
The Resonance Raman Spectrum of Cytosine in Water: Analysis of the Effect of Specific Solute–Solvent Interactions and Non-Adiabatic Couplings
 , , and
, , and 
Abstract
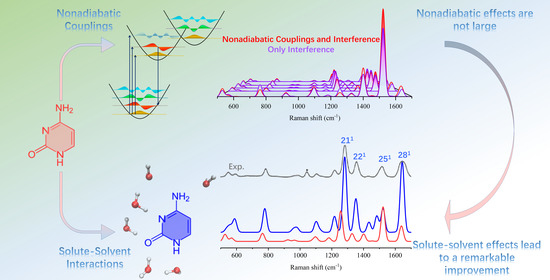
1. Introduction
2. Results
2.1. Absorption Spectra
2.2. Vibrational Resonance Raman Spectra in the Excitation Range Measured in the Experiments
2.2.1. Comparison with Experiments
2.2.2. Effect of Explicit Solvent on vRR
2.2.3. Further Analysis of the Modes Involved in the Most Intense Transitions
2.3. Raman Excitation Profiles
3. Conclusions
4. Materials and Methods
4.1. Theory
4.2. Analytical Correlation Functions for Vibrational Resonant Raman Spectroscopy for “Single-State” Harmonic Systems
4.3. Numerical Computation of the Correlation Functions for Coupled States
4.4. Computation Details
- LVC—included both the effects of the inter-state couplings and of the interference of the different states; computations were performed with Overdia (LVC parameterization) and Quantics (QD), plus local scripts, to combine the correlation functions;
- VGInt —neglected the effects of inter-state couplings, but considered the interference of the different states, i.e., a sum was taken in Equation (1), over all the relevant states m (); computations were performed with FCclasses (in Section S4 of the Supplementary Materials, we show that ML-MCTDH computations with Quantics delivered identical spectra);
- VSum —neglected both the effects of inter-state couplings and interferences; the vRR intensity was thus simply the sum of what was obtained by repeating the computation n times, considering only one ES state m in Equation (1) (); afterwards, the vRR intensities in Equation (3) were summed for all relevant states; computations were performed with FCclasses.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Albrecht, A.C. On the theory of Raman intensities. J. Chem. Phys. 1961, 34, 1476–1484. [Google Scholar] [CrossRef]
- Long, D.A.; Long, D. The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; Wiley Chichester: Chichester, UK, 2002; Volume 8. [Google Scholar]
- Myers, A.B. Resonance Raman intensities and charge-transfer reorganization energies. Chem. Rev. 1996, 96, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Dauber, P. Calculations of resonance Raman spectra of conjugated molecules. J. Chem. Phys. 1977, 66, 5477–5488. [Google Scholar] [CrossRef]
- Lee, S.; Heller, E.J. Time-dependent theory of Raman scattering. J. Chem. Phys. 1979, 71, 4777–4788. [Google Scholar] [CrossRef]
- Owens, N.A.; Pinter, A.; Porter, M.D. Surface-enhanced resonance Raman scattering for the sensitive detection of a tuberculosis biomarker in human serum. J. Raman Spectrosc. 2019, 50, 15–25. [Google Scholar] [CrossRef]
- Li, C.; Ahmed, T.; Ma, M.; Edvinsson, T.; Zhu, J. A facile approach to ZnO/CdS nanoarrays and their photocatalytic and photoelectrochemical properties. Appl. Catal. B Environ. 2013, 138, 175–183. [Google Scholar] [CrossRef]
- Derayea, S.M.; Tsujino, H.; Oyama, Y.; Ishikawa, Y.; Yamashita, T.; Uno, T. Investigation on drug-binding in heme pocket of CYP2C19 with UV–visible and resonance Raman spectroscopies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 209, 209–216. [Google Scholar] [CrossRef]
- Gao, Y.; Martin, T.P.; Thomas, A.K.; Grey, J.K. Resonance raman spectroscopic-and photocurrent imaging of polythiophene/fullerene solar cells. J. Phys. Chem. Lett. 2010, 1, 178–182. [Google Scholar] [CrossRef]
