
Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials
 , ,
, , 
Abstract
1. Introduction
2. Bruton’s Tyrosine Kinase
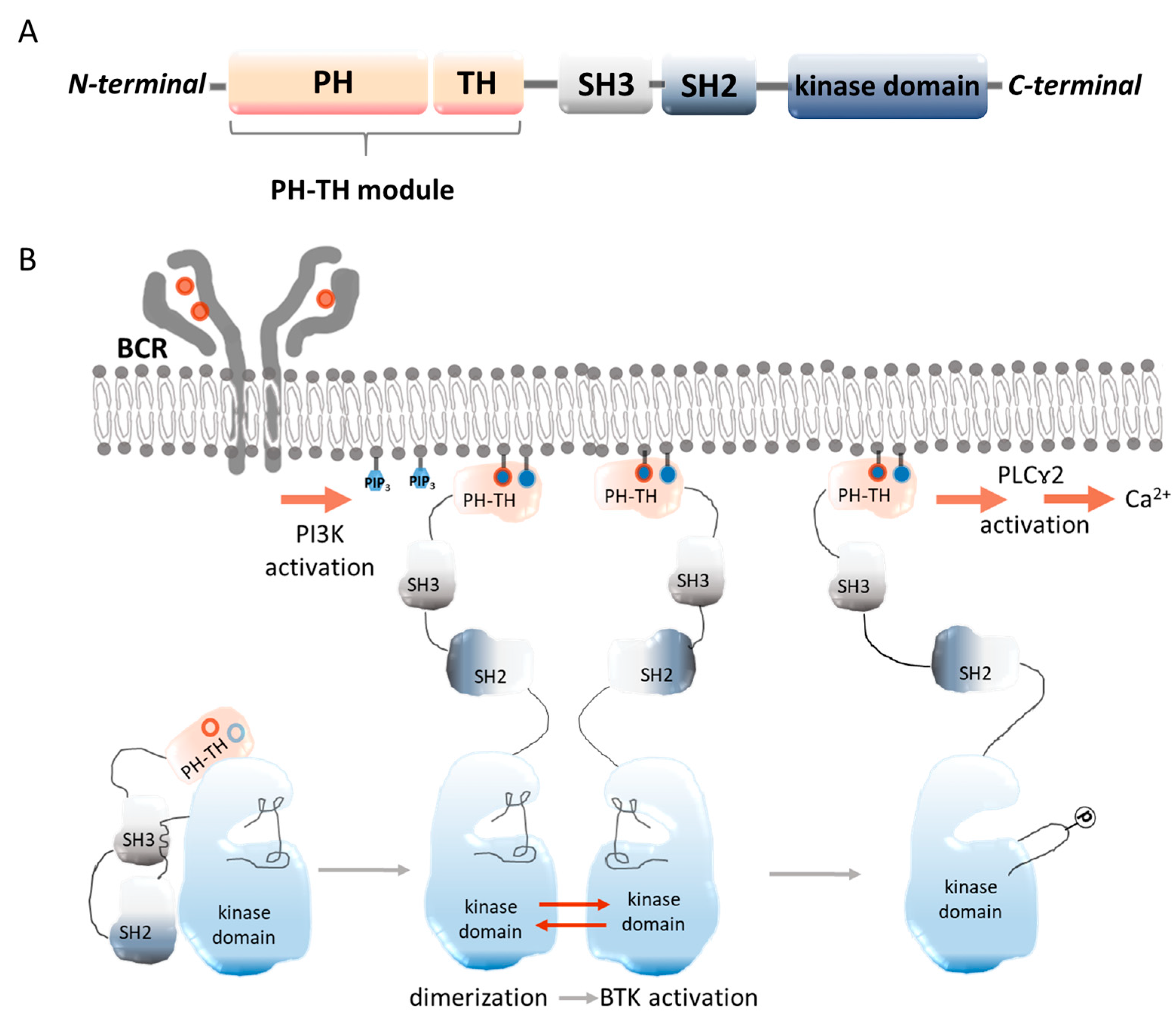
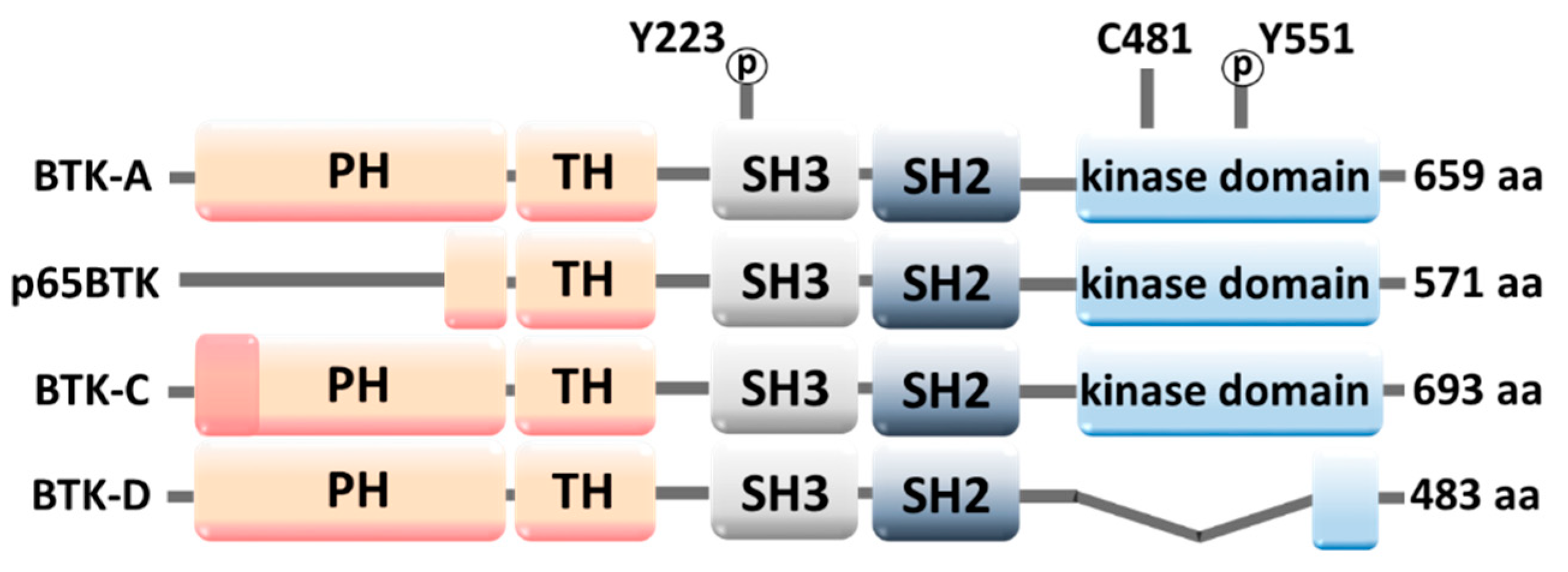
2.1. Molecular Structure of BTK
2.2. Occurrence of BTK and Its Role
2.3. BTK Down- and Upregulation
3. Bruton’s Tyrosine Kinase Inhibitors
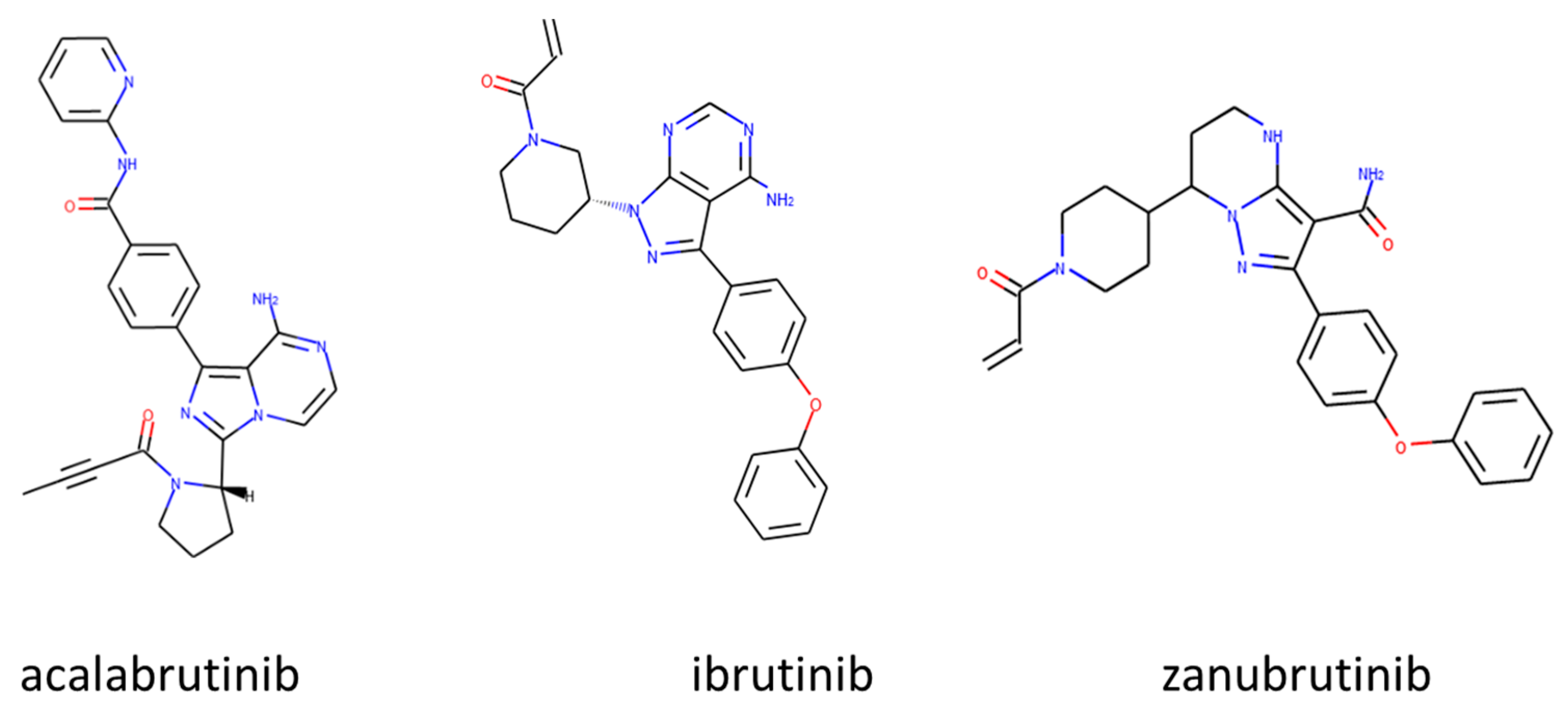
3.1. Approved Irreversible BTKIs
3.1.1. Ibrutinib (Imbruvica®)
3.1.2. Acalabrutinib (Calquence®)
3.1.3. Zanubrutinib (Brukinsa®)
3.1.4. Tirabrutinib (Velexbru®)
3.1.5. Orelabrutinib
3.2. BTK Inhibitors in Clinical Trials
3.2.1. Spebrutinib (CC-292)
3.2.2. Evobrutinib (M2951, MSC-2364447C)
3.2.3. Vecabrutinib (SNS-062)
3.2.4. Pirtobrutinib (LOXO-305)
3.2.5. Fenebrutinib (GDC-0853)
3.3. Combination Therapy
3.4. Dual Inhibitors
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Das, D.; Hong, J. Irreversible Kinase Inhibitors Targeting Cysteine Residues and Their Applications in Cancer Therapy. Mini-Rev. Med. Chem. 2020, 20, 1732–1753. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.L.; Bordallo, H.N.; Mamontov, E. Water Dynamics in Cancer Cells: Lessons from Quasielastic Neutron Scattering. Medicina 2022, 58, 654. [Google Scholar] [CrossRef] [PubMed]
- Szklener, K.; Michalski, A.; Żak, K.; Piwoński, M.; Mańdziuk, S. Ibrutinib in the Treatment of Solid Tumors: Current State of Knowledge and Future Directions. Cells 2022, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Zain, R.; Vihinen, M. Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Front. Immunol. 2021, 12, 694853. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.; Liu, Y.; Xu, Z.; Meng, C.; Yang, D.; Qian, J.; Deng, X.; Zhang, Y.; Ling, Y. Recent Development of BTK-Based Dual Inhibitors in the Treatment of Cancers. Eur. J. Med. Chem. 2022, 233, 114232. [Google Scholar] [CrossRef]
- García-Merino, A. Bruton’s Tyrosine Kinase Inhibitors: A New Generation of Promising Agents for Multiple Sclerosis Therapy. Cells 2021, 10, 2560. [Google Scholar] [CrossRef]
- Wang, Q.; Vogan, E.M.; Nocka, L.M.; Rosen, C.E.; Zorn, J.A.; Harrison, S.C.; Kuriyan, J. Autoinhibition of Bruton’s Tyrosine Kinase (Btk) and Activation by Soluble Inositol Hexakisphosphate. eLife 2015, 4, e06074. [Google Scholar] [CrossRef]
- Chung, J.K.; Nocka, L.M.; Decker, A.; Wang, Q.; Kadlecek, T.A.; Weiss, A.; Kuriyan, J.; Groves, J.T. Switch-like Activation of Bruton’s Tyrosine Kinase by Membrane-Mediated Dimerization. Proc. Natl. Acad. Sci. USA 2019, 166, 10798–10803. [Google Scholar] [CrossRef]
- Wang, X.; Kokabee, L.; Kokabee, M.; Conklin, D.S. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front. Cell Dev. Biol. 2021, 9, 668996. [Google Scholar] [CrossRef]
- Liu, J.; Chen, C.; Wang, D.; Zhang, J.; Zhang, T. Emerging Small-Molecule Inhibitors of the Bruton’s Tyrosine Kinase (BTK): Current Development. Eur. J. Med. Chem. 2021, 217, 113329. [Google Scholar] [CrossRef] [PubMed]
- Ringheim, G.E.; Wampole, M.; Oberoi, K. Bruton’s Tyrosine Kinase (BTK) Inhibitors and Autoimmune Diseases: Making Sense of BTK Inhibitor Specificity Profiles and Recent Clinical Trial Successes and Failures. Front. Immunol. 2021, 12, 662223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Pechersky, Y.; Sagawa, S.; Pan, A.C.; Shaw, D.E. Structural Mechanism for Bruton’s Tyrosine Kinase Activation at the Cell Membrane. Proc. Natl. Acad. Sci. USA 2019, 116, 9390–9399. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Barati, S.; Babamahmoodi, A.; Dastan, F.; Marjani, M. The Possible Role of Bruton Tyrosine Kinase Inhibitors in the Treatment of COVID-19: A Review. Curr. Ther. Res. Clin. Exp. 2022, 96, 100658. [Google Scholar] [CrossRef] [PubMed]
- Messex, J.K.; Liou, G.-Y. Targeting BTK Signaling in the Microenvironment of Solid Tumors as a Feasible Cancer Therapy Option. Cancers 2021, 13, 2198. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s Tyrosine Kinase (BTK) as a Promising Target in Solid Tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Tankiewicz-Kwedlo, A.; Hermanowicz, M.; Pawlak, K.; Czarnomysy, R.; Bielawski, K.; Prokop, I.; Pawlak, D. Erythropoietin Intensifies the Proapoptotic Activity of LFM-A13 in Cells and in a Mouse Model of Colorectal Cancer. Int. J. Mol. Sci. 2018, 19, 1262. [Google Scholar] [CrossRef]
