
An Innovative Structural Rearrangement in Imine Palladacycle Metaloligand Chemistry: From Single-Nuclear to Double-Nuclear Pseudo-Pentacoordinated Complexes

Abstract
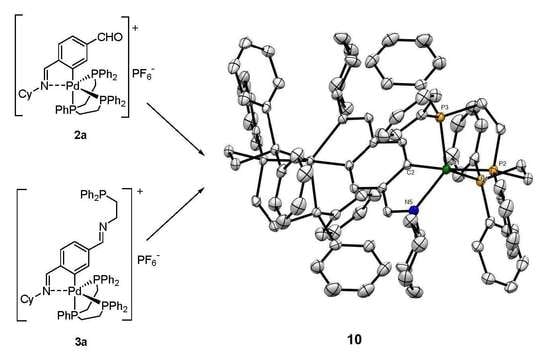
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
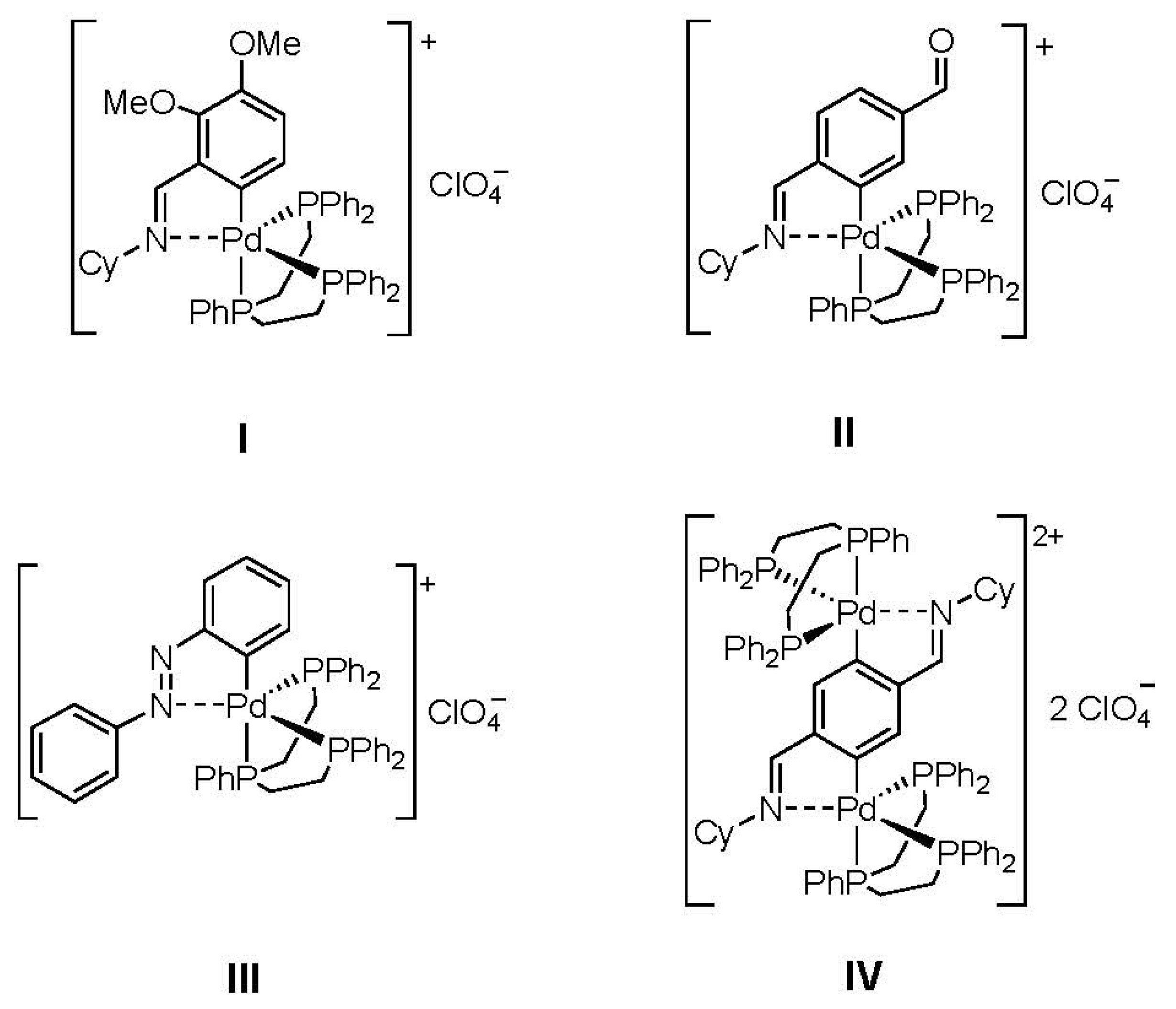
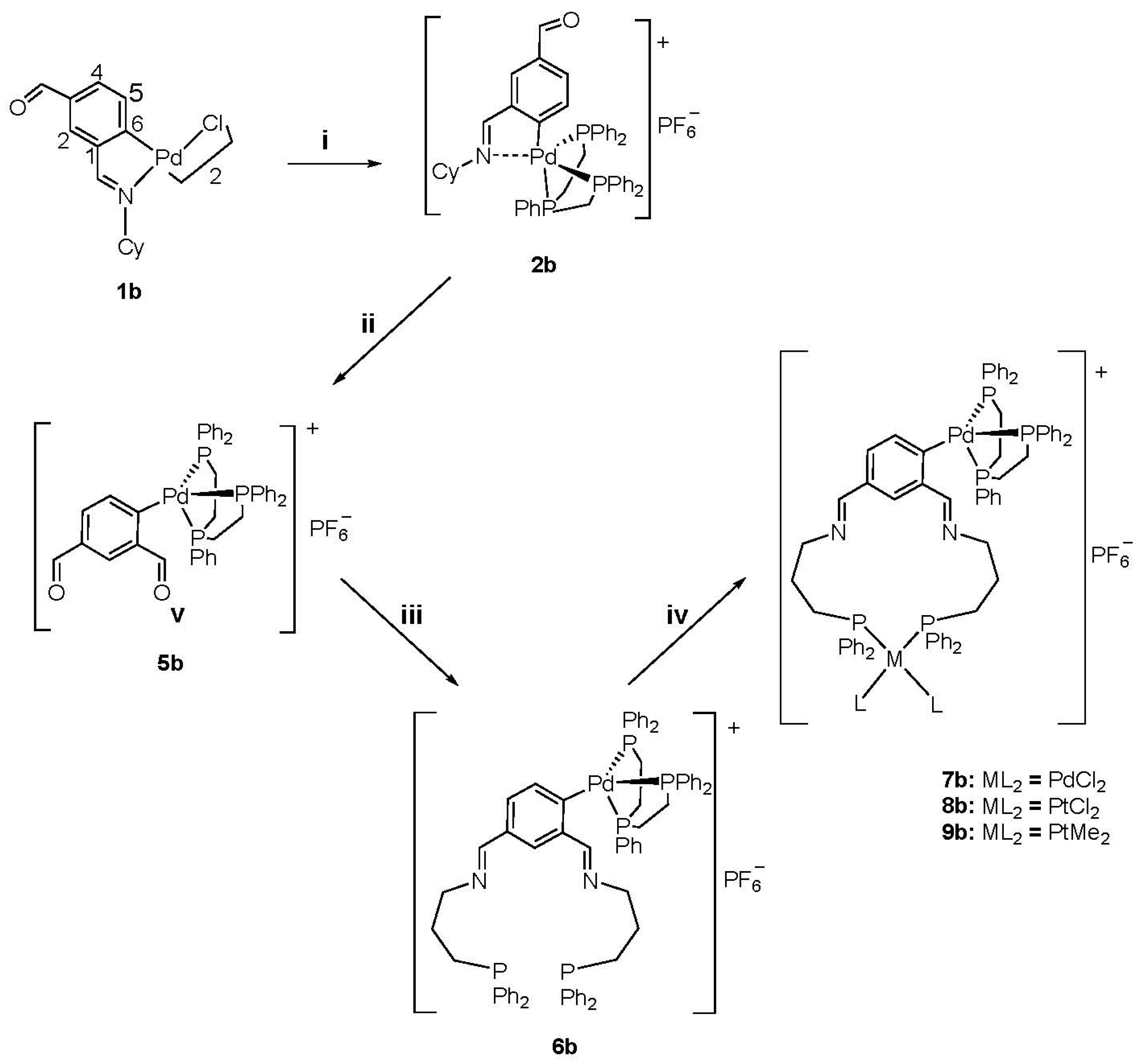
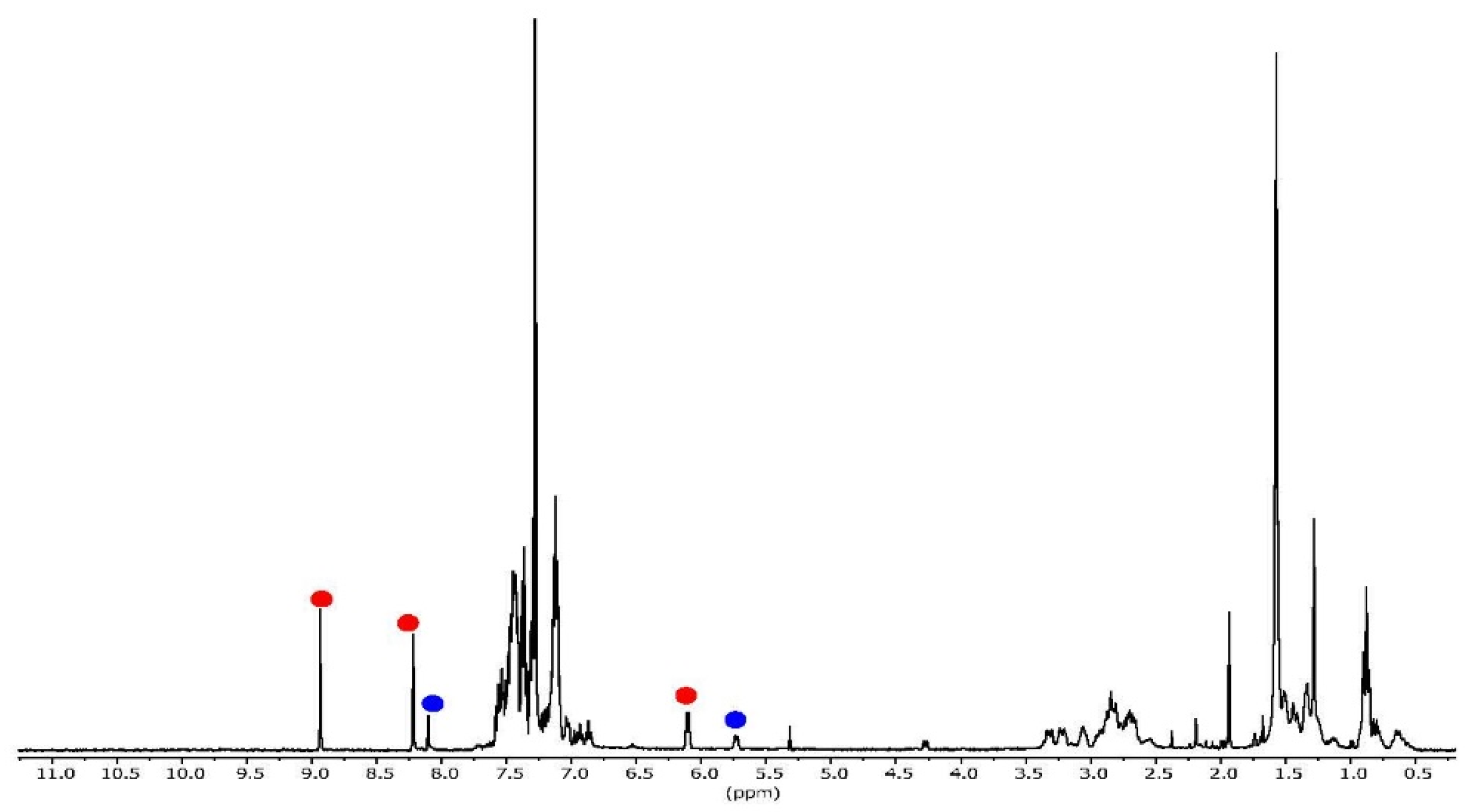
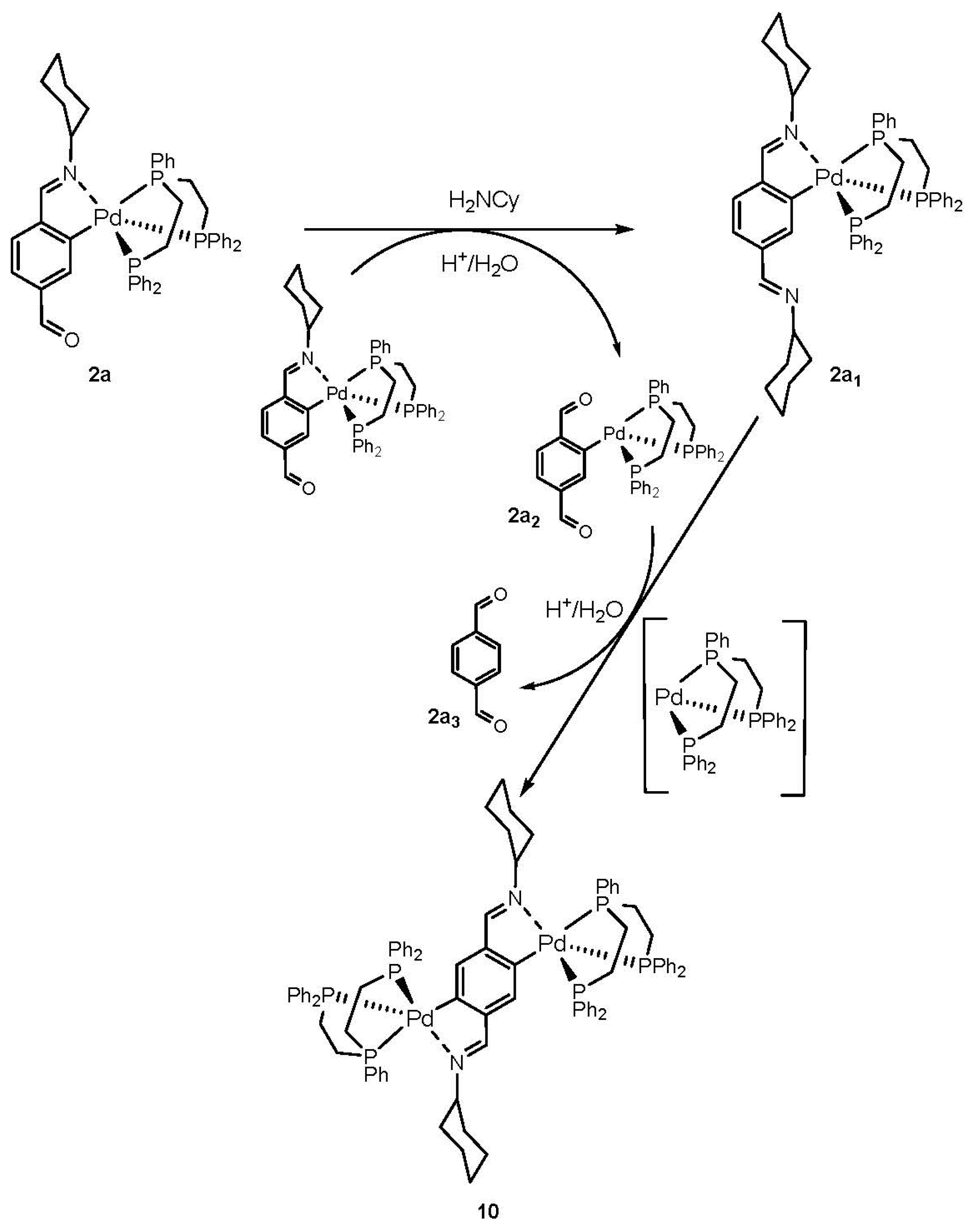
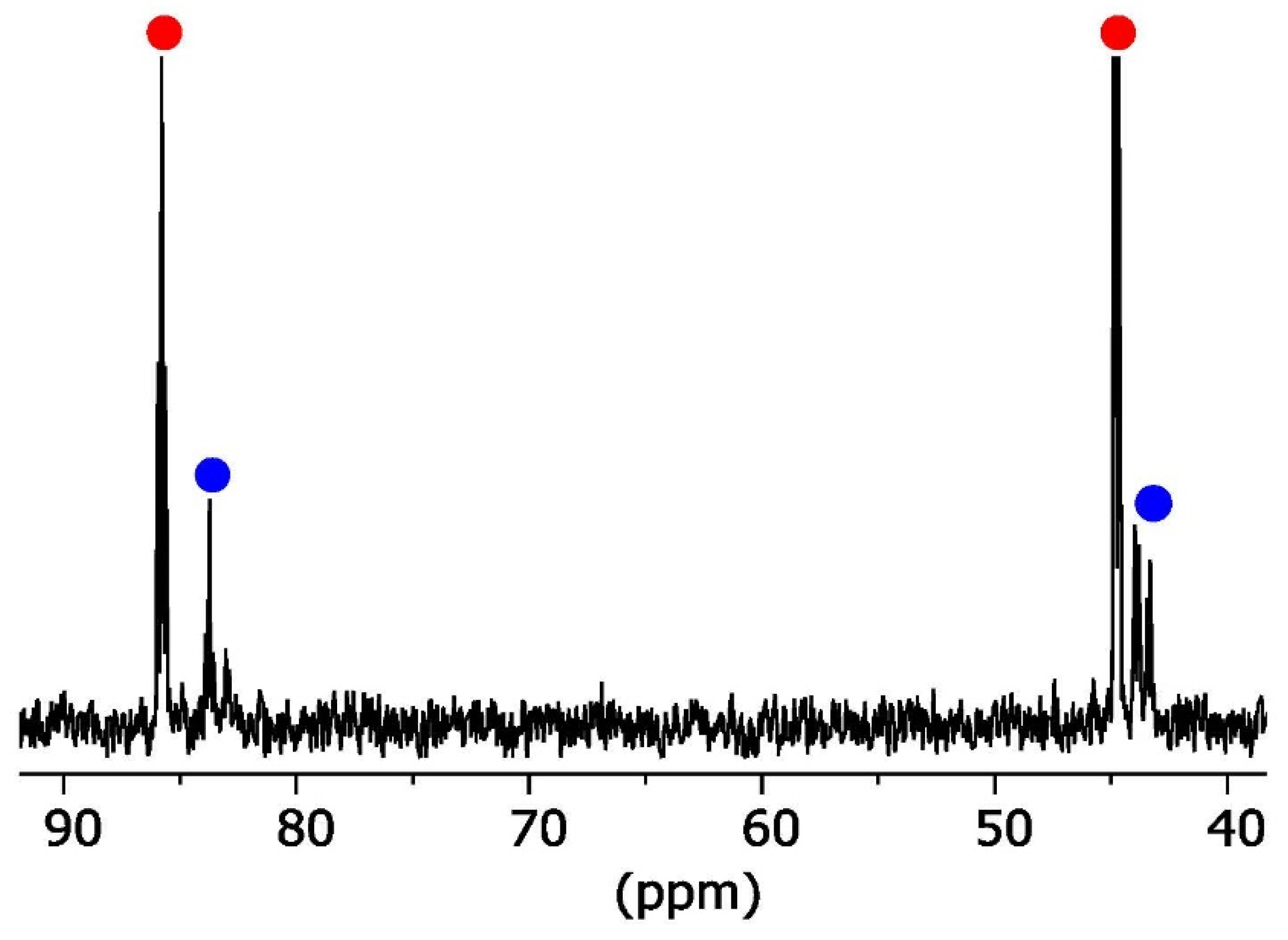
2. Results
3. Experimental Methods
3.1. General Procedures
3.2. Preparation of the Complexes
3.2.1. Synthesis of 3a
3.2.2. Synthesis of 5b
3.2.3. Synthesis of 6b
3.2.4. Synthesis of 7b
3.2.5. Synthesis of 8b
3.2.6. Synthesis of 9b
3.3. Crystal Structure Analysis and Details on Data Collection and Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Albrecht, M. Cyclometalation using d-block transition metals: Fundamental aspects and recent trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef]
- Omae, I. Cyclometalation Reactions: Five-Membered Ring Products as Universal Reagents; Springer: Tokyo, Japan, 2014. [Google Scholar]