- Zedler, L.; Guthmuller, J.; de Moraes, I.R.; Kupfer, S.; Krieck, S.; Schmitt, M.; Popp, J.; Rau, S.; Dietzek, B. Resonance-Raman spectro-electrochemistry of intermediates in molecular artificial photosynthesis of bimetallic complexes. Chem. Commun. 2014, 50, 5227–5229. [Google Scholar] [CrossRef]
- Faust, A.; Amit, Y.; Banin, U. Phonon–Plasmon Coupling and Active Cu Dopants in Indium Arsenide Nanocrystals Studied by Resonance Raman Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 2519–2525. [Google Scholar] [CrossRef]
- Friebel, D.; Louie, M.W.; Bajdich, M.; Sanwald, K.E.; Cai, Y.; Wise, A.M.; Cheng, M.J.; Sokaras, D.; Weng, T.C.; Alonso-Mori, R.; et al. Identification of highly active Fe sites in (Ni, Fe) OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 2015, 137, 1305–1313. [Google Scholar] [CrossRef]
- Heller, E.J.; Sundberg, R.; Tannor, D. Simple aspects of Raman scattering. J. Phys. Chem. 1982, 86, 1822–1833. [Google Scholar] [CrossRef]
- Tannor, D.J.; Heller, E.J. Polyatomic Raman scattering for general harmonic potentials. J. Chem. Phys. 1982, 77, 202–218. [Google Scholar] [CrossRef]
- Tonks, D.L.; Page, J.B. Diagrammatic theory of temperature-dependent resonance Raman scattering from polyatomic systems with general harmonic potential surfaces. J. Chem. Phys. 1988, 88, 738–760. [Google Scholar] [CrossRef]
- Lu, H.M.; Page, J.B. General transform technique including mode mixing and non-Condon coupling in resonance Raman scattering. J. Chem. Phys. 1989, 90, 5315–5326. [Google Scholar] [CrossRef]
- Santoro, F.; Cappelli, C.; Barone, V. Effective Time-Independent Calculations of Vibrational Resonance Raman Spectra of Isolated and Solvated Molecules Including Duschinsky and Herzberg–Teller Effects. J. Chem. Theory Comput. 2011, 7, 1824–1839. [Google Scholar] [CrossRef]
- Ma, H.; Liu, J.; Liang, W. Time-Dependent Approach to Resonance Raman Spectra Including Duschinsky Rotation and Herzberg–Teller Effects: Formalism and Its Realistic Applications. J. Chem. Theory Comput. 2012, 8, 4474–4482. [Google Scholar] [CrossRef]
- Huh, J.; Berger, R. Coherent state-based generating function approach for Franck-Condon transitions and beyond. J. Phys. Conf. Ser. 2012, 380, 012019. [Google Scholar] [CrossRef]
- Baiardi, A.; Bloino, J.; Barone, V. A general time-dependent route to Resonance-Raman spectroscopy including Franck-Condon, Herzberg-Teller and Duschinsky effects. J. Chem. Phys. 2014, 141, 114108. [Google Scholar] [CrossRef]
- Baiardi, A.; Bloino, J.; Barone, V. Accurate simulation of resonance-Raman spectra of flexible molecules: An internal coordinates approach. J. Chem. Theory Comput. 2015, 11, 3267–3280. [Google Scholar] [CrossRef]
- de Souza, B.; Farias, G.; Neese, F.; Izsák, R. Efficient simulation of overtones and combination bands in resonant Raman spectra. J. Chem. Phys. 2019, 150, 214102. [Google Scholar] [CrossRef]
- Cerezo, J.; Santoro, F. FCclasses3: Vibrationally-resolved spectra simulated at the edge of the harmonic approximation. J. Comput. Chem. 2023, 44, 626–643. [Google Scholar] [CrossRef]
- Avila Ferrer, F.J.; Barone, V.; Cappelli, C.; Santoro, F. Duschinsky, Herzberg–Teller, and Multiple Electronic Resonance Interferential Effects in Resonance Raman Spectra and Excitation Profiles. The Case of Pyrene. J. Chem. Theory Comput. 2013, 9, 3597–3611. [Google Scholar] [CrossRef]
- Santoro, F.; Cerezo, J. FCclasses3, A Code for Vibronic Calculations, 2022. Available online: http://www.iccom.cnr.it/en/fcclasses (accessed on 1 February 2023).
