
Study on Pharmacokinetics and Metabolic Profiles of Novel Potential PLK-1 Inhibitors by UHPLC-MS/MS Combined with UHPLC-Q-Orbitrap/HRMS
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis, Characterization and Biological Evaluation of Compound 7a
2.2. UHPLC-MS/MS Method Development and Validation
2.3. Pharmacokinetic Study of Compound 7a
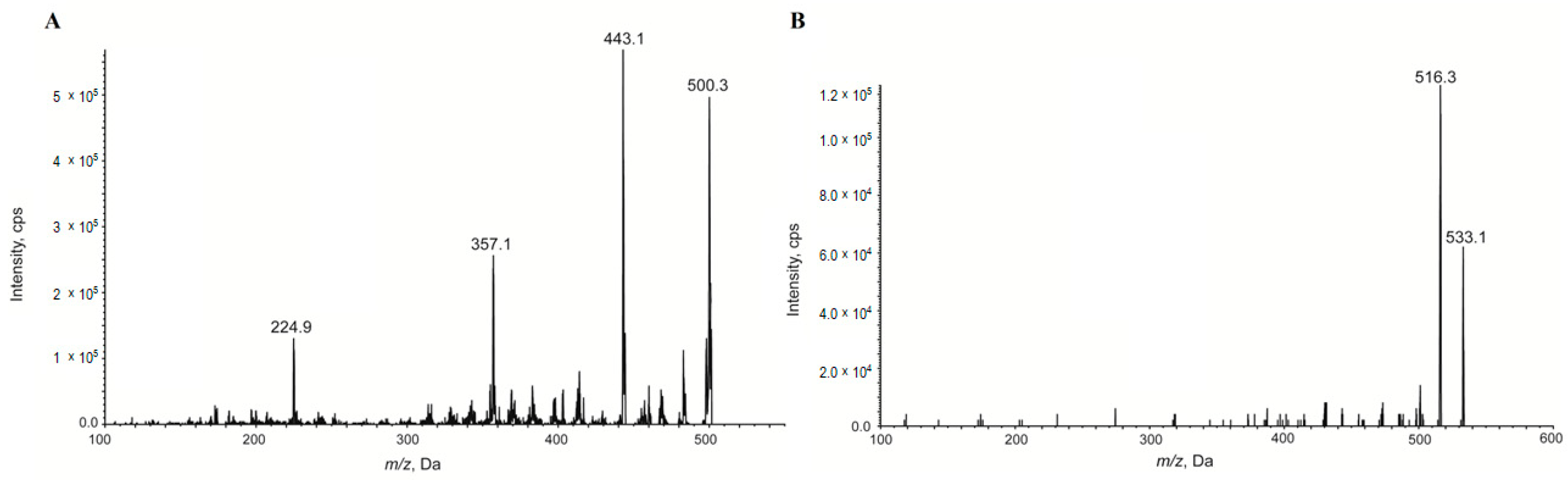
2.4. Metabolism of Compound 7a
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Compound 7a
3.3. Kinase Inhibition Activity
3.4. Cell Proliferation Inhibition Activity
3.5. Method Development and Validation by UHPLC-MS/MS
3.6. Pharmacokinetic Study
3.7. The Metabolism of 7a in Liver Microsomes
3.8. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ben-Salem, S.; Venkadakrishnan, V.B.; Heemers, H.V. Novel insights in cell cycle dysregulation during prostate cancer progression. Endocr. Relat. Cancer 2021, 28, R141–R155. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Bai, X.; Zhang, W.; Ma, M.; Wang, W.; Liang, Y.; Wang, H.; Tian, J.; Yu, P. LPM3770277, a potent novel CDK4/6 degrader, exerts antitumor effect against triple-negative breast cancer. Front. Pharmacol. 2022, 13, 853993. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, I.; Basheer, N.; Hasan, G.M.; Afzal, M.; Hassan, M.I. Polo-like Kinase 1 as an emerging drug target: Structure, function and therapeutic implications. J. Drug Target. 2021, 29, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krssák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Hyun, S.Y.; Hwang, H.I.; Jang, Y.J. Polo-like kinase-1 in DNA damage response. BMB Rep. 2014, 47, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharma, A.R.; Sharma, G.; Chakraborty, C.; Kim, J. PLK-1: Angel or devil for cell cycle progression. Biochim. Biophys. Acta 2016, 1865, 190–203. [Google Scholar] [CrossRef]
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Beria, I.; Ballinari, D.; Bertrand, J.A.; Borghi, D.; Bossi, R.T.; Brasca, M.G.; Cappella, P.; Caruso, M.; Ceccarelli, W.; Ciavolella, A.; et al. Identification of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as a new class of orally and selective Polo-like kinase 1 inhibitors. J. Med. Chem. 2010, 53, 3532–3551. [Google Scholar] [CrossRef]
- Liu, X.; Erikson, R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA. 2003, 100, 5789–5794. [Google Scholar] [CrossRef] [Green Version]
