
Sulphur vs NH Group: Effects on the CO2 Electroreduction Capability of Phenylenediamine-Cp Cobalt Complexes



Abstract
:1. Introduction
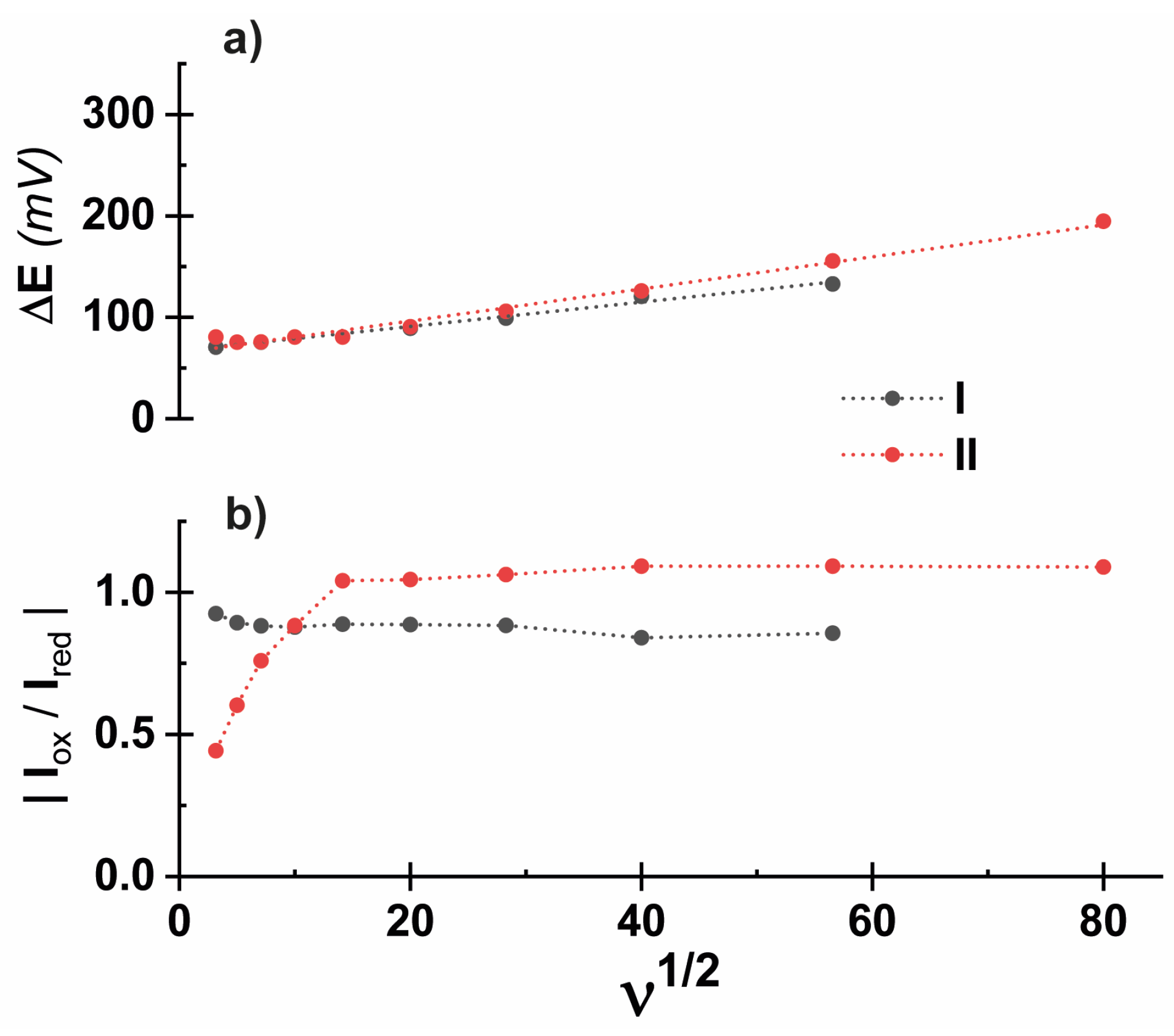
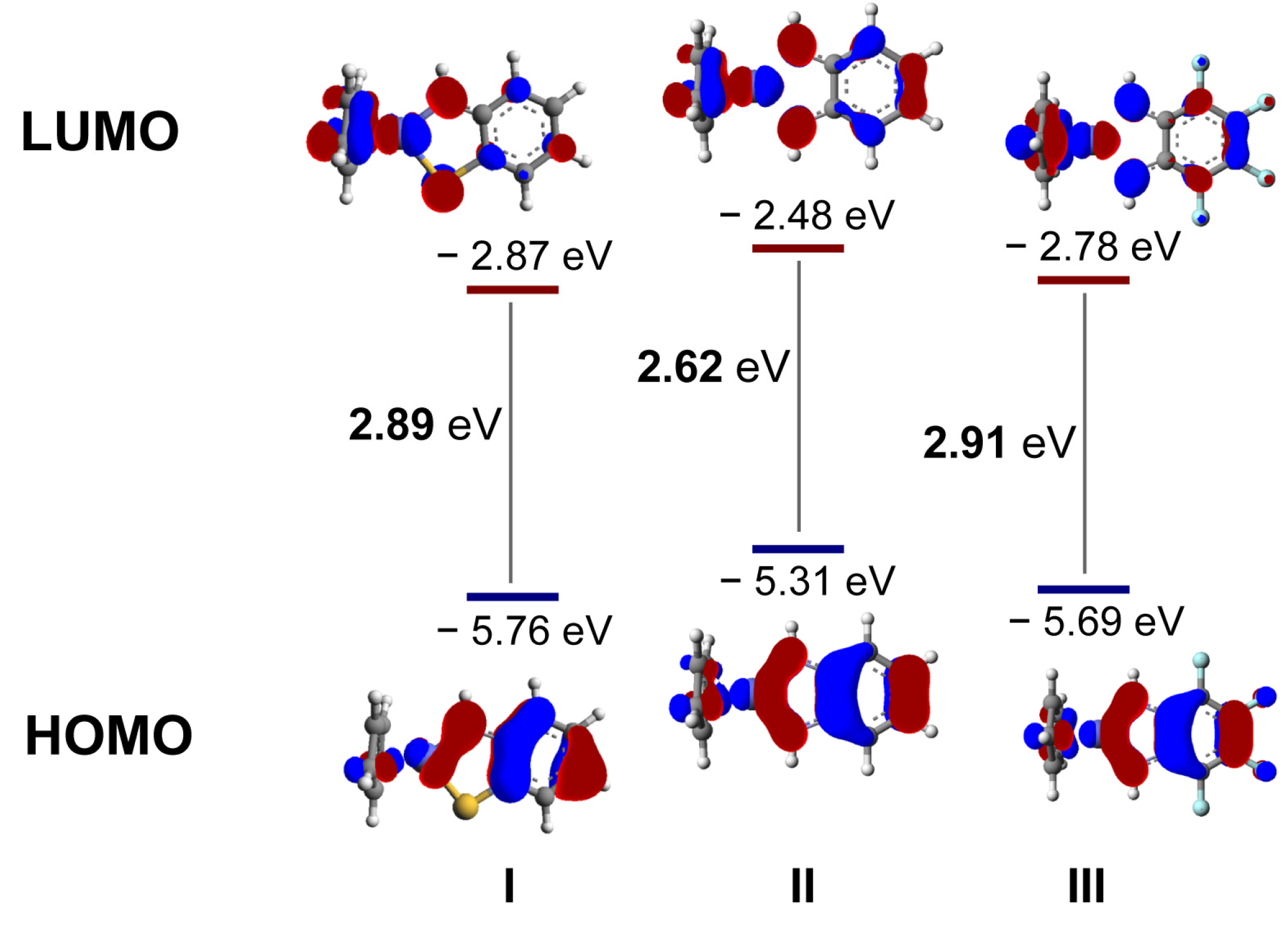
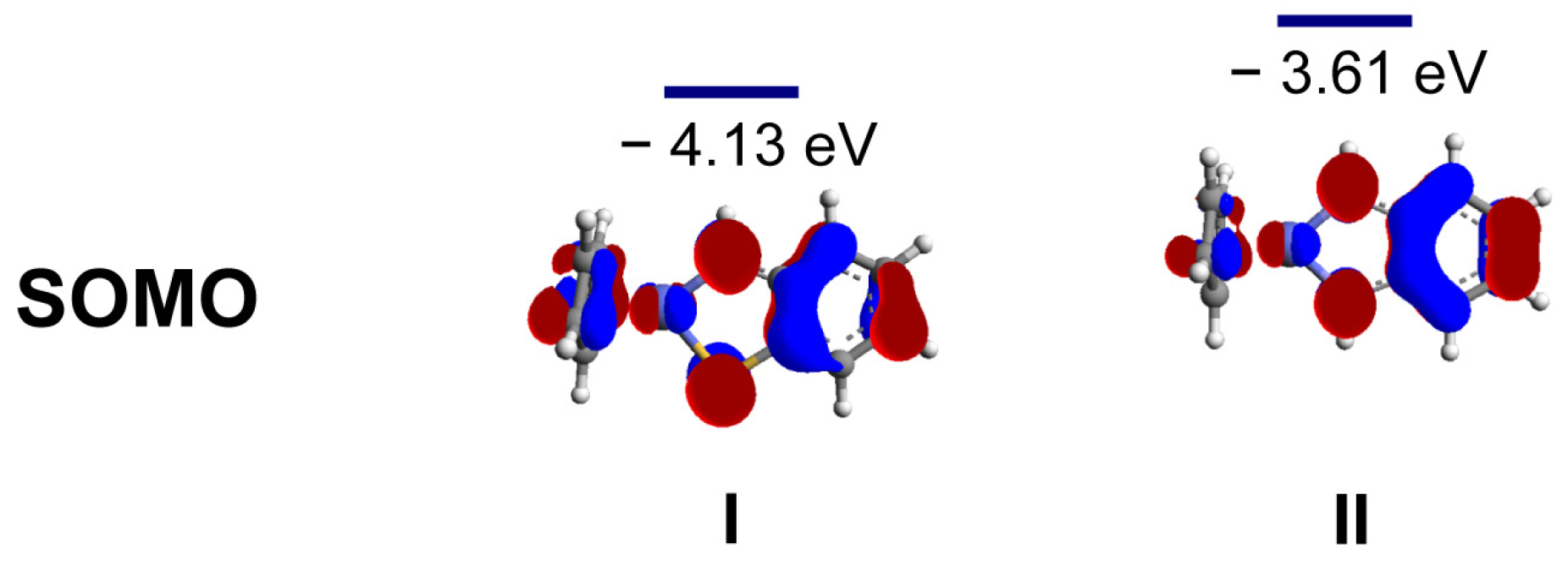
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis
3.2. Electrochemical Methods
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using Carbon Dioxide as a Building Block in Organic Synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abanades, J.C.; Rubin, E.S.; Mazzotti, M.; Herzog, H.J. On the Climate Change Mitigation Potential of CO2 Conversion to Fuels. Energy Environ. Sci. 2017, 10, 2491–2499. [Google Scholar] [CrossRef]
- Bushuyev, O.S.; de Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulié, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Işci, U.; Berlinguette, C.P.; et al. CO2 Electrochemical Catalytic Reduction with a Highly Active Cobalt Phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef] [Green Version]
- Lote, D.A. Literature Survey on Electrochemical Reduction of CO2. Int. J. Electron. Electr. Eng. 2014, 7, 341–346. [Google Scholar]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Orella, M.J.; Román-Leshkov, Y.; Brushett, F.R. Emerging Opportunities for Electrochemical Processing to Enable Sustainable Chemical Manufacturing. Curr. Opin. Chem. Eng. 2018, 20, 159–167. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Mocci, F.; Zhang, X.; Ji, X.; Laaksonen, A. Ionic Liquids for CO2 Electrochemical Reduction. Chin. J. Chem. Eng. 2021, 31, 75–93. [Google Scholar] [CrossRef]
- Franco, F.; Fernández, S.; Lloret-Fillol, J. Advances in the Electrochemical Catalytic Reduction of CO2 with Metal Complexes. Curr. Opin. Electrochem. 2019, 15, 109–117. [Google Scholar] [CrossRef]
- Ishida, H. Electrochemical/Photochemical CO2 Reduction Catalyzed by Transition Metal Complexes. In Carbon Dioxide Chemistry, Capture and Oil Recovery; InTechOpen: London, UK, 2018. [Google Scholar]
