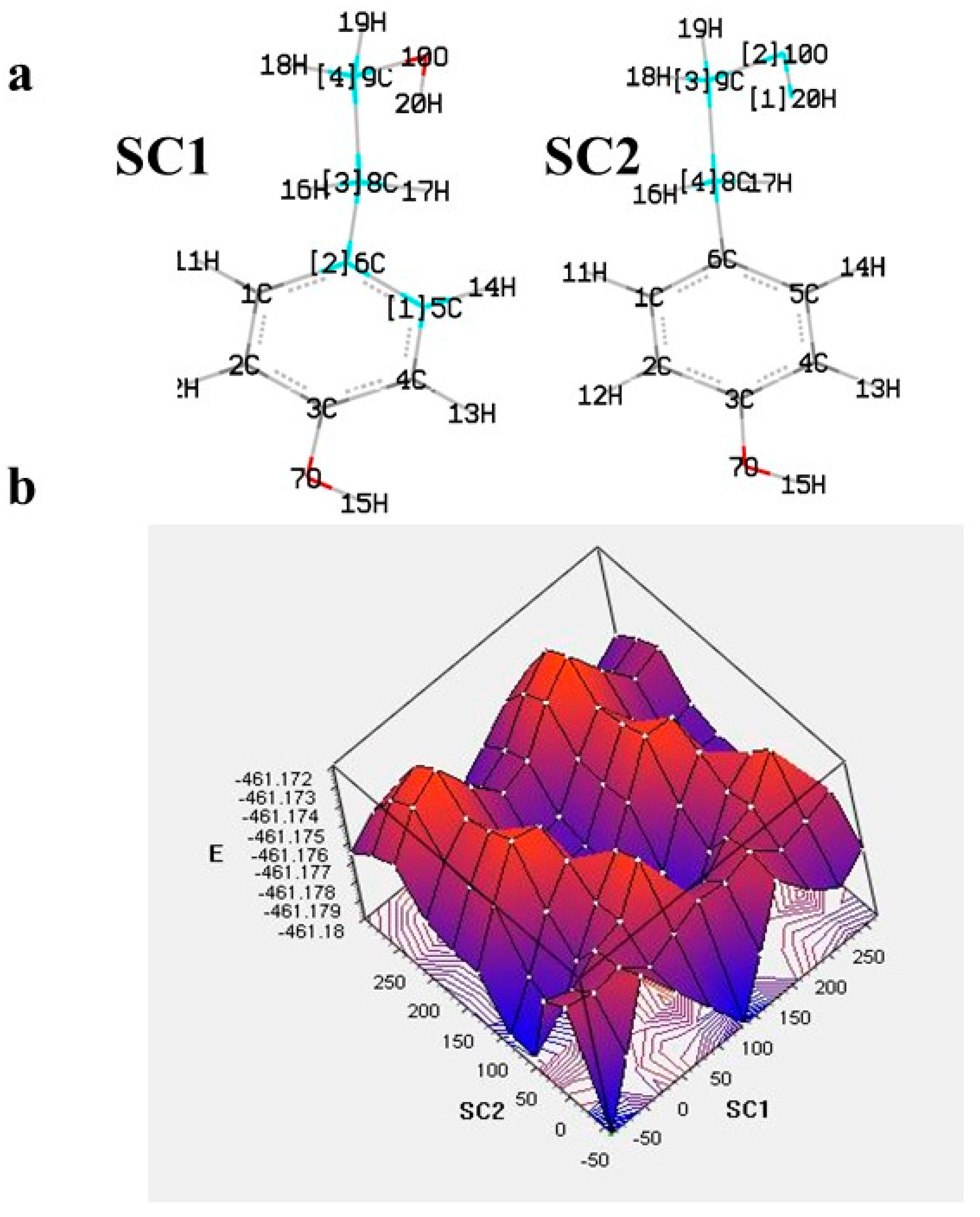
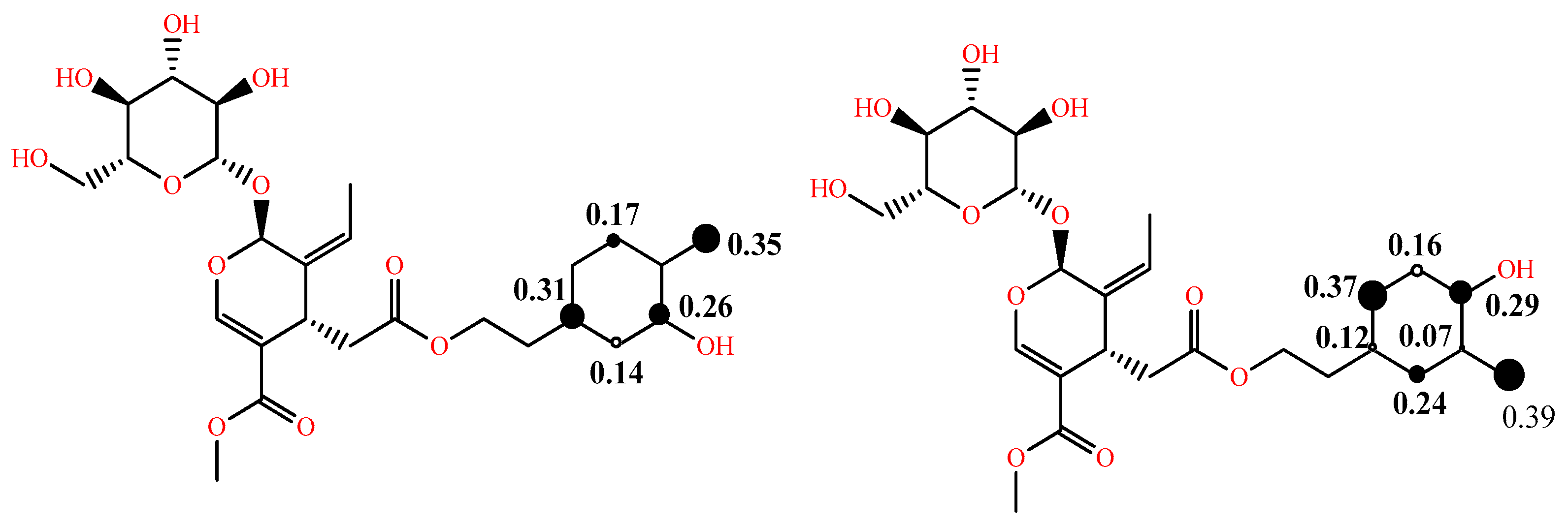
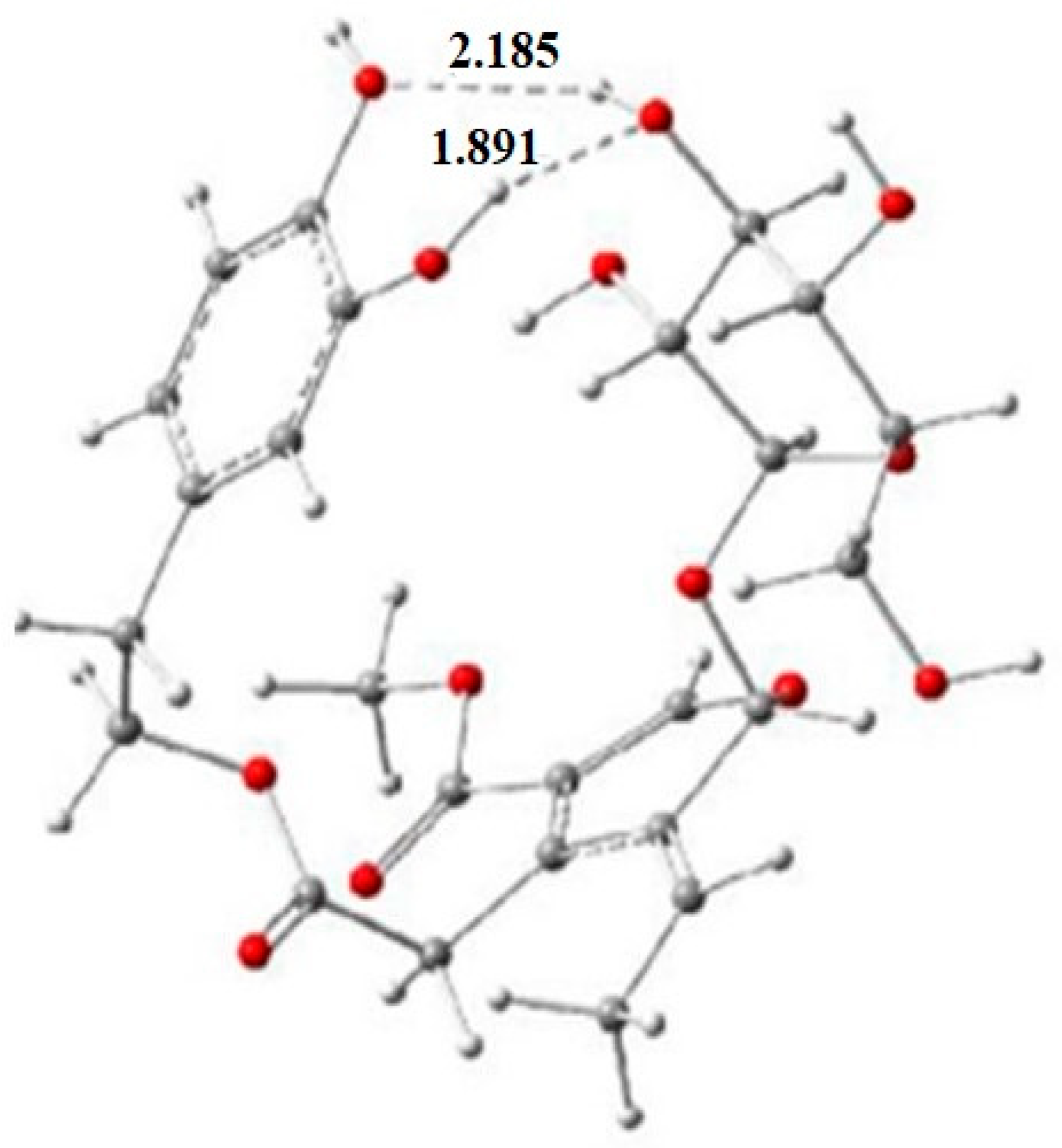
5. Computational Studies on the Radical Scavenging Activity of Olive Secoiridoids and Related Compounds
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Brewer, M. Natural antioxidants: Sources, compounds, mechanisms of action, and potential applications. Compr. Rev. Food Sci. Food Saf. 2011, 10, 221–247. [Google Scholar] [CrossRef]
- Hrelia, S.; Angeloni, C. New mechanisms of action of natural antioxidants in health and disease. Antioxidants 2020, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Salami, S.A.; Guinguina, A.; Agboola, J.O.; Omede, A.A.; Agbonlahor, E.M.; Tayyab, U. Review: In vivo and postmortem effects of feed antioxidants in livestock: A review of the implications on authorization of antioxidant feed additives. Animal 2016, 10, 1375–1390. [Google Scholar] [CrossRef]
- Boskou, D.; Tsimidou, M.; Blekas, G. Polar phenolic compounds. In Olive Oil: Chemistry and Technology, 2nd ed.; Boskou, D., Ed.; AOCS Press: Champaign, IL, USA, 2006; pp. 73–92. [Google Scholar]
- Nenadis, N.; Papoti, V.T.; Tsimidou, M.Z. Bioactive ingredients in olive leaves. In Olives and Olive Oil in Health and Disease Prevention, 2nd ed.; Preedy, V.R., Watson, R.R., Eds.; Academic Press: San Diego, CA, USA, 2021; pp. 65–78. [Google Scholar]
- Zeb, A. Concept, mechanism, and applications of phenolic antioxidants in foods. J. Food Biochem. 2020, 44, e13394. [Google Scholar] [CrossRef]
- Spiegel, M. Current trends in computational quantum chemistry studies on antioxidant radical scavenging activity. J. Chem. Inf. Model. 2022, 62, 2639–2658. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Y.; Li, H.; Deng, Z.; Tsao, R. A review on insoluble-bound phenolics in plant-based food matrix and their contribution to human health with future perspectives. Trends Food Sci. Technol. 2020, 105, 347–362. [Google Scholar] [CrossRef]
