The analytical target profile (ATP) is a useful tool to define a priori quality criteria for results generated by analytical methods. For the ACC studied in this work, the method criteria described below are linked to product specifications based on process capability. In the present work, the following criteria were defined: (i) the retention factor, k, should be at least equal to 1 for all peaks of interest; (ii) the overall chromatographic resolution should be at least equal to 1.5; (iii) the limit of quantitation (LOQ) should be ideally equal to or below 0.1 µg/mL for EDC-urea, 4 µg/mL for succinimide and N-hydroxysuccinimide, and 0.05 µg/mL for 3,2-HOPO-chelator and 3,2-HOPO-TOP-succinimide; (iv) linearity should be not less than R2 = 0.998; and (v) precision should be less than 2% and 6% for UV and MS detection, respectively. Each of these attributes will be measured and compared with expected values.
2.1. Issue #1: Retention and Detection of Succinimide and N-hydroxysuccinimide
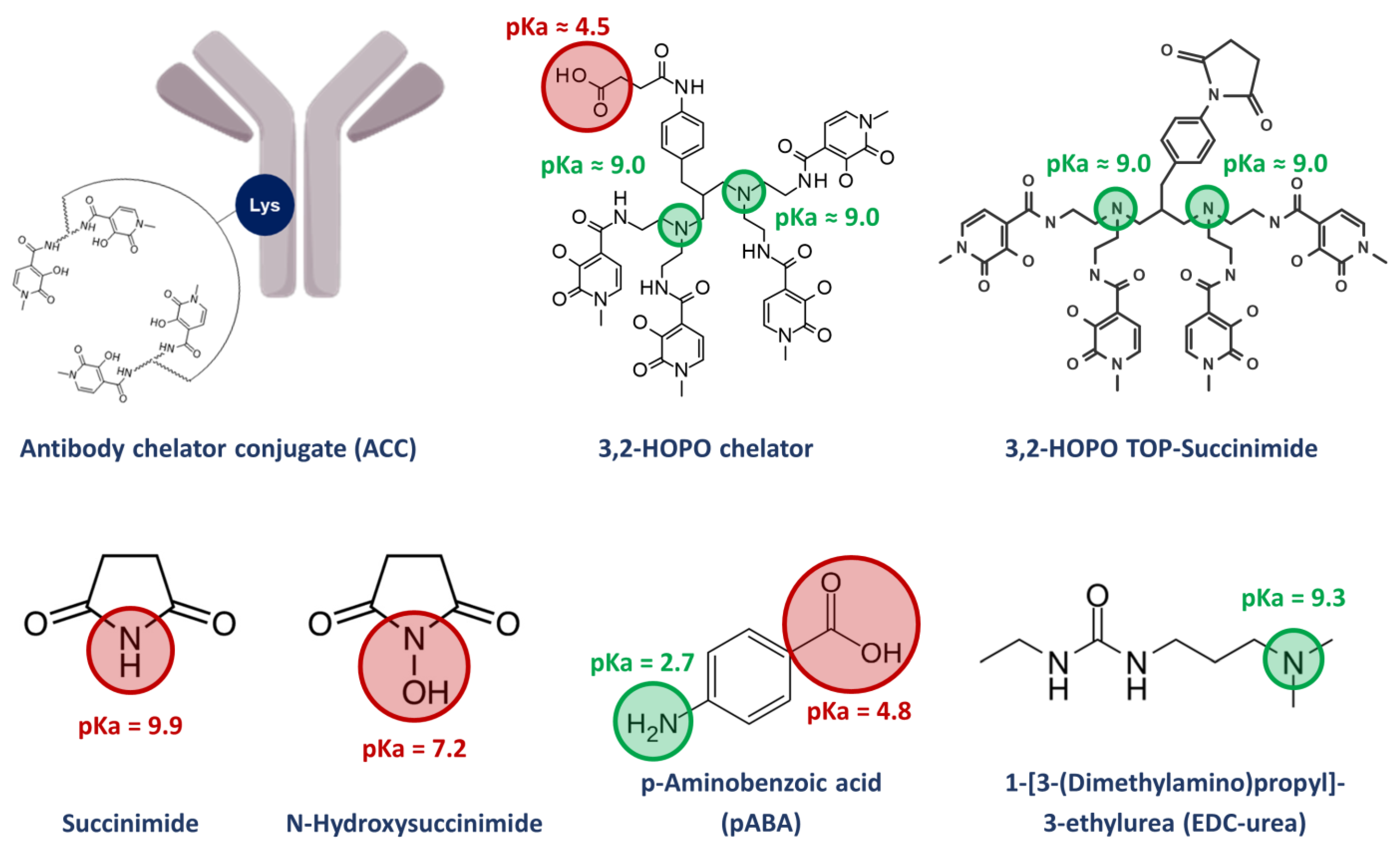
In terms of physico-chemical properties, succinimide has a logP of −0.99 and an acidic pKa of 9.9, while N-hydroxysuccinimide has a logP of −0.99 and an acidic pKa of 7.2. It is well known that RPLC mode is suited for the analysis of compounds having logP ranging from −1 to 6. However, due to the relatively low and very similar logP values of succinimide and N-hydroxysuccinimide, these compounds may not be sufficiently and selectively retained on a regular C18 column (see issue #2 highlighting the potential problems encountered with selectivity). Therefore, alternative column chemistry allowing a highly aqueous mobile phase will be required to achieve a sufficient retention factor (ideally k at least equal to 1, as indicated in the ATP). Ion-pair-RPLC as a possible alternative is hardly applicable, due to the very low ionic character of the compounds at acidic, neutral, or slightly basic conditions. The ionization of these compounds would require the use of highly basic mobile phase conditions, which is hardly compatible with most of the C18 material on the market. Besides the limited retention in RPLC mode, it is also important to notice that succinimide and N-hydroxysuccinimide do not significantly absorb in UV above 200 nm, so this very low wavelength was selected for their detection.