- Liu, X. Targeting Polo-Like Kinases: A promising therapeutic approach for cancer treatment. Transl. Oncol. 2015, 8, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, L.; Wang, J.; Ouyang, L.; Wang, Y. Polo-like Kinase 1 inhibitors in human cancer therapy: Development and therapeutic potential. J. Med. Chem. 2022, 65, 10133–10160. [Google Scholar] [CrossRef]
- Stafford, J.M.; Wyatt, M.D.; McInnes, C. Inhibitors of the PLK1 polo-box domain: Drug design strategies and therapeutic opportunities in cancer. Expert Opin. Drug Discov. 2023, accepted. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Lardon, F.; Deschoolmeester, V.; De Pauw, I.; Vermorken, J.B.; Specenier, P.; Pauwels, P.; Peeters, M.; Wouters, A. Spotlight on Volasertib: Preclinical and clinical evaluation of a promising Plk1 inhibitor. Med. Res. Rev. 2016, 36, 749–786. [Google Scholar] [CrossRef]
- Olmos, D.; Barker, D.; Sharma, R.; Brunetto, A.T.; Yap, T.A.; Taegtmeyer, A.B.; Barriuso, J.; Medani, H.; Degenhardt, Y.Y.; Allred, A.J.; et al. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin. Cancer Res. 2011, 17, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Platzbecker, U.; Chromik, J.; Krönke, J.; Handa, H.; Strickland, S.; Miyazaki, Y.; Wermke, M.; Sakamoto, W.; Tachibana, Y.; Taube, T.; et al. Volasertib as a monotherapy or in combination with azacitidine in patients with myelodysplastic syndrome, chronic myelomonocytic leukemia, or acute myeloid leukemia: Summary of three phase I studies. BMC Cancer 2022, 22, 569. [Google Scholar] [CrossRef]
- Wang, D.; Veo, B.; Pierce, A.; Fosmire, S.; Madhavan, K.; Balakrishnan, I.; Donson, A.; Alimova, I.; Sullivan, K.D.; Joshi, M.; et al. A novel PLK1 inhibitor onvansertib effectively sensitizes MYC-driven medulloblastoma to radiotherapy. Neuro. Oncol. 2022, 24, 414–426. [Google Scholar] [CrossRef]
- Beria, I.; Bossi, R.T.; Brasca, M.G.; Caruso, M.; Ceccarelli, W.; Fachin, G.; Fasolini, M.; Forte, B.; Fiorentini, F.; Pesenti, E.; et al. NMS-P937, a 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivative as potent and selective Polo-like kinase 1 inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 2969–2974. [Google Scholar]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer Ther. 2012, 11, 1006–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagege, A.; Ambrosetti, D.; Boyer, J.; Bozec, A.; Doyen, J.; Chamorey, E.; He, X.; Bourget, I.; Rousset, J.; Saada, E.; et al. The Polo-like kinase 1 inhibitor onvansertib represents a relevant treatment for head and neck squamous cell carcinoma resistant to cisplatin and radiotherapy. Theranostics 2021, 11, 9571–9586. [Google Scholar]