- Zhang, Z.; Vasiliu, T.; Li, F.; Laaksonen, A.; Mocci, F.; Ji, X. Electrochemically Driven Efficient Enzymatic Conversion of CO2 to Formic Acid with Artificial Cofactors. J. CO2 Util. 2021, 52, 101679. [Google Scholar] [CrossRef]
- Collin, J.P.; Sauvage, J.P. Electrochemical Reduction of Carbon Dioxide Mediated by Molecular Catalysts. Coord. Chem. Rev. 1989, 93, 245–268. [Google Scholar] [CrossRef]
- Savéant, J.M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef]
- Windle, C.D.; Reisner, E. Heterogenised Molecular Catalysts for the Reduction of CO2 to Fuels. CHIMIA Int. J. Chem. 2015, 69, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, R.; Hendriksen, D.E. The Binding and Activation of Carbon Monoxide, Carbon Dioxide, and Nitric Oxide and Their Homogeneously Catalyzed Reactions. In Advances in Catalysis; Elsevier: Amsterdam, The Netherlands, 1979; Volume 28, pp. 79–172. [Google Scholar]
- Mascetti, J. Carbon Dioxide Coordination Chemistry and Reactivity of Coordinated CO2. In Carbon Dioxide as Chemical Feedstock; Arasta, M., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 55–88. [Google Scholar]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.D.; Nguyen, A.D.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis. J. Am. Chem. Soc. 2014, 136, 5460–5471. [Google Scholar] [CrossRef]
- Hartl, F.; Rosa, P.; Ricard, L.; le Floch, P.; Záliš, S. Electronic Transitions and Bonding Properties in a Series of Five-Coordinate “16-Electron” Complexes [Mn(CO)3(L2)]− (L2=chelating Redox-Active π-Donor Ligand). Coord. Chem. Rev. 2007, 251, 557–576. [Google Scholar] [CrossRef]
- Song, J.; Klein, E.L.; Neese, F.; Ye, S. The Mechanism of Homogeneous CO2 Reduction by Ni(Cyclam): Product Selectivity, Concerted Proton–Electron Transfer and C–O Bond Cleavage. Inorg. Chem. 2014, 53, 7500–7507. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.M.; Rønne, M.H.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials. Angew. Chem. Int. Ed. 2017, 56, 6468–6472. [Google Scholar] [CrossRef]