First, it is important to note that no RPLC method is available in the literature to simultaneously analyze succinimide and
N-hydroxysuccinimide. In the present work, rather than using a regular C18 column, we decided to test various commercially available RPLC stationary phases having a significant polar surface activity (see
Supplementary Materials: Table S1 for the detailed characteristics of the columns) [
26]. Among the tested columns, there were a fluorophenyl (CSH fluorophenyl), an ether linked phenyl phase (Synergi Polar-RP), a mixed-mode anion-exchange/RPLC column (Premier BEH C18 AX), and five C18 columns with polar endcapping from different providers (Premier HSS T3, Luna Omega polar, Hypersil GOLD aQ, Zorbax SB Aq, and Kinetex Polar). All the tested stationary phases had the same length, but the internal diameter was larger for the Luna Omega Polar and the Kinetex polar (3 mm I.D.), compared to the other ones (2.0–2.1 mm I.D.). Thus, the flow rate was geometrically adjusted at 0.8 mL min
−1 for the 3 mm I.D. columns and at 0.4 mL min
−1 for the 2.0 and 2.1 mm I.D. ones. In terms of particle morphology, most of the columns were packed with fully porous particles ranging from 1.7 to 3.0 µm, while the Kinetex polar was packed with superficially porous particles of 2.6 µm. In all cases, the column void time varied from 0.60 to 0.89 min and was obtained from the baseline disturbance observed at 200 nm in UV. All of these columns were tested under generic mobile phase conditions and further improvements were then brought to the method afterwards.
The analysis of succinimide and
N-hydroxysuccinimide was carried out using purely aqueous acidified mobile phase to maximize retention and achieve sufficient selectivity between excipients and impurities (see case study #2). Obviously, not all RPLC columns can be used under such conditions, due to the well-known phase dewetting phenomenon [
27]. Phase dewetting is highly undesirable; since retention times decrease and are not reproducible, peaks may become distorted and re-equilibration times may be quite long [
28,
29]. To avoid such issue, a phase that is solvated under all mobile phase conditions has to be preferentially used (polar embedded phases, endcapping with a polar functionality, polar bonding…), such as the ones that were selected in this work.