- Laguerre, M.; Bayrasy, C.; Panya, A.; Weiss, J.; McClements, D.J.; Lecomte, J.; Decker, E.A.; Villeneuve, P. What makes good antioxidants in lipid-based systems? The next theories beyond the polar paradox. Crit. Rev. Food Sci. Nutr. 2015, 55, 183–201. [Google Scholar] [CrossRef]
- Dai, J.; Mumper, R.J. Plant phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef] [PubMed]
- Nenadis, N.; Tsimidou, M.Z. Assessing the activity of natural food antioxidants. In Oxidation in Foods and Beverages and Antioxidant Applications; Decker, E., Elias, R., McClements, D.J., Eds.; Woodhead Publishing: Cambridge, UK, 2010; pp. 332–367. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Francisco-Márquez, M.; Alvarez-Idaboy, J.R. Mechanism and kinetics studies on the antioxidant activity of sinapinic acid. Phys. Chem. Chem. Phys. 2011, 13, 11199–11205. [Google Scholar] [CrossRef] [PubMed]
- Martins, N.; Barros, L.; Ferreira, I.C.F.R. In vivo antioxidant activity of phenolic compounds: Facts and gaps. Trends Food Sci. Technol. 2016, 48, 1–12. [Google Scholar] [CrossRef]
- Tsimidou, M.; Nenadis, N.; Zhang, H.Y. Structure radical scavenging activity relationships of flavonoids and phenolic acids. In Natural Antioxidant Phenols. Sources, Structure—Activity Relationship, Current Trends in Analysis and Characterization; Boskou, D., Gerothanassis, I., Kefalas, P., Eds.; Research Signpost: Kerala, India, 2006; pp. 33–51. [Google Scholar]