- Cinar, M.; Hamedani, F.S.; Mo, Z.; Cinar, B.; Amin, H.M.; Alkan, S. Bruton Tyrosine Kinase Is Commonly Overexpressed in Mantle Cell Lymphoma and Its Attenuation by Ibrutinib Induces Apoptosis. Leuk. Res. 2013, 37, 1271–1277. [Google Scholar] [CrossRef]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef]
- Tasso, B.; Spallarossa, A.; Russo, E.; Brullo, C. The Development of Btk Inhibitors: A Five-Year Update. Molecules 2021, 26, 7411. [Google Scholar] [CrossRef]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s Tyrosine Kinase Is Essential for NLRP3 Inflammasome Activation and Contributes to Ischaemic Brain Injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pichulik, T.; Wolz, O.O.; Dang, T.M.; Stutz, A.; Dillen, C.; Delmiro Garcia, M.; Kraus, H.; Dickhöfer, S.; Daiber, E.; et al. Human NACHT, LRR, and PYD Domain–Containing Protein 3 (NLRP3) Inflammasome Activity Is Regulated by and Potentially Targetable through Bruton Tyrosine Kinase. J. Allergy Clin. Immunol. 2017, 140, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Bittner, Z.A.; Liu, X.; Tortola, M.M.; Tapia-Abellán, A.; Shankar, S.; Andreeva, L.; Mangan, M.; Spalinger, M.; Kalbacher, H.; Düwell, P.; et al. BTK Operates a Phospho-Tyrosine Switch to Regulate NLRP3 Inflammasome Activity. J. Exp. Med. 2021, 218, e20201656. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Bieber, M.; Kraft, P.; Weber, A.N.R.; Stoll, G.; Schuhmann, M.K. The NLRP3 Inflammasome Drives Inflammation in Ischemia/Reperfusion Injury after Transient Middle Cerebral Artery Occlusion in Mice. Brain Behav. Immun. 2021, 92, 223–233. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Li, Y.Y.; Xia, L.L.; Wu, Y.G. Bruton’s Tyrosine Kinase Regulates Macrophageinduced Inflammation in the Diabetic Kidney via NLRP3 Inflammasome Activation. Int. J. Mol. Med. 2021, 48, 177. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hauenstein, A.V. The NLRP3 Inflammasome: Mechanism of Action, Role in Disease and Therapies. Mol. Aspects Med. 2020, 76, 100889. [Google Scholar] [CrossRef]
- O’Riordan, C.E.; Purvis, G.S.D.; Collotta, D.; Krieg, N.; Wissuwa, B.; Sheikh, M.H.; Ferreira Alves, G.; Mohammad, S.; Callender, L.A.; Coldewey, S.M.; et al. X-Linked Immunodeficient Mice with No Functional Bruton’s Tyrosine Kinase Are Protected from Sepsis-Induced Multiple Organ Failure. Front. Immunol. 2020, 11, 581758. [Google Scholar] [CrossRef]
- Brunner, C.; Betzler, A.C.; Brown, J.R.; Andreotti, A.H.; Grassilli, E. Editorial: Targeting Bruton Tyrosine Kinase. Front. Cell Dev. Biol. 2022, 10, 909655. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Collino, M.; Aranda-Tavio, H.; Chiazza, F.; O’Riordan, C.E.; Zeboudj, L.; Mohammad, S.; Collotta, D.; Verta, R.; Guisot, N.E.S.; et al. Inhibition of Bruton’s TK Regulates Macrophage NF-ΚB and NLRP3 Inflammasome Activation in Metabolic Inflammation. Br. J. Pharmacol. 2020, 177, 4416–4432. [Google Scholar] [CrossRef]
- Weber, A.N.R. Targeting the NLRP3 Inflammasome via BTK. Front. Cell Dev. Biol. 2021, 9, 630479. [Google Scholar] [CrossRef]
- Grassilli, E.; Pisano, F.; Cialdella, A.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Masiero, L.; Ianzano, L.; Giordano, F.; Cicirelli, V.; et al. A Novel Oncogenic BTK Isoform Is Overexpressed in Colon Cancers and Required for RAS-Mediated Transformation. Oncogene 2016, 35, 4368–4378. [Google Scholar] [CrossRef]
- Grassilli, E.; Cerrito, M.G.; Bonomo, S.; Giovannoni, R.; Conconi, D.; Lavitrano, M. P65BTK Is a Novel Biomarker and Therapeutic Target in Solid Tumors. Front. Cell Dev. Biol. 2021, 9, 690365. [Google Scholar] [CrossRef]
- Lavitrano, M.; Ianzano, L.; Bonomo, S.; Cialdella, A.; Cerrito, M.G.; Pisano, F.; Missaglia, C.; Giovannoni, R.; Romano, G.; McLean, C.M.; et al. BTK Inhibitors Synergise with 5-FU to Treat Drug-Resistant TP53-Null Colon Cancers. J. Pathol. 2020, 250, 134–147. [Google Scholar] [CrossRef]
- Giordano, F.; Vaira, V.; Cortinovis, D.; Bonomo, S.; Goedmakers, J.; Brena, F.; Cialdella, A.; Ianzano, L.; Forno, I.; Cerrito, M.G.; et al. P65BTK Is a Novel Potential Actionable Target in KRAS-Mutated/EGFR-Wild Type Lung Adenocarcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 260. [Google Scholar] [CrossRef]