- Stock, G.; Woywod, C.; Domcke, W.; Swinney, T.; Hudson, B.S. Resonance Raman spectroscopy of the S1 and S2 states of pyrazine: Experiment and first principles calculation of spectra. J. Chem. Phys. 1995, 103, 6851–6860. [Google Scholar] [CrossRef]
- Xu, Q.; Aranda, D.; Yaghoubi Jouybari, M.; Liu, Y.; Wang, M.; Cerezo, J.; Improta, R.; Santoro, F. Nonadiabatic Vibrational Resonance Raman Spectra from Quantum Dynamics Propagations with LVC Models. Application to Thymine. J. Phys. Chem. A 2022, 126, 7468–7479. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Thoss, M. Multilayer formulation of the multiconfiguration time-dependent Hartree theory. J. Chem. Phys. 2003, 119, 1289–1299. [Google Scholar] [CrossRef]
- Manthe, U. Layered discrete variable representations and their application within the multiconfigurational time-dependent Hartree approach. J. Chem. Phys. 2009, 130, 054109. [Google Scholar] [CrossRef]
- Vendrell, O.; Meyer, H.D. Multilayer multiconfiguration time-dependent Hartree method: Implementation and applications to a Henon–Heiles Hamiltonian and to pyrazine. J. Chem. Phys. 2011, 134, 044135. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi Jouybari, M.; Liu, Y.; Improta, R.; Santoro, F. Ultrafast dynamics of the two lowest bright excited states of cytosine and 1-methylcytosine: A quantum dynamical study. J. Chem. Theory Comput. 2020, 16, 5792–5808. [Google Scholar] [CrossRef] [PubMed]
- Green, J.A.; Yaghoubi Jouybari, M.; Asha, H.; Santoro, F.; Improta, R. Fragment Diabatization Linear Vibronic Coupling Model for Quantum Dynamics of Multichromophoric Systems: Population of the Charge-Transfer State in the Photoexcited Guanine–Cytosine Pair. J. Chem. Theory Comput. 2021, 17, 4660–4674. [Google Scholar] [CrossRef]
- Köppel, H.; Domcke, W.; Cederbaum, L. Multimode molecular dynamics beyond the Born-Oppenheimer approximation. Adv. Chem. Phys. 1984, 57, 140. [Google Scholar]
- Köppel, H.; Domcke, W.; Cederbaum, L. The multimode vibronic-coupling approach. In Conical Intersections: Electronic Structure, Dynamics and Spectroscopy; Domcke, W., Yarkony, D., Köppel, H., Eds.; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2004; Chapter 7; pp. 323–367. [Google Scholar]
- Billinghurst, B.E.; Loppnow, G.R. Excited-state structural dynamics of cytosine from resonance Raman spectroscopy. J. Phys. Chem. A 2006, 110, 2353–2359. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Douki, T. DNA Photodamage: From Light Absorption to Cellular Responses and Skin Cancer; Royal Society of Chemistry: London, UK, 2021. [Google Scholar]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Borin, C.A.; Ullrich, S. Photoinduced Phenomena in Nucleic Acids I: Nucleobases in the Gas Phase and in Solvents; Springer International Publishing: Cham, Switzerland, 2015; Volume 355, pp. 1–358. [Google Scholar]
- Martínez Fernández, L.; Santoro, F.; Improta, R. Nucleic Acids as a Playground for the Computational Study of the Photophysics and Photochemistry of Multichromophore Assemblies. Acc. Chem. Res. 2022, 55, 2077–2087. [Google Scholar] [CrossRef]
- Blazej, D.C.; Peticolas, W.L. Ultraviolet Resonant Raman Spectroscopy of Nucleic Acid Components. Proc. Natl. Acad. Sci. USA 1977, 74, 2639. [Google Scholar] [CrossRef] [PubMed]
- Fodor, S.P.A.; Rava, R.P.; Hays, T.R.; Spiro, T.G. Ultraviolet resonance Raman spectroscopy of the nucleotides with 266-, 240-, 218-, and 200-nm pulsed laser excitation. J. Am. Chem. Soc. 1985, 107, 1520–1529. [Google Scholar] [CrossRef]