- Lacy, D.C.; McCrory, C.C.L.; Peters, J.C. Studies of Cobalt-Mediated Electrocatalytic CO2 Reduction Using a Redox-Active Ligand. Inorg. Chem. 2014, 53, 4980–4988. [Google Scholar] [CrossRef]
- Shimoda, T.; Morishima, T.; Kodama, K.; Hirose, T.; Polyansky, D.E.; Manbeck, G.F.; Muckerman, J.T.; Fujita, E. Photocatalytic CO2 Reduction by Trigonal-Bipyramidal Cobalt(II) Polypyridyl Complexes: The Nature of Cobalt(I) and Cobalt(0) Complexes upon Their Reactions with CO2, CO, or Proton. Inorg. Chem. 2018, 57, 5486–5498. [Google Scholar] [CrossRef] [PubMed]
- Nakada, A.; Matsumoto, T.; Chang, H.-C. Redox-Active Ligands for Chemical, Electrochemical, and Photochemical Molecular Conversions. Coord. Chem. Rev. 2022, 473, 214804. [Google Scholar] [CrossRef]
- Sutradhar, M.; Pombeiro, A.J.L.; da Silva, J.A.L. Water Oxidation with Transition Metal Catalysts with Non-Innocent Ligands and Its Mechanisms. Coord. Chem. Rev. 2021, 439, 213911. [Google Scholar] [CrossRef]
- Tarrago, M.; Ye, S.; Neese, F. Electronic Structure Analysis of Electrochemical CO2 Reduction by Iron-Porphyrins Reveals Basic Requirements for Design of Catalysts Bearing Non-Innocent Ligands. Chem. Sci. 2022, 13, 10029–10047. [Google Scholar] [CrossRef]
- Chirik, P.J.; Wieghardt, K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [Google Scholar] [CrossRef]
- Praneeth, V.K.K.; Ringenberg, M.R.; Ward, T.R. Redox-Active Ligands in Catalysis. Angew. Chem. Int. Ed. 2012, 51, 10228–10234. [Google Scholar] [CrossRef]
- Benson, E.E.; Sampson, M.D.; Grice, K.A.; Smieja, J.M.; Froehlich, J.D.; Friebel, D.; Keith, J.A.; Carter, E.A.; Nilsson, A.; Kubiak, C.P. The Electronic States of Rhenium Bipyridyl Electrocatalysts for CO2 Reduction as Revealed by X-Ray Absorption Spectroscopy and Computational Quantum Chemistry. Angew. Chem. Int. Ed. 2013, 52, 4841–4844. [Google Scholar] [CrossRef]