- Vila, J.M.; Pereira, M. The Pd-C Building Block of Palladacycles: A Cornerstone for Stoichiometric C-C and C-X Bond Assemblage. In Palladacycles; Dupont, J., Pfeffer, M., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; pp. 87–108. [Google Scholar]
- Albert, J.; D’Andrea, L.; Granell, J.; Pla-Vilanova, P.; Quirante, J.; Khosa, M.K.; Calvis, C.; Messeguer, R.; Badía, J.; Baldomà, L.; et al. Cyclopalladated and cycloplatinated benzophenone imines: Antitumor, antibacterial and antioxidant activities, DNA interaction and cathepsin B inhibition. J. Inorganic Biochemistry 2014, 140, 80–88. [Google Scholar] [CrossRef]
- Ryabov, A.D. The Exchange of Cyclometalated Ligands. Molecules 2021, 26, 210. [Google Scholar] [CrossRef]
- Ilis, M.; Batalu, D.; Pasuk, I.; Cîrcu, V. Cyclometalated palladium(II) metalomesogens with Schiff bases and N-benzoyl thiourea derivatives as co-ligands. J. Mol. Liq. 2017, 233, 45–51. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, T.; Cao, Y.; Gong, G.; Zhang, Y.; Zhao, H.; Zhao, G. Synthesis and characterization of planar chiral cyclopalladated ferrocenylimines: DNA/HSA interactions and in vitro cytotoxic activity. J. Organomet. Chem. 2018, 871, 1–9. [Google Scholar] [CrossRef]