Under these purely aqueous conditions, the Kinetex Polar provided suitable peak shapes and sufficient selectivity for succinimide and
N-hydroxysuccinimide but was the less retentive stationary phase (
k < 1 for both molecules). However, it is hard to interpret this result, as the polar endcapping employed for this stationary phase is proprietary and no information is available from the provider. The CSH fluorophenyl was also poorly retentive (
k < 1) and provided unsuitable peaks shapes (strong tailing) and limited selectivity. This phase possesses a positively charged surface (along with the fluorophenyl group). It should provide improved peak shapes and weaker retention for bases, while acids should be more strongly retained through the ion exchange mechanism. However, in the present case, both succinimide and
N-hydroxysuccinimide were under their neutral form at the selected pH (~2), and, therefore, the positively charged surface did not increase retention of these two molecules. In addition, the charge transfer retention mechanism produced by the fluorinated ligand was found to be unsatisfactory to retain succinimide and
N-hydroxysuccinimide [
30].
A very similar chromatographic behavior was observed with the Hypersil GOLD aQ and Zorbax SB Aq columns. Retention was better compared to the previously described stationary phases, but still insufficient. Here, again, no information is available from the provider regarding the nature of the polar ligand employed for polar endcapping. The Premier BEH C18 AX column, a mixed-mode stationary phase specifically developed to retain polar acidic analytes, was also found to be insufficiently retentive, mostly due to the fact that the targeted compounds were neutral under the selected mobile phase conditions [
31].
As shown in
Figure 2, the most promising columns to analyze succinimide and
N-hydroxysuccinimide were the Premier HSS T3, Luna Omega Polar, and Synergi Polar RP columns. These three columns offered suitable peak shapes, retention for the first eluted peak (
N-hydroxysuccinimide) with retention factors
k between 0.71 and 1.02, and a baseline separation of the two analytes. The good retention of polar analytes on the HSS T3 was due to the low ligand density (only 1.6 µmol/m
2 vs. >3.0 µmol/m
2 for regular C18 material) promoting the polar interaction with polar endcapped groups at the surface of the stationary phase. For the Luna Omega Polar stationary phase, the retention of succinimide and
N-hydroxysuccinimide was exclusively due to the presence of a proprietary polar endcapping able to interact with these hydrophilic analytes. Among these three columns, the Synergi Polar RP was selected, as it was the only one offering retention factors
k higher than 1 for the two compounds (this was a critical parameter considering the numerous components of the ACC formulation (as illustrated in case study #2) and the poorly selective detection wavelength of 200 nm). This stationary phase has multiple possibilities to interact with the two analytes of interest, thanks to its endcapping (proprietary polar ligand), the presence of a phenyl group, and ether linkage as the polar embedded group, resulting in improved peak shapes and suitable retention of acidic polar analytes.
2.2. Issue #2: Interferences from the Formulation Components
Since a low wavelength of 200 nm was selected to detect succinimide and N-hydroxysuccinimide, and as the retention of these molecules was limited, it was important to consider the possible interferences from some of the components of the ACC formulation (citric acid, sucrose, ethylenediaminetetraacetic acid (EDTA), and p-aminobenzoic acid (pABA)). Based on their UV spectra, all these additives are detectable at 200 nm. Therefore, it was necessary to ensure that they did not interfere with the early eluting analytes (succinimide and N-hydroxysuccinimide).
To evaluate whether the selectivity between excipients and analytes of interest is sufficient, all excipients were individually injected using the previously selected column (Synergi Polar RP). Importantly, a generic gradient was performed in the presence of 0.1% TFA, as it was found that TFA was critical to obtain sharp peaks for 3,2-HOPO chelator and its derivative 3,2-HOPO TOP-succinimide, due to the presence of two tertiary amines with a p
Ka of 9.0 in these two molecules. The gradient started with an isocratic step at 0% MeOH for 5 min to sufficiently retain succinimide and
N-hydroxysuccinimide. Then, the proportion of MeOH was increased from 0% to 100% over 5 min for the elution of EDC-urea. Next, a second isocratic step at 100% MeOH was added for 5 min, allowing for the elution of 3,2-HOPO chelator and its derivative 3,2-HOPO TOP-succinimide. Finally, a re-equilibration of the column at the initial gradient conditions (as described in
Section 3.3) was performed. To analyze all the compounds of interest, various detection conditions were employed to reach the expected limits of detection, namely UV at 200 nm for succinimide and
N-hydroxysuccinimide, UV at 330 nm for the two free chelator species, and MS in selected ion recording (SIR) mode for EDC-urea (m/z 174.1). As previously discussed, succinimide and
N-hydroxysuccinimide were eluted during the initial isocratic step with a purely aqueous mobile phase, while EDC-urea was eluted close to the end of this isocratic step. On the other hand, the two free chelator species were found to be much more hydrophobic, and their elution took place during the final isocratic step at 100% MeOH.