- Mondal, S.; Narayana, C. Role of Explicit Solvation in the Simulation of Resonance Raman Spectra within Short-Time Dynamics Approximation. J. Phys. Chem. B 2019, 123, 8800–8813. [Google Scholar] [CrossRef]
- Billinghurst, B.E.; Oladepo, S.A.; Loppnow, G.R. Initial Excited-State Structural Dynamics of Thymine Derivatives. J. Phys. Chem. B 2012, 116, 10496. [Google Scholar] [CrossRef]
- Rappoport, D.; Shim, S.; Aspuru-Guzik, A. Simplified Sum-Over-States Approach for Predicting Resonance Raman Spectra. Application to Nucleic Acid Bases. J. Phys. Chem. Lett. 2011, 2, 1254. [Google Scholar] [CrossRef]
- Burova, T.G.; Ermolenkov, V.V.; Ten, G.N.; Shcherbakov, R.S.; Baranov, V.I.; Lednev, I.K. Raman spectroscopic study of the tautomeric composition of adenine in water. J. Phys. Chem. A 2011, 115, 10600–10609. [Google Scholar] [CrossRef]
- Sasidharanpillai, S.; Dempster, S.P.; Loppnow, G.R. Initial Excited-State Structural Dynamics of Uridine from Resonance Raman Spectroscopy. J. Raman Spectrosc. 2018, 49, 1487. [Google Scholar] [CrossRef]
- Benevides, J.M.; Overman, S.A.; Thomas, G.J. Raman, Polarized Raman and Ultraviolet Resonance Raman Spectroscopy of Nucleic Acids and Their Complexes. J. Raman Spectrosc. 2005, 36, 279. [Google Scholar] [CrossRef]
- Sasidharanpillai, S.; Loppnow, G.R. Initial Excited-State Structural Dynamics of dT and dA Oligonucleotide Homopentamers Using Resonance Raman Spectroscopy. J. Phys. Chem. B 2019, 123, 3898–3906. [Google Scholar] [CrossRef]
- Di Fonzo, S.; Bottari, C.; Brady, J.W.; Tavagnacco, L.; Caterino, M.; Petraccone, L.; Amato, J.; Giancola, C.; Cesàro, A. Crowding and conformation interplay on human DNA G-quadruplex by ultraviolet resonant Raman scattering. Phys. Chem. Chem. Phys. 2019, 21, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Di Fonzo, S.; Amato, J.; D’Aria, F.; Caterino, M.; D’Amico, F.; Gessini, A.; Brady, J.W.; Cesàro, A.; Pagano, B.; Giancola, C. Ligand binding to G-quadruplex DNA: New insights from ultraviolet resonance Raman spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 8128–8140. [Google Scholar] [CrossRef]
- Amato, J.; Iaccarino, N.; D’Aria, F.; D’Amico, F.; Randazzo, A.; Giancola, C.; Cesàro, A.; Di Fonzo, S.; Pagano, B. Conformational plasticity of DNA secondary structures: Probing the conversion between i-motif and hairpin species by circular dichroism and ultraviolet resonance Raman spectroscopies. Phys. Chem. Chem. Phys. 2022, 24, 7028–7044. [Google Scholar] [CrossRef]
- Moraca, F.; Marzano, S.; D’Amico, F.; Lupia, A.; Di Fonzo, S.; Vertecchi, E.; Salvati, E.; Di Porzio, A.; Catalanotti, B.; Randazzo, A.; et al. Ligand-based drug repurposing strategy identified SARS-CoV-2 RNA G-quadruplex binders. Chem. Commun. 2022, 58, 11913–11916. [Google Scholar] [CrossRef]
- Gómez, S.; Lafiosca, P.; Egidi, F.; Giovannini, T.; Cappelli, C. UV-Resonance Raman Spectra of Systems in Complex Environments: A Multiscale Modeling Applied to Doxorubicin Intercalated into DNA. J. Chem. Inf. Model. 2023, 0, null. [Google Scholar] [CrossRef]
- Fahleson, T.; Kauczor, J.; Norman, P.; Santoro, F.; Improta, R.; Coriani, S. TD-DFT Investigation of the Magnetic Circular Dichroism Spectra of Some Purine and Pyrimidine Bases of Nucleic Acids. J. Phys. Chem. A 2015, 119, 5476–5489. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Pepino, A.J.; Segarra-Martí, J.; Jovaišaitė, J.; Vaya, I.; Nenov, A.; Markovitsi, D.; Gustavsson, T.; Banyasz, A.; Garavelli, M.; et al. Photophysics of Deoxycytidine and 5-Methyldeoxycytidine in Solution: A Comprehensive Picture by Quantum Mechanical Calculations and Femtosecond Fluorescence Spectroscopy. J. Am. Chem. Soc. 2017, 139, 7780–7791. [Google Scholar] [CrossRef] [PubMed]