- Albert, J.; Bosque, R.; Cadena, M.; D’Andrea, L.; Granell, J.; González, A.; Quirante, J.; Calvis, C.; Messeguer, R.; Badía, J.; et al. A new family of doubly cyclopalladated diimines. A remarkable effect of the linker between the metalated units on their cytotoxicity. Organometallics 2014, 33, 2862–2873. [Google Scholar] [CrossRef]
- Ramírez-Rave, S.; Morales-Morales, D.; Grévy, J.M. Microwave assisted Suzuki-Miyaura and Mizoroki-Heck cross-couplings catalyzed by non-symmetric Pd(II) CNS pincers supported by iminophosphorane ligands. Inorg. Chim. Acta 2017, 462, 249–255. [Google Scholar] [CrossRef]
- Arthurs, R.A.; Hughes, D.L.; Richards, C.J. Planar chiral palladacycle precatalysts for asymmetric synthesis. Org. Biomol. Chem. 2020, 18, 5466–5472. [Google Scholar] [CrossRef]
- Ranieri, A.M.; Burt, L.K.; Stagni, S.; Zacchini, S.; Skelton, B.W.; Ogden, M.I.; Bissember, A.C.; Massi, M. Anionic Cyclometalated Platinum(II) Tetrazolato Complexes as Viable Photoredox Catalysts. Organometallics 2019, 38, 1108–1117. [Google Scholar] [CrossRef]
- Alonso, D.A.; Nájera, C.; Pacheco, M.C. Oxime-Derived Palladium Complexes as Very Efficient Catalysts for the Heck-Mizoroki Reaction. Adv. Synth. Catal. 2002, 344, 172–183. [Google Scholar] [CrossRef]