- Zeidan, A.M.; Ridinger, M.; Lin, T.L.; Becker, P.S.; Schiller, G.J.; Patel, P.A.; Spira, A.I.; Tsai, M.L.; Samuëlsz, E.; Silberman, S.L.; et al. A Phase Ib study of onvansertib, a novel oral PLK1 inhibitor, in combination therapy for patients with relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 2020, 26, 6132–6140. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chhabra, G.; Singh, C.K.; Ndiaye, M.A.; Ahmad, N. PLK1 inhibition-based combination therapies for cancer management. Transl. Oncol. 2022, 16, 101332. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.P.; Kim, H.; Bae, H.; Hah, J.M. Design and synthesis of aminopyrimidinyl pyrazole analogs as PLK1 inhibitors using hybrid 3D-QSAR and molecular docking. Pharmaceuticals 2022, 15, 1170. [Google Scholar] [CrossRef]
- Caruso, M.; Valsasina, B.; Ballinari, D.; Bertrand, J.; Brasca, M.G.; Caldarelli, M.; Cappella, P.; Fiorentini, F.; Gianellini, L.M.; Scolaro, A.; et al. 5-(2-amino-pyrimidin-4-yl)-1H-pyrrole and 2-(2-amino-pyrimidin-4-yl)-1,5,6,7-tetrahydro-pyrrolo[3,2-c]pyridin-4-one derivatives as new classes of selective and orally available Polo-like kinase 1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 96–101. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration | Measured Concentration | Precision | Accuracy | |
|---|---|---|---|---|
| (nmol/L) | Mean ± SD (nmol/L) | RSD (%) | RE (%) | |
| Intra-day (n = 6) | 1 | 1.00 ± 0.07 | 6.46 | 0.46 |
| 3 | 3.11 ± 0.22 | 7.19 | 3.67 | |
| 300 | 274 ± 10.7 | 3.91 | −8.62 | |
| 750 | 665 ± 28.9 | 4.34 | −11.3 | |
| Inter-day (n = 18) | 1 | 1.06 ± 0.10 | 10.2 | 5.5 |
| 3 | 2.77 ± 0.31 | 11.1 | −7.67 | |
| 300 | 275 ± 13.5 | 4.91 | −8.39 | |
| 750 | 684 ± 31.1 | 4.55 | −8.76 |
| Analyte | Concentration | Recovery (%) | Matrix Effect (%) | ||
|---|---|---|---|---|---|
| (nmol/L) | Mean ± SD | RSD (%) | Mean ± SD | RSD (%) | |
| 7a (n = 6) | 3.00 | 79.0 ± 3.54 | 5.09 | 51.6 ± 2.50 | 7.88 |
| 300.00 | 76.3 ± 3.20 | 60.0 ± 1.98 | |||
| 750.00 | 81.8 ± 3.76 | 52.7 ± 1.77 | |||
| IS (n = 18) | 40.00 | 107 ± 6.74 | 6.27 | 43.5 ± 0.83 | 3.52 |
| Conditions | Nominal Concentrations (nM) | Measured Concentrations | RSD (%) | RE (%) |
|---|---|---|---|---|
| (nM) | ||||
| Room temperature for 4 h | 3 | 2.59 ± 0.17 | 6.47 | −13.7 |
| 300 | 261 ± 4.94 | 1.89 | −13 | |
| 750 | 649 ± 17.9 | 2.76 | −13.4 | |
| Freeze-thaw for 3 cycles | 3 | 2.73 ± 0.21 | 7.56 | −9.17 |
| 300 | 260 ± 10.8 | 4.16 | −13.4 | |
| 750 | 649 ± 24.2 | 3.74 | −13.5 | |
| Autosampler (4 °C for 24 h) | 3 | 3.05 ± 0.10 | 3.15 | 1.78 |