- Avila Ferrer, F.J.; Santoro, F.; Improta, R. The excited state behavior of cytosine in the gas phase: A TD-DFT study. Computat. Theor. Chem. 2014, 1040–1041, 186–194. [Google Scholar] [CrossRef]
- Zaloudek, F.; Novros, J.S.; Clark, L.B. The electronic spectrum of cytosine. J. Am. Chem. Soc. 1985, 107, 7344–7351. [Google Scholar] [CrossRef]
- Gómez, S.; Egidi, F.; Puglisi, A.; Giovannini, T.; Rossi, B.; Cappelli, C. Unlocking the power of resonance Raman spectroscopy: The case of amides in aqueous solution. J. Mol. Liq. 2022, 346, 117841. [Google Scholar] [CrossRef]
- Gómez, S.; Bottari, C.; Egidi, F.; Giovannini, T.; Rossi, B.; Cappelli, C. Amide Spectral Fingerprints are Hydrogen Bonding-Mediated. J. Phys. Chem. Lett. 2022, 13, 6200–6207. [Google Scholar] [CrossRef]
- Gómez, S.; Rojas-Valencia, N.; Giovannini, T.; Restrepo, A.; Cappelli, C. Ring Vibrations to Sense Anionic Ibuprofen in Aqueous Solution as Revealed by Resonance Raman. Molecules 2022, 27, 442. [Google Scholar] [CrossRef]
- Cerezo, J.; Aranda, D.; Avila Ferrer, F.J.; Prampolini, G.; Santoro, F. Adiabatic-Molecular Dynamics Generalized Vertical Hessian Approach: A Mixed Quantum Classical Method To Compute Electronic Spectra of Flexible Molecules in the Condensed Phase. J. Chem. Theory Comput. 2020, 16, 1215–1231. [Google Scholar] [CrossRef]
- Segalina, A.; Aranda, D.; Green, J.A.; Cristino, V.; Caramori, S.; Prampolini, G.; Pastore, M.; Santoro, F. How the Interplay among Conformational Disorder, Solvation, Local, and Charge-Transfer Excitations Affects the Absorption Spectrum and Photoinduced Dynamics of Perylene Diimide Dimers: A Molecular Dynamics/Quantum Vibronic Approach. J. Chem. Theory Comput. 2022, 18, 3718–3736. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, G.; Teller, E. Fluctuation structure of electron transfer in multiatomic molecules. Z. Phys. Chem. B 1933, 21, 410. [Google Scholar] [CrossRef]
- Aranda, D.; Santoro, F. Vibronic spectra of π-conjugated systems with a multitude of coupled states: A protocol based on linear vibronic coupling models and quantum dynamics tested on hexahelicene. J. Chem. Theory Comput. 2021, 17, 1691–1700. [Google Scholar] [CrossRef]
- Liu, Y.; Aranda, D.; Santoro, F. A computational study of the vibronic effects on the electronic spectra and the photophysics of aza [7] helicene. Phys. Chem. Chem. Phys. 2021, 23, 16551–16563. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jacquemin, D.; Wathelet, V.; Perpete, E.A.; Adamo, C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef] [PubMed]
- Charaf-Eddin, A.; Planchat, A.; Mennucci, B.; Adamo, C.; Jacquemin, D. Choosing a Functional for Computing Absorption and Fluorescence Band Shapes with TD-DFT. J. Chem. Theory Comput. 2013, 9, 2749–2760. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [CrossRef]
- Peach, M.J.G.; Benfield, P.; Helgaker, T.; Tozer, D.J. Excitation energies in density functional theory: An evaluation and a diagnostic test. J. Chem. Phys. 2008, 128, 044118. [Google Scholar] [CrossRef] [PubMed]
- Worth, G.A.; Giri, K.; Richings, G.W.; Beck, M.H.; Jäckle, A.; Meyer, H.D. The QUANTICS Package; University of Birmingham: Birmingham, UK, 2015; Version 1.1. [Google Scholar]
- Worth, G. Quantics: A general purpose package for Quantum molecular dynamics simulations. Comput. Phys. Commun. 2020, 248, 107040. [Google Scholar] [CrossRef]
- Santoro, F.; Green, J.A. Overdia 01, a Fortran 90 Code for Parametrization of Model Hamiltonians Based on a Maximum-Overlap Diabatisation, 2022. Available online: http://www.iccom.cnr.it/en/overdia-en (accessed on 1 February 2023).