- Nenadis, N.; Tsimidou, M.Z. On the use of DFT computations to the radical scavenging activity studies of natural phenolic compounds. In Density Functional Theory: Principles, Applications and Analysis; Morin, J., Pelletier, J.M., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2013; pp. 121–146. [Google Scholar]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Alov, P.; Tsakovska, I.; Pajeva, I. Computational studies of free radical-scavenging properties of phenolic compounds. CTMC 2015, 15, 85–104. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food antioxidants: Chemical insights at the molecular level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Dehkordi, M.M.; Asgarshamsi, M.H. Density functional theory studies of the antioxidants—A Review. J. Mol. Model. 2021, 27, 271. [Google Scholar] [CrossRef]
- Mittal, A.; Vashistha, V.K.; Das, D.K. Recent advances in the antioxidant activity and mechanisms of chalcone derivatives: A computational review. Free Radic. Res. 2022, 56, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Raúl Alvarez-Idaboy, J. Computational strategies for predicting free radical scavengers’ protection against oxidative stress: Where are we and what might follow? Int. J. Quantum Chem. 2019, 119, e25665. [Google Scholar] [CrossRef]
- Lewars, E.G. The concept of the potential energy surface. In Computational Chemistry; Lewars, E.G., Ed.; Springer: Cham, Switzerland, 2016; pp. 9–49. [Google Scholar]
- Semidalas, C.; Semidalas, E.; Matsoukas, M.T.; Nixarlidis, C.; Zoumpoulakis, P. In silico studies reveal the mechanisms behind the antioxidant and anti-inflammatory activities of hydroxytyrosol. Med. Chem. Res. 2016, 25, 2498–2511. [Google Scholar] [CrossRef]
- Nenadis, N.; (Aristotle University of Thessaloniki, School of Chemistry, Thessaloniki, Greece); Pyrka, I.; (Aristotle University of Thessaloniki, School of Chemistry, Thessaloniki, Greece); Tsimidou, M.Z.; (Aristotle University of Thessaloniki, School of Chemistry, Thessaloniki, Greece). Unpublished work, 2023.
- Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why Is the dual descriptor a more accurate local reactivity descriptor than Fukui functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Wang, L.-F.; Zhang, H.-Y. A theoretical investigation on DPPH radical-scavenging mechanism of edaravone. Bioorg. Med. Chem. Lett. 2003, 13, 3789–3792. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Akhtari, K.; Hassanzadeh, H.; Zarei, S.A.; Fakhraei, N.; Hassanzadeh, K. The role of structural C–H compared with phenolic OH sites on the antioxidant activity of oleuropein and its derivatives as a great non-flavonoid family of the olive components: A DFT study. Food Chem. 2014, 164, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Amić, A.; Mastiľák Cagardová, D. DFT study of the direct radical scavenging potency of two natural catecholic compounds. Int. J. Mol. Sci. 2022, 23, 14497. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, A.; Shi, P.; Zhang, H.; Li, Z. Quantum chemical study on the antioxidation mechanism of piceatannol and isorhapontigenin toward hydroxyl and hydroperoxyl radicals. PLoS ONE 2015, 10, e0133259. [Google Scholar] [CrossRef]
- Halliwell, B.; Aeschbach, R.; Löliger, J.; Aruoma, O.I. The characterization of antioxidants. Food Chem. Toxicol. 1995, 33, 601–617. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Francisco-Márquez, M.; Medina, M.E. A Quantum chemical study on the free radical scavenging activity of tyrosol and hydroxytyrosol. Theor. Chem. Acc. 2012, 131, 1173. [Google Scholar] [CrossRef]
- Bakalbassis, E.G.; Chatzopoulou, A.; Melissas, V.S.; Tsimidou, M.; Tsolaki, M.; Vafiadis, A. Ab initio and density functional theory studies for the explanation of the antioxidant activity of certain phenolic acids. Lipids 2001, 36, 181–191. [Google Scholar] [CrossRef]
- Bakalbassis, E.G.; Lithoxoidou, A.T.; Vafiadis, A.P. Theoretical calculation of accurate absolute and relative gas- and liquid-phase O−H bond dissociation enthalpies of 2-mono- and 2,6-disubstituted phenols, using DFT/B3LYP. J. Phys. Chem. A 2003, 107, 8594–8606. [Google Scholar] [CrossRef]
- Wayner, D.D.M.; Lusztyk, E.; Page, D.; Ingold, K.U.; Mulder, P.; Laarhoven, L.J.J.; Aldrich, H.S. Effects of solvation on the enthalpies of reaction of tert-butoxyl radicals with phenol and on the calculated O-H bond strength in phenol. J. Am. Chem. Soc. 1995, 117, 8737–8744. [Google Scholar] [CrossRef]
- Borges dos Santos, R.M.; Martinho Simões, J.A. Energetics of the O–H bond in phenol and substituted phenols: A critical evaluation of literature data. J. Phys. Chem. Ref. Data 1998, 27, 707–739. [Google Scholar] [CrossRef]
- Sharopov, F.S.; Wink, M.; Setzer, W.N. Radical scavenging and antioxidant activities of essential oil components—An experimental and computational investigation. Nat. Prod. Commun. 2015, 10, 153–156. [Google Scholar] [CrossRef]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John Wiley and Sons, Inc.: New York, NY, USA, 2001; pp. 206–215. [Google Scholar]
- Spiegel, M.; Gamian, A.; Sroka, Z. A Statistically supported antioxidant activity DFT benchmark—The effects of Hartree–Fock exchange and basis set selection on accuracy and resources uptake. Molecules 2021, 26, 5058. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M. Dielectric Continuum methods for quantum chemistry. WIREs Comput. Mol. Sci. 2021, 11, e1519. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A Computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef] [PubMed]
- Litwinienko, G.; Ingold, K.U. Solvent effects on the rates and mechanisms of reaction of phenols with free radicals. Acc. Chem. Res. 2007, 40, 222–230. [Google Scholar] [CrossRef]
- Guerra, M.; Amorati, R.; Pedulli, G.F. Water Effect on the O−H dissociation enthalpy of para-substituted phenols: A DFT Study. J. Org. Chem. 2004, 69, 5460–5467. [Google Scholar] [CrossRef]
- Kozlowski, D.; Marsal, P.; Steel, M.; Mokrini, R.; Duroux, J.L.; Lazzaroni, R.; Trouillas, P. Theoretical investigation of the formation of a new series of antioxidant depsides from the radiolysis of flavonoid compounds. Radiat. Res. 2007, 168, 243–252. [Google Scholar] [CrossRef]
- Nenadis, N.; Samara, E.; Mantzouridou, F.T. On the role of the carboxyl group to the protective effect of O-dihydroxybenzoic acids to Saccharomyces cerevisiae cells upon induced oxidative stress. Antioxidants 2022, 11, 161. [Google Scholar] [CrossRef]
- International Olive Council [IOC]. World and EU Olive Oil Figures (Production). Available online: https://www.internationaloliveoil.org/wp-content/uploads/2021/12/HO-W901-17-12-2021-P.pdf (accessed on 8 December 2022).