- Basile, D.; Gerratana, L.; Buonadonna, A.; Garattini, S.K.; Perin, T.; Grassilli, E.; Miolo, G.; Cerrito, M.G.; Belluco, C.; Bertola, G.; et al. Role of Bruton’s Tyrosine Kinase in Stage III Colorectal Cancer. Cancers 2019, 11, 880. [Google Scholar] [CrossRef]
- Kokabee, M.; Wang, X.; Voorand, E.; Alin, E.; Kokabee, L.; Khan, F.; Desrosiers, S.; Conklin, D.S. Palmitoylation of the Alternative Amino Terminus of the BTK-C Isoform Controls Subcellular Distribution and Signaling. Cancer Genom. Proteom. 2022, 19, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, J.; Sevinsky, C.J.; Kokabee, L.; Khan, F.; Sun, Y.; Conklin, D.S. Bruton’s Tyrosine Kinase Inhibitors Prevent Therapeutic Escape in Breast Cancer Cells. Mol. Cancer Ther. 2016, 15, 2198–2208. [Google Scholar] [CrossRef]
- Betzler, A.C.; Strobel, H.; Abou Kors, T.; Ezić, J.; Lesakova, K.; Pscheid, R.; Azoitei, N.; Sporleder, J.; Staufenberg, A.-R.; Drees, R.; et al. BTK Isoforms P80 and P65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression. Cancers 2023, 15, 310. [Google Scholar] [CrossRef]
- Liu, S.C.; Wu, Y.C.; Huang, C.M.; Hsieh, M.S.; Huang, T.Y.; Huang, C.S.; Hsu, T.N.; Huang, M.S.; Lee, W.H.; Yeh, C.T.; et al. Inhibition of Bruton’s Tyrosine Kinase as a Therapeutic Strategy for Chemoresistant Oral Squamous Cell Carcinoma and Potential Suppression of Cancer Stemness. Oncogenesis 2021, 10, 20. [Google Scholar] [CrossRef]
- Li, T.; Deng, Y.; Shi, Y.; Tian, R.; Chen, Y.; Zou, L.; Kazi, J.U.; Rönnstrand, L.; Feng, B.; Chan, S.O.; et al. Bruton’s Tyrosine Kinase Potentiates ALK Signaling and Serves as a Potential Therapeutic Target of Neuroblastoma. Oncogene 2018, 37, 6180–6194. [Google Scholar] [CrossRef]
- Pikatan, N.W.; Liu, Y.L.; Bamodu, O.A.; Hsiao, M.; Hsu, W.M.; Haryana, S.M.; Sutaryo; Chao, T.Y.; Yeh, C.T. Aberrantly Expressed Bruton’s Tyrosine Kinase Preferentially Drives Metastatic and Stem Cell-like Phenotypes in Neuroblastoma Cells. Cell. Oncol. 2020, 43, 1067–1084. [Google Scholar] [CrossRef] [PubMed]
- McCay, J.; Gribben, J.G. The Role of BTK Inhibitors on the Tumor Microenvironment in CLL. Leuk. Lymphoma 2022, 63, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, D.; Groth, D.; Wright, H.; Bonilla, F.A.; Fuleihan, R.L.; Cunningham-Rundles, C.; Sullivan, K.E.; Ochs, H.D.; Marsh, R.; Feuille, E. X-Linked Agammaglobulinemia: Infection Frequency and Infection-Related Mortality in the USIDNET Registry. J. Clin. Immunol. 2022, 42, 827–836. [Google Scholar] [CrossRef]
- Kim, J.M.; Park, J.; Noh, E.M.; Song, H.K.; Kang, S.Y.; Jung, S.H.; Kim, J.S.; Park, B.H.; Lee, Y.R.; Youn, H.J. Bruton’s Agammaglobulinemia Tyrosine Kinase (Btk) Regulates TPA-Induced Breast Cancer Cell Invasion via PLCγ2/PKCβ/NF-ΚB/AP-1-Dependent Matrix Metalloproteinase-9 Activation. Oncol. Rep. 2021, 45, 56. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, J.E.; Smith, C.I.E.; Nore, B.F. Silencing of Bruton’s Tyrosine Kinase (Btk) Using Short Interfering RNA Duplexes (SiRNA). FEBS Lett. 2002, 527, 274–278. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Tankiewicz-Kwedlo, A.; Hermanowicz, J.M.; Domaniewski, T.; Pawlak, K.; Rusak, M.; Pryczynicz, A.; Surazynski, A.; Kaminski, T.; Kazberuk, A.; Pawlak, D. Simultaneous Use of Erythropoietin and LFM-A13 as a New Therapeutic Approach for Colorectal Cancer. Br. J. Pharmacol. 2018, 175, 743–762. [Google Scholar] [CrossRef] [PubMed]
- Rozkiewicz, D.; Hermanowicz, J.M.; Tankiewicz-Kwedlo, A.; Sieklucka, B.; Pawlak, K.; Czarnomysy, R.; Bielawski, K.; Surazynski, A.; Kalafut, J.; Przybyszewska, A.; et al. The Intensification of Anticancer Activity of LFM-A13 by Erythropoietin as a Possible Option for Inhibition of Breast Cancer. J. Enzyme Inhib. Med. Chem. 2020, 35, 1697–1711. [Google Scholar] [CrossRef]
- Pan, Y.; Chiu, Y.H.; Chiu, S.C.; Cho, D.Y.; Lee, L.M.; Wen, Y.C.; Whang-Peng, J.; Hsiao, C.H.; Shih, P.H. Inhibition of Bruton’s Tyrosine Kinase Suppresses Cancer Stemness and Promotes Carboplatin-Induced Cytotoxicity against Bladder Cancer Cells. Anticancer Res. 2020, 40, 6093–6099. [Google Scholar] [CrossRef]