| 300 | 280 ± 11.9 | 4.26 | −6.67 | |
| 750 | 701 ± 46.5 | 6.63 | −6.58 | |
| Freeze (−20 °C for 7 days) | 3 | 2.55 ± 0.13 | 5.08 | −14.9 |
| 300 | 272 ± 9.67 | 3.55 | −9.33 | |
| 750 | 685 ± 34.7 | 5.08 | −8.71 |
| T1/2 (h) | Tmax(h) | Cmax (nmol/L) | AUC0–24h | F | ||
|---|---|---|---|---|---|---|
| (h* nmol/L) | (%) | |||||
| 5 mg/kg | M | 4.4 ± 0.9 | 3.0 ± 2.6 | 87 ± 45 | 565 ± 132 | 23.90% |
| F | 4.6 ± 1.4 | 0.8 ± 1.0 | 102 ± 25 | 469 ± 64 | 19.90% | |
| mean | 4.5 ± 1.1 | 1.9 ± 2.1 | 95 ± 33 | 517 ± 106 | 21.90% | |
| 30 mg/kg | M | 3.7 ± 1.3 | 2.0 ± 1.9 | 435 ± 81 | 3221 ± 1039 | 22.70% |
| F | 6.1 ± 0.9 | 2.3 ± 1.5 | 428 ± 90 | 3162 ± 1021 | 22.30% | |
| mean | 5.1 ± 1.6 | 2.2 ± 1.6 | 432 ± 77 | 3192 ± 922 | 22.50% |
| Symbol | RT (min) | Elemental Composition | Observed m/z | Theoretical m/z | Error (ppm) | Mass Shift | Metabolic Pathway | RLM (%) | HLM (%) |
|---|---|---|---|---|---|---|---|---|---|
| parent | 16.28 | C24H24F3N7O2 | 500.2016 | 500.2022 | −1.20 | 0 | parent | 5.34 | 43.73 |
| M1 | 14.98 | C23H22F3N7O3 | 502.1809 | 502.1814 | −1.00 | 1.9793 | +O-CH2 | 19.51 | 1.03 |
| M2 | 16.15 | C23H22F3N7O2 | 486.1860 | 486.1865 | −1.03 | −14.0156 | -CH2 | 6.53 | 34.76 |
| M3 | 17.13 | C24H24F3N7O3 | 516.1965 | 516.1971 | −1.16 | 15.9949 | +O☐ | 68.62 | 20.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Lei, H.; Lu, J.; Wang, W.; Liu, C.; Wang, Y.; Yang, Y.; Tian, J.; Zhang, J. Study on Pharmacokinetics and Metabolic Profiles of Novel Potential PLK-1 Inhibitors by UHPLC-MS/MS Combined with UHPLC-Q-Orbitrap/HRMS. Molecules 2023, 28, 2550. https://doi.org/10.3390/molecules28062550
Wang L, Lei H, Lu J, Wang W, Liu C, Wang Y, Yang Y, Tian J, Zhang J. Study on Pharmacokinetics and Metabolic Profiles of Novel Potential PLK-1 Inhibitors by UHPLC-MS/MS Combined with UHPLC-Q-Orbitrap/HRMS. Molecules. 2023; 28(6):2550. https://doi.org/10.3390/molecules28062550
Chicago/Turabian StyleWang, Lin, Hui Lei, Jing Lu, Wenyan Wang, Chunjiao Liu, Yunjie Wang, Yifei Yang, Jingwei Tian, and Jianzhao Zhang. 2023. "Study on Pharmacokinetics and Metabolic Profiles of Novel Potential PLK-1 Inhibitors by UHPLC-MS/MS Combined with UHPLC-Q-Orbitrap/HRMS" Molecules 28, no. 6: 2550. https://doi.org/10.3390/molecules28062550
APA StyleWang, L., Lei, H., Lu, J., Wang, W., Liu, C., Wang, Y., Yang, Y., Tian, J., & Zhang, J. (2023). Study on Pharmacokinetics and Metabolic Profiles of Novel Potential PLK-1 Inhibitors by UHPLC-MS/MS Combined with UHPLC-Q-Orbitrap/HRMS. Molecules, 28(6), 2550. https://doi.org/10.3390/molecules28062550