- International Olive Council [IOC]. World and EU Olive Oil Figures (Production). Available online: https://www.internationaloliveoil.org/wp-content/uploads/2021/12/HO-CE901-17-12-2021-P.pdf (accessed on 8 December 2022).
- Servili, M.; Esposto, S.; Taticchi, A.; Urbani, S.; Di Maio, I.; Veneziani, G.; Selvaggini, R. New approaches to virgin olive oil quality, technology, and by-products valorization. Eur. J. Lipid Sci. Technol. 2015, 117, 1882–1892. [Google Scholar] [CrossRef]
- Herrero, M.; Temirzoda, T.N.; Segura-Carretero, A.; Quirantes, R.; Plaza, M.; Ibañez, E. New possibilities for the valorization of olive oil by-products. J. Chromatogr. A 2011, 1218, 7511–7520. [Google Scholar] [CrossRef] [PubMed]
- Tsimidou, M.Z. Virgin olive oil (VOO) and other olive tree products as sources of α-tocopherol: Updating and perspective ch. 1. In Tocopherol: Sources, Uses and Health Benefits; Catala, A., Ed.; Nova Science Publications: Hauppauge, NY, USA, 2012; pp. 1–21. [Google Scholar]
- EFSA Panel on Dietetic Products, Nutrition, and Allergies (NDA). Scientific Opinion on dietary reference values for vitamin E as α-tocopherol. EFSA J. 2015, 13, 4149. [Google Scholar]
- Tsimidou, M.Z.; Mastralexi, A.; Özdikicierler, O. Cold pressed olive oils. In Cold Pressed Oils, Green Technology, Bioactive Compounds, Functionality and Applications; Ramadan, M.F., Ed.; Academic Press: San Diego, CA, USA, 2020; pp. 547–556. [Google Scholar]
- Mohamed, R.; Pineda, M.; Aguilar, M. Antioxidant capacity of extracts from wild and crop plants of the Mediterranean region. J. Food Sci. 2007, 72, S059–S063. [Google Scholar] [CrossRef]
- Ryan, D.; Robards, K. Critical review. Phenolic compounds in olives. Analyst 1998, 123, 31R–44R. [Google Scholar] [CrossRef]
- Talhaoui, N.; Taamalli, A.; Gómez-Caravaca, A.M.; Fernández-Gutiérrez, A.; Segura-Carretero, A. Phenolic compounds in olive leaves: Analytical determination, biotic and abiotic influence, and health benefits. Food Res. Int. 2015, 77, 92–108. [Google Scholar] [CrossRef]
- Zahi, M.R.; Zam, W.; El Hattab, M. State of knowledge on chemical, biological and nutritional properties of olive mill wastewater. Food Chem. 2022, 381, 132238. [Google Scholar] [CrossRef] [PubMed]
- Bendini, A.; Cerretani, L.; Carrasco-Pancorbo, A.; Gómez-Caravaca, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Lercker, G. Phenolic molecules in virgin olive oils: A survey of their sensory properties, health effects, antioxidant activity and analytical methods. An overview of the last decade. Molecules 2007, 12, 1679–1719. [Google Scholar] [CrossRef] [PubMed]
- International Olive Council [IOC]. Determination of Biophenols in Olive Oils by HPLC. COI/T.20/Doc. No. 29; International Olive Council [IOC]: Madrid, Spain, 2009; Available online: https://www.internationaloliveoil.org/wp-content/uploads/2019/11/COI-T.20-Doc.-No-29-Rev-1-2017.pdf (accessed on 8 December 2022).