- Eifert, C.; Wang, X.; Kokabee, L.; Kourtidis, A.; Jain, R.; Gerdes, M.J.; Conklin, D.S. A Novel Isoform of the B Cell Tyrosine Kinase BTK Protects Breast Cancer Cells from Apoptosis. Genes Chromosomes Cancer 2013, 52, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Kokabee, L.; Wang, X.; Sevinsky, C.J.; Wang, W.L.W.; Cheu, L.; Chittur, S.V.; Karimipoor, M.; Tenniswood, M.; Conklin, D.S. Bruton’s Tyrosine Kinase Is a Potential Therapeutic Target in Prostate Cancer. Cancer Biol. Ther. 2015, 16, 1604–1615. [Google Scholar] [CrossRef]
- Grassilli, E.; Cerrito, M.G. Emerging Actionable Targets to Treat Therapy-Resistant Colorectal Cancers. Cancer Drug Resist. 2022, 5, 36–63. [Google Scholar] [CrossRef]
- Chong, I.Y.; Aronson, L.; Bryant, H.; Gulati, A.; Campbell, J.; Elliott, R.; Pettitt, S.; Wilkerson, P.; Lambros, M.B.; Reis-Filho, J.S.; et al. Mapping Genetic Vulnerabilities Reveals BTK as a Novel Therapeutic Target in Oesophageal Cancer. Gut 2018, 67, 1780–1792. [Google Scholar] [CrossRef]
- Wang, J.D.; Chen, X.Y.; Ji, K.W.; Tao, F. Targeting Btk with Ibrutinib Inhibit Gastric Carcinoma Cells Growth. Am. J. Transl. Res. 2016, 8, 3003–3012. [Google Scholar]
- Zhu, Z.; Ling, L.; Qi, L.; Chong, Y.; Xue, L. Bruton’s Tyrosine Kinase (BTK) Inhibitor (Ibrutinib)-Suppressed Migration and Invasion of Prostate Cancer. OncoTargets Ther. 2020, 13, 4113–4122. [Google Scholar] [CrossRef]
- Zucha, M.A.; Wu, A.T.H.; Lee, W.H.; Wang, L.S.; Lin, W.W.; Yuan, C.C.; Yeh, C.T. Bruton’s Tyrosine Kinase (Btk) Inhibitor Ibrutinib Suppresses Stem-like Traits in Ovarian Cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef] [PubMed]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes From Patients With Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Vizcaya, A.; Fasano, S.; Isenberg, D.A. Bruton’s Tyrosine Kinase Inhibitors: A New Therapeutic Target for the Treatment of SLE? Immunotargets Ther. 2020, 9, 105–110. [Google Scholar] [CrossRef]
- Fang, X.; Liu, C.; Zhang, K.; Yang, W.; Wu, Z.; Shen, S.; Ma, Y.; Lu, X.; Chen, Y.; Lu, T.; et al. Discovery of Orally Active 1,4,5,6,8-Pentaazaacenaphthylens as Novel, Selective, and Potent Covalent BTK Inhibitors for the Treatment of Rheumatoid Arthritis. Eur. J. Med. Chem. 2023, 246, 114940. [Google Scholar] [CrossRef]
- Kifle, Z.D. Bruton Tyrosine Kinase Inhibitors as Potential Therapeutic Agents for COVID-19: A Review. Metabol. Open 2021, 11, 100116. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Han, W.; Wang, Y.; Liu, Y.; Zhang, Y.; Zhou, D.; Xiang, L. Inhibition of BTK Protects Lungs from Trauma-Hemorrhagic Shock-Induced Injury in Rats. Mol. Med. Rep. 2017, 16, 192–200. [Google Scholar] [CrossRef]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Davis, S.A.; Matthay, M.A.; Kurdowska, A.K. Inhibiting Bruton’s Tyrosine Kinase Rescues Mice from Lethal Influenza-Induced Acute Lung Injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L52–L58. [Google Scholar] [CrossRef]
- de Porto, A.P.; Liu, Z.; de Beer, R.; Florquin, S.; de Boer, O.J.; Hendriks, R.W.; van der Poll, T.; de Vos, A.F. Btk Inhibitor Ibrutinib Reduces Inflammatory Myeloid Cell Responses in the Lung during Murine Pneumococcal Pneumonia. Mol. Med. 2019, 25, 3. [Google Scholar] [CrossRef]
- Zhang, D.; Gong, H.; Meng, F. Recent Advances in Btk Inhibitors for the Treatment of Inflammatory and Autoimmune Diseases. Molecules 2021, 26, 4907. [Google Scholar] [CrossRef]
- Langrish, C.L.; Bradshaw, J.M.; Francesco, M.R.; Owens, T.D.; Xing, Y.; Shu, J.; LaStant, J.; Bisconte, A.; Outerbridge, C.; White, S.D.; et al. Preclinical Efficacy and Anti-Inflammatory Mechanisms of Action of the Bruton Tyrosine Kinase Inhibitor Rilzabrutinib for Immune-Mediated Disease. J. Immunol. 2021, 206, 1454–1468. [Google Scholar] [CrossRef]
- Owens, T.D.; Brameld, K.A.; Verner, E.J.; Ton, T.; Li, X.; Zhu, J.; Masjedizadeh, M.R.; Bradshaw, J.M.; Hill, R.J.; Tam, D.; et al. Discovery of Reversible Covalent Bruton’s Tyrosine Kinase Inhibitors PRN473 and PRN1008 (Rilzabrutinib). J. Med. Chem. 2022, 65, 5300–5316. [Google Scholar] [CrossRef]
- Compound Report Card. Available online: https://www.ebi.ac.uk/chembl/compound_report_card/CHEMBL3936761/ (accessed on 28 December 2022).