- Grice, K.A.; Saucedo, C. Electrocatalytic Reduction of CO2 by Group 6 M(CO)6 Species without “Non-Innocent” Ligands. Inorg. Chem. 2016, 55, 6240–6246. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(Bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Melis, N.; Mocci, F.; Vacca, A.; Pilia, L. Novel Homogeneous Selective Electrocatalysts for CO 2 Reduction: An Electrochemical and Computational Study of Cyclopentadienyl-Phenylendiamino-Cobalt Complexes. Sustain. Energy Fuels 2020, 4, 5609–5617. [Google Scholar] [CrossRef]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef]
- Kaim, W. The Shrinking World of Innocent Ligands: Conventionaland Non-Conventional Redox-Active Ligands. Eur. J. Inorg. Chem. 2012, 2012, 343–348. [Google Scholar] [CrossRef]
- Fourmigué, M.; Cauchy, T.; Nomura, M. Experimental and Theoretical Evaluation of Magnetic Coupling in Organometallic Radicals: The Eloquent Case of Face-to-Face Cp⋯Cp Interactions. CrystEngComm 2009, 11, 1491. [Google Scholar] [CrossRef] [Green Version]
- Pilia, L.; Shuku, Y.; Dalgleish, S.; Hofmann, D.W.M.; Melis, N.; Awaga, K.; Robertson, N. Effect of Fluorination on the Crystal and Electronic Structure of Organometallic Cyclopentadienyl-Phenylenediamino-Cobalt Complexes. J. Organomet. Chem. 2020, 918, 121277. [Google Scholar] [CrossRef]
- Spielvogel, K.D.; Coughlin, E.J.; Petras, H.; Luna, J.A.; Benson, A.; Donahue, C.M.; Kibasa, A.; Lee, K.; Salacinski, R.; Bart, S.C.; et al. The Influence of Redox-Innocent Donor Groups in Tetradentate Ligands Derived from o -Phenylenediamine: Electronic Structure Investigations with Nickel. Inorg. Chem. 2019, 58, 12756–12774. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Sarmah, A.; Mukherjee, C. Where Is the Unpaired Electron Density? A Combined Experimental and Theoretical Finding on the Geometric and Electronic Structures of the Co(III) and Mn(IV) Complexes of the Unsymmetrical Non-Innocent Pincer ONS Ligand. Dalton Trans. 2022, 51, 16723–16732. [Google Scholar] [CrossRef] [PubMed]