- Lucio-Martínez, F.; Adrio, L.A.; Polo-Ces, P.; Ortigueira, J.M.; Fernández, J.J.; Adams, H.; Pereira, M.T.; Vila, J.M. Palladacycle catalysis: An innovation to the Suzuki-Miyaura cross-coupling reaction. Dalt. Trans. 2016, 45, 17598–17601. [Google Scholar] [CrossRef]
- Pretorius, R.; McDonald, A.; da Costa, L.R.B.; Müller-Bunz, H.; Albrecht, M. Palladium(II), Rhodium(I), and Iridium(I) Complexes Containing O-Functionalized 1,2,3-Triazol-5-ylidene Ligands. Eur. J. Inorg. Chem. 2019, 2019, 4263–4272. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Brossmer, C.; Öfele, K.; Reisinger, C.-P.; Priermeier, T.; Beller, M.; Fischer, H. Palladacycles as Structurally Defined Catalysts for the Heck Olefination of Chloro- and Bromoarenes. Angew. Chem. Int. Ed. Engl. 1995, 34, 1844–1848. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Böhm, V.P.W.; Reisinger, C.P. Application of palladacycles in Heck type reactions. J. Organomet. Chem. 1999, 576, 23–41. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc. Chem. Commun. 1979, 19, 866–867. [Google Scholar] [CrossRef]