- Ghanbari, R.; Anwar, F.; Alkharfy, K.M.; Gilani, A.-H.; Saari, N. Valuable nutrients and functional bioactives in different parts of olive (Olea Europaea L.)—A review. Int. J. Mol. Sci. 2012, 13, 3291–3340. [Google Scholar] [CrossRef]
- Tapia-Quirós, P.; Montenegro-Landívar, M.F.; Reig, M.; Vecino, X.; Cortina, J.L.; Saurina, J.; Granados, M. Recovery of polyphenols from agri-food by-products: The olive oil and winery industries cases. Foods 2022, 11, 362. [Google Scholar] [CrossRef]
- Abbattista, R.; Ventura, G.; Calvano, C.D.; Cataldi, T.R.I.; Losito, I. Bioactive compounds in waste by-products from olive oil production: Applications and structural characterization by mass spectrometry techniques. Foods 2021, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Tsimidou, M.Z.; Nenadis, N.; Servili, M.; García-González, D.L.; Gallina Toschi, T. Why tyrosol derivatives have to be quantified in the calculation of “Olive oil polyphenols” content to support the health claim provisioned in the EC Reg. 432/2012. Eur. J. Lipid Sci. Technol. 2018, 120, 1800098. [Google Scholar] [CrossRef]
- Rodrıguez-Pérez, C.; Quirantes-Piné, R.; Lozano-Sanchez, J.; Menéndez, J.; Segura-Carretero, A. Composition and analysis of functional components of olive leaves. In Olives and Olive Oil as Functional Foods: Bioactivity, Chemistry and Processing; Kiritsakis, A.K., Shahidi, F., Eds.; Wiley: Hoboken, NJ, USA, 2017; pp. 383–400. [Google Scholar]
- Boskou, D. Olive fruit, table olives, and olive oil bioactive constituents. In Olive and Olive Oil Bioactive Constituents; Boskou, D., Ed.; AOCS Press: Urbana, IL, USA, 2015; pp. 1–30. [Google Scholar]
- Boskou, D.; Camposeo, S.; Clodoveo, M.L. Table olives as sources of bioactive compounds. In Olive and Olive Oil Bioactive Constituents; Boskou, D., Ed.; AOCS Press: Urbana, IL, USA, 2015; pp. 217–259. [Google Scholar]
- Malheiro, R.; Rodrigues, N.; Pereira, J.A. Olive oil phenolic composition as affected by geographic origin, olive cultivar, and cultivation systems. In Olive and Olive Oil Bioactive Constituents; Boskou, D., Ed.; AOCS Press: Urbana, IL, USA, 2015; pp. 93–121. [Google Scholar]
- Rodríguez, G.; Lama, A.; Rodríguez, R.; Jiménez, A.; Guillén, R.; Fernández-Bolaños, J. Olive stone an attractive source of bioactive and valuable compounds. Bioresour. Technol. 2008, 99, 5261–5269. [Google Scholar] [CrossRef]
- Gouvinhas, I.; Machado, N.; Sobreira, C.; Domínguez-Perles, R.; Gomes, S.; Rosa, E.; Barros, A. Critical review on the significance of olive phytochemicals in plant physiology and human health. Molecules 2017, 22, 1986. [Google Scholar] [CrossRef]
- Diamantakos, P.; Velkou, A.; Killday, K.B.; Gimisis, T.; Melliou, E.; Magiatis, P. Oleokoronal y oleomisional: Los nuevos components fenólicos mayores del aceite de oliva extra virgen. Olivae Rev. Of. Del Cons. Oleícola Int. 2015, 122, 23–34. [Google Scholar]
- Antoniadi, L.; Angelis, A.; Stathopoulos, P.; Bata, E.-M.; Papoutsaki, Z.; Halabalaki, M.; Skaltsounis, L.A. Oxidized forms of olive oil secoiridoids: Semisynthesis, identification and correlation with quality parameters. Planta Med. 2022, 88, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Tsimidou, M. Polyphenols and quality of virgin olive oil in retrospect. Ital. J. Food Sci. 1998, 10, 99–116. [Google Scholar]
- Tsimidou, M.Z. Analytical methodologies: Phenolic compounds related to olive oil taste issues. In Handbook of Olive Oil; Aparicio, R., Harwood, J., Eds.; Springer US: Boston, MA, USA, 2013; pp. 311–333. [Google Scholar]
- Boskou, D.; Blekas, G.; Tsimidou, M. Phenolic compounds in olive oil and olives. Curr. Top. Nutraceutical Res. 2005, 3, 125–136. [Google Scholar]
- European Commission. Regulation (EU) No 432/2012 of 16 May 2012. Establishing a list of permitted health claims made on foods, other than those referring to the reduction of disease risk and to children’s development and health. Off. J. Eur. Union 2012, L136, 1–40. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2012:136:0001:0040:en:PDF (accessed on 8 December 2022).