- Ferrer, F.J.A.; Santoro, F. Comparison of vertical and adiabatic harmonic approaches for the calculation of the vibrational structure of electronic spectra. Phys. Chem. Chem. Phys. 2012, 14, 13549–13563. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Time-dependent density functional theory for molecules in liquid solutions. J. Chem. Phys. 2001, 115, 4708–4717. [Google Scholar] [CrossRef]
- Aikens, C.M.; Gordon, M.S. Incremental Solvation of Nonionized and Zwitterionic Glycine. J. Am. Chem. Soc. 2006, 128, 12835–12850. [Google Scholar] [CrossRef] [PubMed]
- HongWei, K.; Li, R.; Xin, X.; YiJing, Y. Density functional theory study of 1:1 glycine-water complexes in the gas phase and in solution. Sci. China Chem. 2010, 53, 383–395. [Google Scholar]
- Martínez-Fernández, L.; Pepino, A.J.; Segarra-Martí, J.; Banyasz, A.; Garavelli, M.; Improta, R. Computing the Absorption and Emission Spectra of 5-Methylcytidine in Different Solvents: A Test-Case for Different Solvation Models. J. Chem. Theory Comput. 2016, 12, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Petrone, A.; Cerezo, J.; Ferrer, F.J.A.; Donati, G.; Improta, R.; Rega, N.; Santoro, F. Absorption and Emission Spectral Shapes of a Prototype Dye in Water by Combining Classical/Dynamical and Quantum/Static Approaches. J. Phys. Chem. A 2015, 119, 5426–5438. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, T.; Egidi, F.; Cappelli, C. Molecular spectroscopy of aqueous solutions: A theoretical perspective. Chem. Soc. Rev. 2020, 49, 5664–5677. [Google Scholar] [CrossRef]
- Bazsó, G.; Tarczay, G.; Fogarasi, G.; Szalay, P.G. Tautomers of cytosine and their excited electronic states: A matrix isolation spectroscopic and quantum chemical study. Phys. Chem. Phys. 2011, 13, 6799–6807. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STATE | CAM-B3LYP | |||
| Sym. | Egf(eV) | δOPA | Descr. | |
| S () | A′ | 5.15 | 0.12 | |
| S (n) | A″ | 5.70 | 0.0028 | n |
| S () | A′ | 5.94 | 0.21 | |
| S () | A″ | 6.25 | 0.0054 | |
| S (n) | A″ | 6.30 | 0.0007 | n+n + |
| S () | A′ | 6.48 | 0.38 | |
| S (n) | A″ | 6.57 | 0.0002 | n-n |
| S () | A″ | 6.71 | 0.0038 | |
| S () | A′ | 6.89 | 0.40 | |
| STATE | PBE0 | |||
| Sym. | Egf(eV) | δOPA | Descr. | |
| S () | A′ | 4.98 | 0.092 | |
| S (n) | A″ | 5.44 | 0.0025 | n +n |
| S () | A′ | 5.65 | 0.15 | |
| S (n) | A″ | 5.78 | 0.0002 | n-n |
| S () | A″ | 6.13 | 0.0053 | |
| S (n) | A″ | 6.17 | 0.0001 | n |
| S () | A′ | 6.30 | 0.21 | |
| S () | A″ | 6.62 | 0.0028 | |
| S (n) | A″ | 6.66 | 0.0025 | n |
| STATE | CAM-B3LYP | ||
| Egf(eV) | δOPA | Char. | |