- Suzuki, A. Carbon-carbon bonding made easy. Chem. Commun. 2005, 38, 4759–4763. [Google Scholar] [CrossRef]
- Albert, J.; D’Andrea, L.; Granell, J.; Tavera, R.; Font-Bardia, M.; Solans, X. Synthesis and reactivity towards carbon monoxide of an optically active endo five-membered ortho-cyclopalladated imine: X-ray molecular structure of trans-(μ-Cl)2[Pd(κ2-C,N-(R)-C6H4-CH{double bond, long}N-CHMe-Ph)]2. J. Organomet. Chem. 2007, 692, 3070–3080. [Google Scholar] [CrossRef]
- Vila, J.M.; Gayoso, M.; Pereira, M.T.; López, M.; Alonso, G.; Fernández, J.J. Cyclometallated complexes of PdII and MnI with N,N-terephthalylidenebis(cyclohexylamine). J. Organomet. Chem. 1993, 445, 287–294. [Google Scholar] [CrossRef]
- Vila, J.M.; Gayoso, M.; Pereira, M.T.; López-Torres, M.; Fernández, J.J.; Fernández, A.; Ortigueira, J.M. Synthesis and characterization of cyclometallated complexes of palladium(II) and manganese(I) with bidentate Schiff bases. J. Organomet. Chem. 1996, 506, 165–174. [Google Scholar]
- Dickmu, G.C.; Smoliakova, I.P. Cyclopalladated complexes containing an (sp3)C–Pd bond. Coord. Chem. Rev. 2020, 409, 213203. [Google Scholar] [CrossRef]