- Nenadis, N.; Wang, L.-F.; Tsimidou, M.Z.; Zhang, H.-Y. Radical scavenging potential of phenolic compounds encountered in O. Europaea products as indicated by calculation of bond dissociation enthalpy and ionization potential values. J. Agric. Food Chem. 2005, 53, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Gikas, E.; Bazoti, F.N.; Tsarbopoulos, A. Conformation of oleuropein, the major bioactive compound of Olea europea. J. Mol. Struct. THEOCHEM 2007, 821, 125–132. [Google Scholar] [CrossRef]
- Souilem, S.; Treesuwan, W.; Kobayashi, I.; Khalid, N.; Bouallagui, Z.; Neves, M.A.; Uemura, K.; Isoda, H.; Sayadi, S.; Nakajima, M. Simulation of oleuropein structural conformation in vacuum, water and triolein–water systems using molecular dynamics. Food Res. Int. 2016, 88, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Galiano, V.; Villalaín, J. Oleuropein aglycone in lipid bilayer membranes. A molecular dynamics study. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2849–2858. [Google Scholar] [CrossRef]
- Aree, T.; Jongrungruangchok, S. Structure—Antioxidant activity relationship of β-cyclodextrin inclusion complexes with olive tyrosol, hydroxytyrosol and oleuropein: Deep insights from X-Ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym. 2018, 199, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Baysal, G.; Kasapbaşı, E.E.; Yavuz, N.; Hür, Z.; Genç, K.; Genç, M. Determination of theoretical calculations by DFT method and investigation of antioxidant, antimicrobial properties of olive leaf extracts from different regions. J. Food Sci. Technol. 2021, 58, 1909–1917. [Google Scholar] [CrossRef]
- Li, M.-J.; Liu, L.; Fu, Y.; Guo, Q.-X. Accurate bond dissociation enthalpies of popular antioxidants predicted by the ONIOM-G3B3 method. J. Mol. Struct. THEOCHEM 2007, 815, 1–9. [Google Scholar] [CrossRef]
- Erkoç, F.; Keskin, N.; Erkoç, Ş. Theoretical investigation of hydroxytyrosol and its radicals. J. Mol. Struct. THEOCHEM 2003, 625, 87–94. [Google Scholar] [CrossRef]
- Leopoldini, M.; Marino, T.; Russo, N.; Toscano, M. Antioxidant properties of phenolic compounds: H-atom versus electron transfer mechanism. J. Phys. Chem. A 2004, 108, 4916–4922. [Google Scholar] [CrossRef]
- Rezaei-Sadabady, R.; Zarghami, N.; Barzegar, A.; Eidi, A.; Akbarzadeh, A.; Rezaei-Tavirani, M. Studies of the relationship between structure and antioxidant activity in interesting systems, including tyrosol, hydroxytyrosol derivatives indicated by quantum chemical calculations. Soft 2013, 2, 13–18. [Google Scholar] [CrossRef]
- Dávalos, J.Z.; Valderrama-Negrón, A.C.; Barrios, J.R.; Freitas, V.L.S.; Ribeiro da Silva, M.D.M.C. Energetic and structural properties of two phenolic antioxidants: Tyrosol and hydroxytyrosol. J. Phys. Chem. A 2018, 122, 4130–4137. [Google Scholar] [CrossRef]
- Nenadis, N.; Siskos, D. Radical scavenging activity characterization of synthetic isochroman-derivatives of hydroxytyrosol: A gas-phase DFT approach. Food Res. Int. 2015, 76, 506–510. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.N.; Meyer, A.S. The problems of using one-dimensional methods to evaluate multifunctional food and biological antioxidants. J. Sci. Food Agric. 2000, 80, 1925–1941. [Google Scholar] [CrossRef]
- Paiva-Martins, F.; Gordon, M.H.; Gameiro, P. Activity and location of olive phenolic antioxidants. Chem. Phys. Lipids 2003, 124, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Nenadis, N.; Tsimidou, M. Observations on the estimation of scavenging activity of phenolic compounds using rapid 1, 1-diphenyl-2-picrylhydrazyl (DPPH•) tests. J. Am. Oil Chem. Soc. 2002, 79, 1191–1195. [Google Scholar] [CrossRef]
- Gordon, M.H.; Paiva-Martins, F.; Almeida, M. Antioxidant activity of hydroxytyrosol acetate compared with that of other olive oil polyphenols. J. Agric. Food Chem. 2001, 49, 2480–2485. [Google Scholar] [CrossRef]
- Owen, R.W.; Giacosa, A.; Hull, W.E.; Haubner, R.; Spiegelhalder, B.; Bartsch, H. The antioxidant/anticancer potential of phenolic compounds isolated from olive oil. Eur. J. Cancer 2000, 36, 1235–1247. [Google Scholar] [CrossRef]
- Tuck, K.L.; Hayball, P.J.; Stupans, I. Structural characterization of the metabolites of hydroxytyrosol, the principal phenolic component in olive oil, in rats. J. Agric. Food Chem. 2002, 50, 2404–2409. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Wang, L.-F. Are allylic hydrogens in catechins more abstractable than catecholic hydrogens? J. Am. Oil Chem. Soc. 2002, 79, 943–944. [Google Scholar] [CrossRef]
- Boulebd, H. DFT Study of the antiradical properties of some aromatic compounds derived from antioxidant essential oils: C–H bond vs. O–H bond. Free Radic. Res. 2019, 53, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Nenadis, N.; Papapostolou, M.; Tsimidou, M.Z. Suggestions on the contribution of methyl eugenol and eugenol to bay laurel (Laurus nobilis L.) essential oil preservative activity through radical scavenging. Molecules 2021, 26, 2342. [Google Scholar] [CrossRef]
- Chimi, H.; Cillard, J.; Cillard, P.; Rahmani, M. Peroxyl and hydroxyl radical scavenging activity of some natural phenolic antioxidants. J. Am. Oil Chem. Soc. 1991, 68, 307–312. [Google Scholar] [CrossRef]
- Czerwińska, M.; Kiss, A.K.; Naruszewicz, M. A comparison of antioxidant activities of oleuropein and its dialdehydic derivative from olive oil, oleacein. Food Chem. 2012, 131, 940–947. [Google Scholar] [CrossRef]
- GROMACS. Available online: https://www.gromacs.org/ (accessed on 8 December 2022).