- Compound Report Card. Available online: https://www.ebi.ac.uk/chembl/compound_report_card/CHEMBL1873475/ (accessed on 28 December 2022).
- Compound Report Card. Available online: https://www.ebi.ac.uk/chembl/compound_report_card/CHEMBL3707348/ (accessed on 28 December 2022).
- IMBRUVICA® (Ibrutinib): Prescribing Information 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/217003s000lbl.pdf (accessed on 28 December 2022).
- Saleh, L.M.; Wang, W.; Herman, S.E.M.; Saba, N.S.; Anastas, V.; Barber, E.; Corrigan-Cummins, M.; Farooqui, M.; Sun, C.; Sarasua, S.M.; et al. Ibrutinib Downregulates a Subset of MiRNA Leading to Upregulation of Tumor Suppressors and Inhibition of Cell Proliferation in Chronic Lymphocytic Leukemia. Leukemia 2017, 31, 340–349. [Google Scholar] [CrossRef]
- Xia, S.; Liu, X.; Cao, X.; Xu, S. T-Cell Expression of Bruton’s Tyrosine Kinase Promotes Autoreactive T-Cell Activation and Exacerbates Aplastic Anemia. Cell. Mol. Immunol. 2020, 17, 1042–1052. [Google Scholar] [CrossRef]
- Zhu, S.; Gokhale, S.; Jung, J.; Spirollari, E.; Tsai, J.; Arceo, J.; Wu, B.W.; Victor, E.; Xie, P. Multifaceted Immunomodulatory Effects of the BTK Inhibitors Ibrutinib and Acalabrutinib on Different Immune Cell Subsets—Beyond B Lymphocytes. Front. Cell. Dev. Biol. 2021, 9, 727531. [Google Scholar] [CrossRef]
- Emerson, D.A.; Rolig, A.S.; Redmond, W.L. Enhancing the Generation of Eomeshi CD8þ T Cells Augments the Efficacy of OX40- And CTLA-4-targeted Immunotherapy. Cancer Immunol. Res. 2021, 9, 430–440. [Google Scholar] [CrossRef]
- Liu, X.J.; Liu, X.; Pang, X.J.; Yuan, X.-Y.; Yu, G.X.; Li, Y.R.; Guan, Y.F.; Zhang, Y.B.; Song, J.; Zhang, Q.R.; et al. Progress in the Development of Small Molecular Inhibitors of the Bruton’s Tyrosine Kinase (BTK) as a Promising Cancer Therapy. Bioorganic Med. Chem. 2021, 47, 116358. [Google Scholar] [CrossRef]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9, 630942. [Google Scholar] [CrossRef]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2022 Update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef] [PubMed]
- Alu, A.; Lei, H.; Han, X.; Wei, Y.; Wei, X. BTK Inhibitors in the Treatment of Hematological Malignancies and Inflammatory Diseases: Mechanisms and Clinical Studies. J. Hematol. Oncol. 2022, 15, 1–35. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Ashraf, A.; Farooq, T.B.; Anjum, A.; Rehman, S.; Akbar, A.; Kanate, A.; Dean, R.; Ahmed, M.Q.; Tariq, M.J.; et al. Novel Targeted Therapies for Chronic Lymphocytic Leukemia in Elderly Patients: A Systematic Review. Clin. Lymphoma Myeloma Leuk. 2020, 20, e414–e426. [Google Scholar] [CrossRef]
- Roskoski, R. Small Molecule Inhibitors Targeting the EGFR/ErbB Family of Protein-Tyrosine Kinases in Human Cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, S.; Gustine, J.N.; Flynn, C.A.; Leventoff, C.; Little, M.; White, T.; Meid, K.; Treon, S.P.; Castillo, J.J. Dose Reductions in Patients with Waldenström Macroglobulinaemia Treated with Ibrutinib. Br. J. Haematol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Gertz, M.A. Waldenström Macroglobulinemia: 2023 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2023, 98, 348–358. [Google Scholar] [CrossRef]
- Bouclet, F.; Krzisch, D.; Leblond, V.; Tomowiak, C.; Laribi, K.; Ysebaert, L.; Tournilhac, O.; Dartigeas, C.; Leprêtre, S.; Jondreville, L. Waldenström Disease: News and Perspectives in 2022. Bull. Cancer 2022, 110, S0007–S4551. [Google Scholar]
- Castillo, J.J.; Buske, C.; Trotman, J.; Sarosiek, S.; Treon, S.P. Bruton Tyrosine Kinase Inhibitors in the Management of Waldenström Macroglobulinemia. Am. J. Hematol. 2022, 98, 338–347. [Google Scholar] [CrossRef]
- Keam, S.J. Ibrutinib: Pediatric First Approval. Pediatr. Drugs 2022, 25, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinoshita, T.; Sukbuntherng, J.; Chang, B.Y.; Elias, L. Ibrutinib Inhibits ERBB Receptor Tyrosine Kinases and HER2-Amplified Breast Cancer Cell Growth. Mol. Cancer Ther. 2016, 15, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of Myeloid-Derived Suppressor Cells in Cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Varikuti, S.; Singh, B.; Volpedo, G.; Ahirwar, D.K.; Jha, B.K.; Saljoughian, N.; Viana, A.G.; Verma, C.; Hamza, O.; Halsey, G.; et al. Ibrutinib Treatment Inhibits Breast Cancer Progression and Metastasis by Inducing Conversion of Myeloid-Derived Suppressor Cells to Dendritic Cells. Br. J. Cancer 2020, 122, 1005–1013. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truit, M.; Olson, P.; et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Hong, Y.; Wang, S.; Chen, P.; Gu, A.; Guo, X.; Zhao, P. Ibrutinib, a Bruton’s Tyrosine Kinase Inhibitor, Exhibits Antitumoral Activity and Induces Autophagy in Glioblastoma. J. Exp. Clin. Cancer Res. 2017, 36, 96. [Google Scholar] [CrossRef] [PubMed]
- Paydas, S. Management of Adverse Effects/Toxicity of Ibrutinib. Crit. Rev. Oncol. Hematol. 2019, 136, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, H.; Khattab, A.; Faisal, M.S.; Chilkulwar, A.; Albrethsen, M.; Sadashiv, S.; Fazal, S. Case Series of Unique Adverse Events Related to the Use of Ibrutinib in Patients with B-Cell Malignancies—A Single Institution Experience and a Review of Literature. J. Oncol. Pharm. Pract. 2019, 25, 1265–1270. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, H.; Yang, M.; Xu, C.; Eskazan, A.E. Adverse Drug Events Associated with Ibrutinib for the Treatment of Elderly Patients with Chronic Lymphocytic Leukemia: A Systematic Review and Meta-Analysis of Randomized Trials. Medicine 2019, 98, e16915. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.Y.; Zhou, S.N.; Pan, W.T.; Sun, J.; Yang, L.Q.; Zhang, L.; Qiu, M.Z.; Yang, D.J. A Multi-Kinase Inhibitor APG-2449 Enhances the Antitumor Effect of Ibrutinib in Esophageal Squamous Cell Carcinoma via EGFR/FAK Pathway Inhibition. Biochem. Pharmacol. 2021, 183, 114318. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Elkholy, K.H.; Wani, N.A.; Li, D.; Hu, P.; Barajas, J.M.; Yu, L.; Zhang, X.; Jacob, S.T.; Khan, W.N.; et al. Ibrutinib Potentiates Antihepatocarcinogenic Efficacy of Sorafenib by Targeting EGFR in Tumor Cells and BTK in Immune Cells in the Stroma. Mol. Cancer. Ther. 2020, 19, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Huang, Y.; Zhang, B.; Lin, N. The Effect of Ibrutinib on Radiosensitivity in Pancreatic Cancer Cells by Targeting EGFR/AKT/MTOR Signaling Pathway. Biomed. Pharmacother. 2020, 128, 110133. [Google Scholar] [CrossRef]