- Thombal, R.S.; Feoktistova, T.; González-Montiel, G.A.; Cheong, P.H.Y.; Lee, Y.R. Palladium-catalyzed synthesis of β-hydroxy compounds: Via a strained 6,4-palladacycle from directed C-H activation of anilines and C-O insertion of epoxides. Chem. Sci. 2020, 11, 7260–7265. [Google Scholar] [CrossRef]
- Albert, J.; Gómez, M.; Granell, J.; Sales, J.; Soláns, X. Five- and Six-Membered Exo-Cyclopalladated Compounds of N-Benzylideneamines. Synthesis and X-ray Crystal Structure of [PdBr{p-MeOC6H3(CH2)2N=CH(2,6-Cl2C6H3)}(PPh3)] and [PdBr{p-MeOC6H3(CH2)2N=CH(2,6-Cl2C6H3(PEt3)2]. Organometallics 1990, 9, 1405–1413. [Google Scholar]
- López-Torres, M.; Fernández, A.; Fernández, J.J.; Suárez, A.; Pereira, M.T.; Ortigueira, J.M.; Vila, J.M.; Admas, H. Mono- and Dinuclear Five-coordinate Cyclometalated Palladium(II) Compounds. Inorg. Chem. 2001, 40, 4583–4587. [Google Scholar]
- Bedford, R.B.; Betham, M.; Butts, C.P.; Coles, S.J.; Cutajar, M.; Gelbrich, T.; Hursthouse, M.B.; Scullyd, P.N.; Wimperisc, S. Five-coordinate Pd(II) orthometallated triarylphosphite complexes. Dalton Trans. 2007, 459–466. [Google Scholar]