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-J.; Liu, L.; Fu, Y.; Guo, Q.-X. Development of an ONIOM-G3B3 method to accurately predict C−H and N−H bond dissociation enthalpies of ribonucleosides and deoxyribonucleosides. J. Phys. Chem. B 2005, 109, 13818–13826. [Google Scholar] [CrossRef] [PubMed]
- Saija, A.; Trombetta, D.; Tomaino, A.; Cascio, R.L.; Princi, P.; Uccella, N.; Castelli, F. In vitro evaluation of the antioxidant activity and biomembrane interaction of the plant phenols oleuropein and hydroxytyrosol. Int. J. Pharm. 1998, 166, 123–133. [Google Scholar] [CrossRef]
- Guiso, M.; Marra, C.; Arcos, R.R. An investigation on dihydroxy-isochromans in extra virgin olive oil. Nat. Prod. Res. 2008, 22, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Mateos, R.; Madrona, A.; Pereira-Caro, G.; Domínguez, V.; Cert, R.M.; Parrado, J.; Sarriá, B.; Bravo, L.; Espartero, J.L. Synthesis and antioxidant evaluation of isochroman-derivatives of hydroxytyrosol: Structure–activity relationship. Food Chem. 2015, 173, 313–320. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Index | Formula * |
|---|---|
| Bond dissociation enthalpy (BDE) | Hr + Hh − Hp |
| Adiabatic ionization potential (IP) | Ecr − Ep |
| Proton dissociation enthalpy (PDE) | Hr + Hpr − Hcr |
| Proton affinity (PA) | Ha + Hpr − Hp |
| Electron transfer energy (ETE) ** | Er − Ea |
| Structure and Numbering | Pentyl Ethanoate | Water |
|---|---|---|
 hydroxytyrosol | HAT(1a) = 61.53 | HAT(1a) = 49.69 |
| HAT(2a) = 37.69 | HAT(2a) = 49.69 | |
| HAT(7) = 0.06 | HAT(7) = 0.03 | |
| HAT(8) = 0.14 | HAT(8) = 0.10 | |
| RAF*(1) = 0.55 | RAF(1) = 0.20 | |
| RAF(2) = 0.00 | RAF(2) = 0.12 | |
| RAF(3) = 0.00 | RAF(3) = 0.01 | |
| RAF(4) = 0.03 | RAF(4) = 0.16 | |
| RAF(5) = 0.00 | RAF(5) = 0.01 | |
| RAF(6) = 0.00 | RAF(6) = 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nenadis, N.; Pyrka, I.; Tsimidou, M.Z. The Contribution of Theoretical Prediction Studies to the Antioxidant Activity Assessment of the Bioactive Secoiridoids Encountered in Olive Tree Products and By-Products. Molecules 2023, 28, 2267. https://doi.org/10.3390/molecules28052267
Nenadis N, Pyrka I, Tsimidou MZ. The Contribution of Theoretical Prediction Studies to the Antioxidant Activity Assessment of the Bioactive Secoiridoids Encountered in Olive Tree Products and By-Products. Molecules. 2023; 28(5):2267. https://doi.org/10.3390/molecules28052267
Chicago/Turabian StyleNenadis, Nikolaos, Ioanna Pyrka, and Maria Z. Tsimidou. 2023. "The Contribution of Theoretical Prediction Studies to the Antioxidant Activity Assessment of the Bioactive Secoiridoids Encountered in Olive Tree Products and By-Products" Molecules 28, no. 5: 2267. https://doi.org/10.3390/molecules28052267
APA StyleNenadis, N., Pyrka, I., & Tsimidou, M. Z. (2023). The Contribution of Theoretical Prediction Studies to the Antioxidant Activity Assessment of the Bioactive Secoiridoids Encountered in Olive Tree Products and By-Products. Molecules, 28(5), 2267. https://doi.org/10.3390/molecules28052267