- Hershkovitz-Rokah, O.; Pulver, D.; Lenz, G.; Shpilberg, O. Ibrutinib Resistance in Mantle Cell Lymphoma: Clinical, Molecular and Treatment Aspects. Br. J. Haematol. 2018, 181, 306–319. [Google Scholar] [CrossRef]
- Liu, L.; Shi, B.; Wang, X.; Xiang, H. Strategies to Overcome Resistance Mutations of Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. Future Med. Chem. 2018, 10, 343–356. [Google Scholar] [CrossRef]
- Agarwal, R.; Dawson, M.A.; Dreyling, M.; Tam, C.S. Understanding Resistance Mechanisms to BTK and BCL2 Inhibitors in Mantle Cell Lymphoma: Implications for Design of Clinical Trials. Leuk. Lymphoma 2018, 59, 2769–2781. [Google Scholar] [CrossRef]
- Sun, S.-L.; Wu, S.-H.; Kang, J.-B.; Ma, Y.-Y.; Chen, L.; Cao, P.; Chang, L.; Ding, N.; Xue, X.; Li, N.-G.; et al. Medicinal Chemistry Strategies for the Development of Bruton’s Tyrosine Kinase Inhibitors against Resistance. J. Med. Chem. 2022, 65, 7415–7437. [Google Scholar] [CrossRef]
- Nakhoda, S.; Vistarop, A.; Wang, Y.L. Resistance to Bruton Tyrosine Kinase Inhibition in Chronic Lymphocytic Leukaemia and Non-Hodgkin Lymphoma. Br. J. Haematol. 2022, 200, 137–149. [Google Scholar] [CrossRef]
- Stephens, D.M.; Byrd, J.C. Resistance to Bruton Tyrosine Kinase Inhibitors: The Achilles Heel of Their Success Story in Lymphoid Malignancies. Blood 2021, 138, 1099–1109. [Google Scholar] [CrossRef]
- Ye, H.; Huang, S.; Liu, Y.; Chen, Z.; Wang, M.; Jiang, V.C. Dual Targeting of PI3K and BCL-2 Overcomes Ibrutinib Resistance in Aggressive Mantle Cell Lymphoma. J. Cell. Mol. Med. 2022, 26, 3068–3073. [Google Scholar] [CrossRef]
- George, B.; Mullick Chowdhury, S.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers 2020, 12, 1328. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.M.; Byrd, J.C. Next-Generation Bruton Tyrosine Kinase Inhibitors. J. Clin. Oncol. 2020, 38, 2937–2940. [Google Scholar] [CrossRef]
- Iskierka-Jażdżewska, E.; Obracaj, A.; Urbaniak, M.; Robak, T. New Treatment Options for Newly-Diagnosed and Relapsed Chronic Lymphocytic Leukemia. Curr. Treat. Options Oncol. 2022, 23, 775–795. [Google Scholar] [CrossRef]
- Timofeeva, N.; Gandhi, V. Ibrutinib Combinations in CLL Therapy: Scientific Rationale and Clinical Results. Blood Cancer J. 2021, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Wang, J.; Hong, J. Next-Generation Bruton’s Tyrosine Kinase (BTK) Inhibitors Potentially Targeting BTK C481S Mutation- Recent Developments and Perspectives. Curr. Top Med. Chem. 2022, 22, 1674–1691. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.; Liu, Y.; Wang, C.; Xu, Z.; Zhang, Y.; Liu, Y.; Zhao, G.; Ling, Y. Review of the Development of BTK Inhibitors in Overcoming the Clinical Limitations of Ibrutinib. Eur. J. Med. Chem. 2022, 229, 114009. [Google Scholar] [CrossRef]
- CALQUENCE® (Acalabrutinib): Prescribing Information. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/210259s000lbl.pdf (accessed on 28 December 2022).
- Lipsky, A.H.; Lamanna, N. Novel Combination Approaches with Targeted Agents in Frontline Chronic Lymphocytic Leukemia. Cancer 2022, 129, 18–31. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Nuri Yenerel, M.; Illés, A.; Kay, N.; et al. Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J. Clin. Oncol. 2021, 39, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 Study of the Selective BTK Inhibitor Zanubrutinib in B-Cell Malignancies and Safety and Efficacy Evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef] [PubMed]
- BRUKINSA® (Zanubrutinib): Prescribing Information. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213217s005lbl.pdf (accessed on 28 December 2022).
- Syed, Y.Y. Zanubrutinib: First Approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Song, Y.; Sun, M.; Qi, J.; Xu, W.; Zhou, J.; Li, D.; Li, J.; Qiu, L.; Du, C.; Guo, H.; et al. A Two-Part, Single-Arm, Multicentre, Phase I Study of Zanubrutinib, a Selective Bruton Tyrosine Kinase Inhibitor, in Chinese Patients with Relapsed/Refractory B-Cell Malignancies. Br. J. Haematol. 2022, 198, 62–72. [Google Scholar] [CrossRef]
- Tam, C.S.; Dimopoulos, M.; Garcia-Sanz, R.; Trotman, J.; Opat, S.; Roberts, A.W.; Owen, R.; Song, Y.; Xu, W.; Zhu, J.; et al. Pooled Safety Analysis of Zanubrutinib Monotherapy in Patients with B-Cell Malignancies. In Proceedings of the Blood Advances. Am. Soc. Hematol. 2022, 6, 1296–1308. [Google Scholar]
- Brown, J.R.; Eichhorst, B.; Hillmen, P.; Jurczak, W.; Kaźmierczak, M.; Lamanna, N.; O’Brien, S.M.; Tam, C.S.; Qiu, L.; Zhou, K.; et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 388, 319–332. [Google Scholar] [CrossRef]
- Muñoz, J.; Paludo, J.; Sarosiek, S.; Castillo, J.J. Coming of Age for BTK Inhibitor Therapy: A Review of Zanubrutinib in Waldenström Macroglobulinemia. Cells 2022, 11, 3287. [Google Scholar] [CrossRef]
- Dhillon, S. Tirabrutinib: First Approval. Drugs 2020, 80, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, N.; Rai, S.; Munakata, W.; Suzuki, K.; Handa, H.; Shibayama, H.; Endo, T.; Terui, Y.; Iwaki, N.; Fukuhara, N.; et al. Two-Year Outcomes of Tirabrutinib Monotherapy in Waldenström’s Macroglobulinemia. Cancer Sci. 2022, 113, 2085–2096. [Google Scholar] [CrossRef]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.P.; Cull, G.; Owen, R.G.; Marlton, P.; Ewahlin, B.; Sanz, R.G.; et al. A Randomized Phase 3 Trial of Zanubrutinib vs Ibrutinib in Symptomatic Waldenström Macroglobulinemia: The ASPEN Study. Blood 2020, 136, 2038–2050. [Google Scholar] [CrossRef]
- Dhillon, S. Orelabrutinib: First Approval. Drugs 2021, 81, 503–507. [Google Scholar] [CrossRef]
- Gu, D.; Li, J.; Miao, Y. Evaluating Orelabrutinib as a Novel Treatment Option for Relapsed/Refractory Chronic Lymphocytic Leukemia in China. Expert. Opin. Pharmacother. 2022, 23, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Witkowska, M.; Smolewski, P. The Role of Bruton’s Kinase Inhibitors in Chronic Lymphocytic Leukemia: Current Status and Future Directions. Cancers 2022, 14, 771. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, L.; Xue, Y.; Zhou, S.; Li, Y. An Open-label, Phase 1, Randomized, Three Treatments, Three-period, Crossover, Relative Bioavailability Study of CC-292, a Potent and Orally Available Inhibitor of Bruton Tyrosine Kinase. J. Clin. Pharm. Ther. 2022, 47, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Ribrag, V.; Chavez, J.C.; Boccomini, C.; Kaplan, J.; Chandler, J.C.; Santoro, A.; Corradini, P.; Flinn, I.W.; Advani, R.; Cassier, P.A.; et al. Phase Ib Study of Combinations of Avadomide (CC-122), CC-223, CC-292, and Rituximab in Patients with Relapsed/Refractory Diffuse Large B-cell Lymphoma. EJHaem 2022, 3, 139–153. [Google Scholar] [CrossRef]