- Kistes, I.; Gelman, D. Carbometalated Complexes Possessing Tripodal Pseudo-C3-Symmetric Triptycene-Based Ligands. Organometallics 2018, 37, 526–529. [Google Scholar]
- Samiee, S.; Shiralinia, A.; Hoviezi, E.; Gable, E.H. Mono- and dinuclear oxime palladacycles bearing diphosphine ligands: An unusual coordination mode for dppe. Appl. Organomet. Chem. 2019, 33, e5098. [Google Scholar]
- Onoue, H.; Moritani, I. Ortho-Metalation reactions of N-phenylbenzaldimine and its related compounds by palladium (II) acetate. J. Organomet. Chem. 1972, 43, 431–436. [Google Scholar] [CrossRef]
- Onoue, H.; Minami, K.; Nakagawa, K. Aromatic metalation reactions by palladium (II) and platinum (II) on aromatic aldoximes and ketoximes. Bull. Chem. Soc. Jpn. 1970, 43, 3480–3485. [Google Scholar] [CrossRef]
- Ustynyuk, Y.A.; Chertkov, V.A.; Barinov, I.V. The interaction of nickelocene with benzal anilines. J. Organomet. Chem. 1971, 29, C53–C54. [Google Scholar]
- Cecconi, F.; Ghilardi, C.A.; Midollini, S.; Moneti, S.; Orlandini, A.; Scapacci, G. Palladium complexes with the tripodal phosphine tris-(2-diphenylphos-phinoethyl) amine. Synthesis and structure of trigonal, tetrahedral, trigonal bipyramidal, and square planar complexes. J. Chem. Soc. Dalton Trans. 1989, 2, 211–216. [Google Scholar]