- Combariza, M.Y.; Vachet, R.W. Gas-Phase Ion-Molecule Reactions of Transition Metal Complexes: The Effect of Different Coordination Spheres on Complex Reactivity. J. Am. Soc. Mass Spectrom. 2002, 13, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.B. Practical Hardness Scales for Metal Ion Complexes. Inorg. Chim. Acta 2002, 339, 27–33. [Google Scholar] [CrossRef]
- Roy, S.; Sharma, B.; Pécaut, J.; Simon, P.; Fontecave, M.; Tran, P.D.; Derat, E.; Artero, V. Molecular Cobalt Complexes with Pendant Amines for Selective Electrocatalytic Reduction of Carbon Dioxide to Formic Acid. J. Am. Chem. Soc. 2017, 139, 3685–3696. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Jia, H.; Kobiro, K.; Cabelli, D.E.; Muckerman, J.T.; Fujita, E. Nickel(Ii) Macrocycles: Highly Efficient Electrocatalysts for the Selective Reduction of CO2 to CO. Energy Environ. Sci. 2012, 5, 9502. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Do, T.H.; Haiges, R.; Takase, M.K.; Marinescu, S.C. Proton-Assisted Reduction of CO2 by Cobalt Aminopyridine Macrocycles. J. Am. Chem. Soc. 2016, 138, 5765–5768. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Passard, G.; Robert, M.; Savéant, J.-M. Pendant Acid–Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0)Porphyrins Bearing Prepositioned Phenol Functionalities. J. Am. Chem. Soc. 2014, 136, 11821–11829. [Google Scholar] [CrossRef]
- Costentin, C.; Passard, G.; Savéant, J.M. Benchmarking of Homogeneous Electrocatalysts: Overpotential, Turnover Frequency, Limiting Turnover Number. J. Am. Chem. Soc. 2015, 137, 5461–5467. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules. In Density Functional Theory of Atoms and Molecules; Dordrecht: New York, NY, USA, 1989; p. 25. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Nicholson, R.S.; Shain, I. Theory of Stationary Electrode Polarography. Anal. Chem. 1964, 36, 1212. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts. ChemElectroChem 2014, 1, 1226–1236. [Google Scholar] [CrossRef]