| S () | 5.25 | 0.19 | |
| S () | 5.98 | 0.15 | |
| S (n) | 6.07 | 0.028 | n +n |
| S () | 6.31 | 0.27 | |
| S () | 6.47 | 0.0054 | |
| S (n) | 6.71 | 0.0043 | n +n |
| S () | 6.84 | 0.54 | |
| S () | 6.96 | 0.0075 | |
| S (n) | 6.97 | 0.025 | n -n |
| STATE | PBE0 | ||
| Egf(eV) | δOPA | Char. | |
| S () | 5.11 | 0.15 | |
| S () | 5.69 | 0.11 | |
| S (n) | 5.78 | 0.022 | n +n |
| S () | 6.08 | 0.16 | |
| S () | 6.22 | 0.0028 | |
| S (n) | 6.26 | 0.0001 | n +n |
| S (n) | 6.48 | 0.0013 | n +n |
| S (CT) | 6.54 | 0.0048 | CT |
| S () | 6.64 | 0.0054 | |
| Mode | CAM-B3LYP | |||||
| Cytosine | Cytosine·6H2O | |||||
| ω | Δ1 | Δ2 | ω | Δ1 | Δ2 | |
| 21 | 1256 | 0.98 | 0.27 | 1281 | −0.92 | −0.42 |
| 22 | 1327 | −0.43 | 0.13 | 1351 | 0.54 | −0.15 |
| 23 | 1400 | −0.43 | −0.80 | 1433 | −0.33 | −0.49 |
| 24 | 1454 | 0.09 | −0.07 | 1483 | 0.41 | 0.69 |
| 25 | 1523 | 0.91 | 1.39 | 1522 | 0.47 | 0.81 |
| 28 | 1639 | −0.51 | −0.20 | 1644 | −0.69 | −0.16 |
| Mode | PBE0 | |||||
| Cytosine | Cytosine·6H2O | |||||
| ω | Δ1 | Δ2 | ω | Δ1 | Δ2 | |
| 21 | 1265 | −0.71 | −0.32 | 1290 | 0.66 | 0.34 |
| 22 | 1323 | 0.40 | 0.58 | 1346 | 0.52 | 0.63 |
| 23 | 1400 | 0.47 | 0.19 | 1437 | −0.49 | −0.36 |
| 24 | 1457 | −0.23 | −0.60 | 1487 | 0.31 | 0.38 |
| 25 | 1520 | −1.00 | −0.90 | 1522 | 0.45 | 0.89 |
| 28 | 1644 | 0.60 | −0.36 | 1640 | −0.62 | 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Q.; Liu, Y.; Wang, M.; Cerezo, J.; Improta, R.; Santoro, F. The Resonance Raman Spectrum of Cytosine in Water: Analysis of the Effect of Specific Solute–Solvent Interactions and Non-Adiabatic Couplings. Molecules 2023, 28, 2286. https://doi.org/10.3390/molecules28052286
Xu Q, Liu Y, Wang M, Cerezo J, Improta R, Santoro F. The Resonance Raman Spectrum of Cytosine in Water: Analysis of the Effect of Specific Solute–Solvent Interactions and Non-Adiabatic Couplings. Molecules. 2023; 28(5):2286. https://doi.org/10.3390/molecules28052286
Chicago/Turabian StyleXu, Qiushuang, Yanli Liu, Meishan Wang, Javier Cerezo, Roberto Improta, and Fabrizio Santoro. 2023. "The Resonance Raman Spectrum of Cytosine in Water: Analysis of the Effect of Specific Solute–Solvent Interactions and Non-Adiabatic Couplings" Molecules 28, no. 5: 2286. https://doi.org/10.3390/molecules28052286
APA StyleXu, Q., Liu, Y., Wang, M., Cerezo, J., Improta, R., & Santoro, F. (2023). The Resonance Raman Spectrum of Cytosine in Water: Analysis of the Effect of Specific Solute–Solvent Interactions and Non-Adiabatic Couplings. Molecules, 28(5), 2286. https://doi.org/10.3390/molecules28052286