- Armarego, W.L. Purification of Laboratory Chemicals; Butterworth-Heinemann: Amsterdam, The Netherlands, 2017; ISBN 978-185-617-567-8. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
al Janabi, B.; Reigosa, F.; Alberdi, G.; Ortigueira, J.M.; Vila, J.M. An Innovative Structural Rearrangement in Imine Palladacycle Metaloligand Chemistry: From Single-Nuclear to Double-Nuclear Pseudo-Pentacoordinated Complexes. Molecules 2023, 28, 2328. https://doi.org/10.3390/molecules28052328
al Janabi B, Reigosa F, Alberdi G, Ortigueira JM, Vila JM. An Innovative Structural Rearrangement in Imine Palladacycle Metaloligand Chemistry: From Single-Nuclear to Double-Nuclear Pseudo-Pentacoordinated Complexes. Molecules. 2023; 28(5):2328. https://doi.org/10.3390/molecules28052328
Chicago/Turabian Styleal Janabi, Basma, Francisco Reigosa, Gemma Alberdi, Juan M. Ortigueira, and José M. Vila. 2023. "An Innovative Structural Rearrangement in Imine Palladacycle Metaloligand Chemistry: From Single-Nuclear to Double-Nuclear Pseudo-Pentacoordinated Complexes" Molecules 28, no. 5: 2328. https://doi.org/10.3390/molecules28052328