- Hopkins Leseberg, J.A.; Lionetti, D.; Day, V.W.; Blakemore, J.D. Electrochemical Kinetic Study of [Cp*Rh] Complexes Supported by Bis(2-Pyridyl)Methane Ligands. Organometallics 2021, 40, 266–277. [Google Scholar] [CrossRef]
- Gonell, S.; Assaf, E.A.; Duffee, K.D.; Schauer, C.K.; Miller, A.J.M.M. Kinetics of the Trans Effect in Ruthenium Complexes Provide Insight into the Factors That Control Activity and Stability in CO2 Electroreduction. J. Am. Chem. Soc. 2020, 142, 8980–8999. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, J.D.; Kubiak, C.P. The Homogeneous Reduction of CO2 by [Ni(Cyclam)]+: Increased Catalytic Rates with the Addition of a CO Scavenger. J. Am. Chem. Soc. 2015, 137, 3565–3573. [Google Scholar] [CrossRef] [PubMed]
- Chapovetsky, A.; Welborn, M.; Luna, J.M.; Haiges, R.; Miller, T.F.; Marinescu, S.C. Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction. ACS Cent. Sci. 2018, 4, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsen, J.B.; Rønne, M.H.; Daasbjerg, K.; Skrydstrup, T. Are Amines the Holy Grail for Facilitating CO2 Reduction? Angew. Chem. Int. Ed. 2021, 60, 9174–9179. [Google Scholar] [CrossRef]
- D’Andrade, B.W.; Datta, S.; Forrest, S.R.; Djurovich, P.; Polikarpov, E.; Thompson, M.E. Relationship between the Ionization and Oxidation Potentials of Molecular Organic Semiconductors. Org. Electron. 2005, 6, 11–20. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Atomic Dipole Moment Corrected Hirshfeld Population Method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- King, R.B. Organometallic Chemistry of the Transition Metals. XI. Some New Cyclopentadienyl Derivatives of Cobalt and Rhodium. Inorg. Chem. 1966, 5, 82–87. [Google Scholar] [CrossRef]
- Heck, R.F. Cyclopentadienylcobalt Derivatives of Chelating Aromatic Ligands. Inorg. Chem. 1968, 7, 1513–1516. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A. ArgusLab 4.0.1. Available online: http://www.arguslab.com/arguslab.com/ArgusLab.html (accessed on 6 March 2020).