- Brown, J.R.; Harb, W.A.; Hill, B.T.; Gabrilove, J.; Sharman, J.P.; Schreeder, M.T.; Barr, P.M.; Foran, J.M.; Miller, T.P.; Burger, J.A.; et al. Phase I Study of Single-Agent CC-292, a Highly Selective Bruton’s Tyrosine Kinase Inhibitor, in Relapsed/Refractory Chronic Lymphocytic Leukemia. Haematologica 2016, 101, e295–e298. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.H.; Kivitz, A.J.; Ma, J.; Korish, S.; Sutherland, D.; Li, L.; Azaryan, A.; Kosek, J.; Adams, M.; Capone, L.; et al. Spebrutinib (CC-292) Affects Markers of B Cell Activation, Chemotaxis, and Osteoclasts in Patients with Rheumatoid Arthritis: Results from a Mechanistic Study. Rheumatol. Ther. 2020, 7, 101–119. [Google Scholar] [CrossRef]
- Montalban, X.; Wallace, D.; Genovese, M.C.; Tomic, D.; Parsons-Rich, D.; le Bolay, C.; Kao, A.H.; Guehring, H. Characterisation of the Safety Profile of Evobrutinib in over 1000 Patients from Phase II Clinical Trials in Multiple Sclerosis, Rheumatoid Arthritis and Systemic Lupus Erythematosus: An Integrated Safety Analysis. J. Neurol. Neurosurg. Psychiatry 2023, 94, 1–9. [Google Scholar] [CrossRef]
- Haselmayer, P.; Camps, M.; Liu-Bujalski, L.; Nguyen, N.; Morandi, F.; Head, J.; O’Mahony, A.; Zimmerli, S.C.; Bruns, L.; Bender, A.T.; et al. Efficacy and Pharmacodynamic Modeling of the BTK Inhibitor Evobrutinib in Autoimmune Disease Models. J. Immunol. 2019, 202, 2888–2906. [Google Scholar] [CrossRef]
- Papasouliotis, O.; Mitchell, D.; Girard, P.; Dyroff, M. Population Pharmacokinetic and Pharmacodynamic Modeling of Evobrutinib in Healthy Adult Participants. Clin. Transl. Sci. 2022, 15, 2899–2908. [Google Scholar] [CrossRef]
- Papasouliotis, O.; Mitchell, D.; Girard, P.; Dangond, F.; Dyroff, M. Determination of a Clinically Effective Evobrutinib Dose: Exposure–Response Analyses of a Phase II Relapsing Multiple Sclerosis Study. Clin. Transl. Sci. 2022, 15, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Dörner, T.; Pisetsky, D.S.; Sanchez-Guerrero, J.; Patel, A.C.; Parsons-Rich, D.; le Bolay, C.; Drouin, E.E.; Kao, A.H.; Guehring, H.; et al. Efficacy and Safety of the Bruton’s Tyrosine Kinase Inhibitor Evobrutinib in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Dose-Ranging Trial. ACR Open Rheumatol. 2022, 5, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.L.; Cheah, C.Y. Non-Covalent BTK Inhibitors-The New BTKids on the Block for B-Cell Malignancies. J. Pers. Med. 2021, 11, 764. [Google Scholar] [CrossRef]
- Aslan, B.; Hubner, S.E.; Fox, J.A.; Taverna, P.; Wierda, W.G.; Kornblau, S.M.; Gandhi, V. Vecabrutinib Inhibits B-Cell Receptor Signal Transduction in Chronic Lymphocytic Leukemia Cell Types with Wild-Type or Mutant Bruton Tyrosine Kinase. Haematologica 2022, 107, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.N.; Pinilla-Ibarz, J.; Gladstone, D.E.; Patel, K.; Sharman, J.P.; Wierda, W.G.; Choi, M.Y.; O’Brien, S.M.; Shadman, M.; Davids, M.S.; et al. Phase Ib Dose-Escalation Study of the Selective, Noncovalent, Reversible Bruton’s Tyrosine Kinase Inhibitor Vecabrutinib in B-Cell Malignancies. Haematologica 2022, 107, 984–987. [Google Scholar] [CrossRef]
- Jebaraj, B.M.C.; Müller, A.; Dheenadayalan, R.P.; Endres, S.; Roessner, P.M.; Seyfried, F.; Walliser, C.; Wist, M.; Qi, J.; Tausch, E.; et al. Evaluation of Vecabrutinib as a Model for Noncovalent BTK/ITK Inhibition for Treatment of Chronic Lymphocytic Leukemia. Blood 2022, 139, 859–875. [Google Scholar] [CrossRef]
- Hou, J.Z.; Ye, J.C.; Pu, J.J.; Liu, H.; Ding, W.; Zheng, H.; Liu, D. Novel Agents and Regimens for Hematological Malignancies: Recent Updates from 2020 ASH Annual Meeting. J. Hematol. Oncol. 2021, 14, 66. [Google Scholar] [CrossRef]
- Aslan, B.; Kismali, G.; Iles, L.R.; Manyam, G.C.; Ayres, M.L.; Chen, L.S.; Gagea, M.; Bertilaccio, M.T.S.; Wierda, W.G.; Gandhi, V. Pirtobrutinib Inhibits Wild-Type and Mutant Bruton’s Tyrosine Kinase-Mediated Signaling in Chronic Lymphocytic Leukemia. Blood Cancer J. 2022, 12, 80. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in Relapsed or Refractory B-Cell Malignancies (BRUIN): A Phase 1/2 Study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Cohen, J.B.; Shah, N.N.; Alencar, A.J.; Gerson, J.N.; Patel, M.R.; Fakhri, B.; Jurczak, W.; Tan, X.N.; Lewis, K.L.; Fenske, T.; et al. MCL-133 Pirtobrutinib, a Highly Selective, Non-Covalent (Reversible) BTK Inhibitor in Previously Treated Mantle Cell Lymphoma: Updated Results From the Phase 1/2 BRUIN Study. Clin. Lymphoma Myeloma Leuk. 2022, 22, S394–S395. [Google Scholar] [CrossRef]
- Eyre, T.A.; Shah, N.N.; Dreyling, M.; Jurczak, W.; Wang, Y.; Cheah, C.Y.; Song, Y.; Gandhi, M.; Chay, C.; Sharman, J.; et al. BRUIN MCL-321: Phase III Study of Pirtobrutinib versus Investigator Choice of BTK Inhibitor in BTK Inhibitor Naïve Mantle Cell Lymphoma. Future Oncol. 2022, 18, 3961–3969. [Google Scholar] [CrossRef]
- Munshi, M.; Liu, X.; Kofides, A.; Tsakmaklis, N.; Gustine, J.; Sarosiek, S.; Flynn, C.A.; Meid, K.; White, T.P.; Leventoff, C.; et al. Pirtobrutinib (LOXO-305) Is Active and Overcomes ERK Related Pro-Survival Signaling in Ibrutinib Resistant, BTK Cys481 Mutant Expressing WM and ABC DLBCL Lymphoma Cells Driven By Activating MYD88 Mutations. Blood 2021, 138, 2261. [Google Scholar] [CrossRef]
- Ito, R.; Eyre, T.A.; Shah, N.N.; Le Gouill, S.; Dreyling, M.; Vandenberghe, E.; Jurczak, W.; Wang, Y.; Cheah, C.Y.; Gandhi, M.; et al. MCL-135 BRUIN MCL-321, a Phase 3 Open-Label, Randomized Study of Pirtobrutinib Versus Investigator Choice of BTK Inhibitor in Patients With Previously Treated, BTK Inhibitor Naïve Mantle Cell Lymphoma (Trial in Progress). Clin. Lymphoma Myeloma Leuk. 2022, 22, S395–S396. [Google Scholar] [CrossRef]
- Study of BTK Inhibitor LOXO-305 Versus Approved BTK Inhibitor Drugs in Patients With Mantle Cell Lymphoma (MCL)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04662255 (accessed on 17 January 2023).