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Gerschel, P.; Warm, K.; Farquhar, E.R.; Englert, U.; Reback, M.L.; Siegmund, D.; Ray, K.; Apfel, U.-P. Sulfur Substitution in a Ni(Cyclam) Derivative Results in Lower Overpotential for CO2 Reduction and Enhanced Proton Reduction. Dalton Trans. 2019, 48, 5923–5932. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Tsukakoshi, Y.; Kotani, H.; Ishizuka, T.; Kojima, T. Visible-Light-Driven Photocatalytic CO2 Reduction by a Ni(II) Complex Bearing a Bioinspired Tetradentate Ligand for Selective CO Production. J. Am. Chem. Soc. 2017, 139, 6538–6541. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O1 (V) | R1 (V) | E1/2 (V) | ΔE (mV) | |
|---|---|---|---|---|
| I | −1.32 | −1.41 | −1.36 | 90 |
| II | −1.66 | −1.74 | −1.70 | 80 |
| R1 (V) | Ep/2 (V) | R2 (V) | E (V) | ICO2/IN2 | |
|---|---|---|---|---|---|
| I | −1.35 | −1.29 | −2.34 | −2.34 | 7.16 |
| −2.52 | −2.52 | 9.41 | |||
| II | −1.78 | −1.72 | −2.52 | −2.54 | 4.12 |
| I | II | |||||
|---|---|---|---|---|---|---|
| Ecat (V) | −2.34 | −2.52 | −2.54 | |||
| H2O (%v/v) | 0 | 9.09 | 0 | 9.09 | 0 | 9.09 |
| ICO2/IN2 | 7.16 | 16.89 | 9.41 | 22.73 | 4.12 | 24.40 |
| (Icat/Ip)2 | 5.48 | 42.25 | 10.26 | 83.90 | 3.32 | 280.07 |
| IHER/I[a] | 1.02 | 1.37 | 1.09 | |||
| I | II | |||||
|---|---|---|---|---|---|---|
| Red− | Neutr | f− | Red− | Neutr | f− | |
| Co | −0.105 | −0.097 | −0.008 | −0.136 | −0.077 | −0.059 |
| N | −0.326 | −0.241 | −0.086 | −0.350 | −0.266 | −0.083 |
| C (Cp) | −0.159 | −0.105 | −0.054 | −0.169 | −0.131 | −0.038 |
| S | −0.455 | −0.228 | −0.226 | |||
| Sum | −1.682 | −1.091 | −0.591 | −1.682 | −1.267 | −0.415 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melis, N.; Mocci, F.; Vacca, A.; Pilia, L. Sulphur vs NH Group: Effects on the CO2 Electroreduction Capability of Phenylenediamine-Cp Cobalt Complexes. Molecules 2023, 28, 2364. https://doi.org/10.3390/molecules28052364
Melis N, Mocci F, Vacca A, Pilia L. Sulphur vs NH Group: Effects on the CO2 Electroreduction Capability of Phenylenediamine-Cp Cobalt Complexes. Molecules. 2023; 28(5):2364. https://doi.org/10.3390/molecules28052364
Chicago/Turabian StyleMelis, Nicola, Francesca Mocci, Annalisa Vacca, and Luca Pilia. 2023. "Sulphur vs NH Group: Effects on the CO2 Electroreduction Capability of Phenylenediamine-Cp Cobalt Complexes" Molecules 28, no. 5: 2364. https://doi.org/10.3390/molecules28052364
APA StyleMelis, N., Mocci, F., Vacca, A., & Pilia, L. (2023). Sulphur vs NH Group: Effects on the CO2 Electroreduction Capability of Phenylenediamine-Cp Cobalt Complexes. Molecules, 28(5), 2364. https://doi.org/10.3390/molecules28052364