- Qiu, H.; Ali, Z.; Bender, A.; Caldwell, R.; Chen, Y.Y.; Fang, Z.; Gardberg, A.; Glaser, N.; Goettsche, A.; Goutopoulos, A.; et al. Discovery of Potent and Selective Reversible Bruton’s Tyrosine Kinase Inhibitors. Bioorg. Med. Chem. 2021, 40, 116163. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A.; et al. Fenebrutinib Versus Placebo or Adalimumab in Rheumatoid Arthritis: A Randomized, Double-Blind, Phase II Trial. Arthritis Rheumatol. 2020, 72, 1435–1446. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Commerford, S.R.; Okrah, K.; et al. First-in-Human Phase 1 Study of the BTK Inhibitor GDC-0853 in Relapsed or Refractory B-Cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef]
- Metz, M.; Sussman, G.; Gagnon, R.; Staubach, P.; Tanus, T.; Yang, W.H.; Lim, J.J.; Clarke, H.J.; Galanter, J.; Chinn, L.W.; et al. Fenebrutinib in H1 Antihistamine-Refractory Chronic Spontaneous Urticaria: A Randomized Phase 2 Trial. Nat. Med. 2021, 27, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.; Furie, R.; Jones, N.S.; Guibord, P.; Galanter, J.; Lee, C.; McGregor, A.; Toth, B.; Rae, J.; Hwang, O.; et al. Efficacy, Safety, and Pharmacodynamic Effects of the Bruton’s Tyrosine Kinase Inhibitor Fenebrutinib (GDC-0853) in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2021, 73, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Faia, K.; White, K.; Murphy, E.; Proctor, J.; Pink, M.; Kosmider, N.; McGovern, K.; Kutok, J. The Phosphoinositide-3 Kinase (PI3K)-δ,γ Inhibitor, Duvelisib Shows Preclinical Synergy with Multiple Targeted Therapies in Hematologic Malignancies. PLoS ONE 2018, 13, e0200725. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.M.; Michaud, L.; Whiting, K.; Nakajima, R.; Nichols, C.; de Frank, S.; Hamlin, P.A.; Matasar, M.J.; Gerecitano, J.F.; Drullinsky, P.; et al. Phase I/Ib Study of the Efficacy and Safety of Buparlisib and Ibrutinib Therapy in MCL, FL, and DLBCL with Serial Cell-Free DNA Monitoring. Clin. Cancer Res. 2022, 28, 45–56. [Google Scholar] [CrossRef]
- Zhu, J.; He, Y.; Li, J.; Ding, N.; Wang, X.; Song, Y. Combination of Enzastaurin and Ibrutinib Synergistically Induces Anti-Tumor Effects in Diffuse Large B Cell Lymphoma. Blood 2018, 132, 1666. [Google Scholar] [CrossRef]
- Yahiaoui, A.; Meadows, S.A.; Sorensen, R.A.; Cui, Z.-H.; Keegan, K.S.; Brockett, R.; Chen, G.; Quéva, C.; Li, L.; Tannheimer, S.L. PI3Kδ Inhibitor Idelalisib in Combination with BTK Inhibitor ONO/GS-4059 in Diffuse Large B Cell Lymphoma with Acquired Resistance to PI3Kδ and BTK Inhibitors. PLoS ONE 2017, 12, e0171221. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Xie, Y.; Ying, Z.; Liu, W.; Ping, L.; Zhang, C.; Pan, Z.; Ding, N.; Song, Y.; et al. The MTOR Kinase Inhibitor Everolimus Synergistically Enhances the Anti-Tumor Effect of the Bruton’s Tyrosine Kinase (BTK) Inhibitor PLS-123 on Mantle Cell Lymphoma. Int. J. Cancer 2018, 142, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Grassilli, E.; Cerrito, M.G.; Lavitrano, M. BTK, the New Kid on the (Oncology) Block? Front. Oncol. 2022, 12, 944538. [Google Scholar] [CrossRef]
- Li, C.; Fan, C.; Lu, S.; Qiu, Q.; Gao, X.; Yan, X.; Wang, S.; Zhao, B.; Liu, X.; Song, Y.; et al. Targeting Ibrutinib to Tumor-Infiltrating T Cells with a Sialic Acid Conjugate-Modified Phospholipid Complex for Improved Tumor Immunotherapy. Mol. Pharm. 2022, 20, 438–450. [Google Scholar] [CrossRef]
- Garg, N.; Padron, E.J.; Rammohan, K.W.; Goodman, C.F. Bruton’s Tyrosine Kinase Inhibitors: The Next Frontier of B-Cell-Targeted Therapies for Cancer, Autoimmune Disorders, and Multiple Sclerosis. J. Clin. Med. 2022, 11, 6139. [Google Scholar] [CrossRef]
- Shirley, M. Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies: Their Use and Differential Features. Target Oncol. 2022, 17, 69–84. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Synonyms | Type of Inhibitor | Indications | Year of Approval | Activity (IC50) |
|---|---|---|---|---|---|
| Ibrutinib Imbruvica® | CRA-032765 PC-32765 PCI-32765 PCI-32765-00 | Irreversible | MCL, CLL, SLL, WM, MZL, GVHD | 2013 | BTK IC50 = 0.47 nM |
| ITK IC50 = 55 nM | |||||
| TEC IC50 = 3.2 nM | |||||
| Acalabrutinib Calquence® | ACP-196 | Irreversible | CLL, SLL, MCL | 2017 | BTK IC50 = 2.5 nM |
| ITK IC50 > 20,000 nM | |||||
| TEC IC50 = 37 nM | |||||
| Zanubrutinib Brukinsa® | BGB-3111 | Irreversible | NHL, CLL, MCL | 2019 | BTK IC50 = 0.3 nM |
| ITK IC50 = 56 nM | |||||
| TEC IC50 = 2 nM | |||||
| Tirabrutinib Velexbru® | ONO-4059 | Irreversible | CNS lymphoma, WM, CLL | 2020 | BTK IC50 = 6.8 nM |
| ITK IC50 > 20,000 nM | |||||
| TEC IC50 = 48 nM | |||||
| Orelabrutinib | ICP-022 | Irreversible | MCL, CLL, SLL | 2020 | BTK IC50 = 1.6 nM |
| Name | Synonyms | Type of Inhibitor | Indications | Activity (IC50) |
|---|---|---|---|---|
| Spebrutinib | CC-292 AVL-292 | Irreversible | CLL, NHL | BTK IC50 = 9.2 nM |
| ITK IC50 = 1050 nM | ||||
| TEC IC50 = 8.4 nM | ||||
| Evobrutinib | M2951 MSC-2364447C | Irreversible | RA, MS | BTK IC50 = 8.9 nM |
| Vecabrutinib | SNS-062 | Reversible | CLL, SLL | BTK IC50 = 1.9 nM |
| Pirtobrutinib | LOXO-305 | Reversible | CLL, MCL | BTK IC50 = 3.15 nM |
| ITK IC50 > 5000 nM | ||||
| TEC IC50 = 1234 nM | ||||
| Fenebrutinib | GDC-0853 | Reversible | RA, SLE, CSU | BTK IC50 = 2.3 nM |
| ITK IC50 = 1000 nM | ||||
| TEC IC50 = 1000 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozkiewicz, D.; Hermanowicz, J.M.; Kwiatkowska, I.; Krupa, A.; Pawlak, D. Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials. Molecules 2023, 28, 2400. https://doi.org/10.3390/molecules28052400
Rozkiewicz D, Hermanowicz JM, Kwiatkowska I, Krupa A, Pawlak D. Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials. Molecules. 2023; 28(5):2400. https://doi.org/10.3390/molecules28052400
Chicago/Turabian StyleRozkiewicz, Dariusz, Justyna Magdalena Hermanowicz, Iwona Kwiatkowska, Anna Krupa, and Dariusz Pawlak. 2023. "Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials" Molecules 28, no. 5: 2400. https://doi.org/10.3390/molecules28052400
APA StyleRozkiewicz, D., Hermanowicz, J. M., Kwiatkowska, I., Krupa, A., & Pawlak, D. (2023). Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials. Molecules, 28(5), 2400. https://doi.org/10.3390/molecules28052400