Under the selected conditions, most of the excipients were not problematic as they were either excluded from the pores of the column (elution before the baseline disturbance corresponding to
t0), not retained (eluted at
t0), or hardly detectable in UV at a wavelength of 200 nm at the concentration level of the formulation. However, the radiostabilizer pABA was retained on the Synergi Polar RP column due to the presence of an aromatic ring that can interact with the ether-phenyl bonding at the surface of the stationary phase, and eluted slightly before 5 min, far from the elution zone of succinimide and
N-hydroxysuccinimide (see
Figure 3A). However, pABA was eluted in the tail of the EDC-urea peak. Even if the SIR of EDC-urea was selective (m/z 174.1), pABA possesses an aniline group which can be ionized in the positive ESI mode. Therefore, we observed a competition for ionization between EDC-urea and pABA in the electrospray ionization source (matrix effects), leading to peak deformation observed in
Figure 3A and inaccurate quantitation.
Additionally, citric acid and EDTA were observed to be slightly retained under the selected conditions and eluted just before the peak of
N-hydroxysuccinimide. An interesting solution to improve the separation of citric acid, EDTA, and
N-hydroxysuccinimide was to tune the amount of TFA and mobile phase pH. Indeed, EDTA is a poly-acid with four different carboxylic acid groups having p
Ka of 0, 1.5, 2.0, and 2.66, and citric acid is a triprotic acid with p
Ka of 3.1, 4.7, and 6.4. On the other hand,
N-hydroxysuccinimide has a significantly higher acidic p
Ka of 7.2. Based on these acid-basic properties, a slight increase of the mobile phase pH should provide a retention decrease for EDTA and citric acid as conditions are created so that the second and third protons can be donated, while the retention of
N-hydroxysuccinimide should not be affected as its p
Ka is too far from the mobile phase pH of 2.0. In a first instance, different mobile phases differing in the proportion of TFA (i.e., 0.15%, 0.1%, 0.075%, and 0.03% TFA) were evaluated (data not shown). It appeared that the impact of this change was quite limited, mostly due to a narrow change in mobile phase pH produced. Nevertheless, the resolution between EDTA and
N-hydroxysuccinimide (the two most critical compounds) was improved when using the aqueous mobile phase with 0.075% TFA resulting in a mobile phase pH of approx. 2.15. Therefore, this TFA proportion was kept for the rest of the study. To further improve selectivity between
N-hydroxysuccinimide and the two interfering substances (citric acid and EDTA), the mobile phase was further adjusted to various pH comprised between 2.15 and 3.0, by adding ammonium hydroxide. Finally, the best separation was obtained when fixing the pH at 2.6, as illustrated in
Figure 3B, where succinimide and
N-hydroxysuccinimide were clearly separated from the interfering compounds.
In addition, the pH increase also impacted the elution of pABA, which possess a basic p
Ka of 2.7. pABA was therefore more retained at pH 2.6 (
Figure 3B) vs. pH 2.0 (
Figure 3A), while EDC-urea was not affected by this pH change (basic p
Ka of 9.3), overcoming the interference effects in MS detection. Interestingly, the peak shape of EDC-urea was also drastically improved due to the presence of ammonium hydroxide in the mobile phase (ammonium ions can mask residual silanols at the column surface, reducing undesirable secondary ionic interactions).
2.3. Issue #3: Analysis of Impurities in Presence of ACC at High Concentration
An additional constraint that was considered during the development of this method was the presence of ACC at high concentration. Indeed, it was necessary to inject the ACC product at 6 mg mL
−1 to reach sufficient LOD/LOQ for the impurities. The stationary phase selected to analyse the impurities was obviously ideally suited for the analysis of ACC (molecular weight in the range of 150 kDa, hydrodynamic radius around 5–7 nm), as the pore size was extremely small (80 Å) [
32]. As shown in
Figure 4A, an increase in column backpressure by about 50 bar after only 30 injections of ACC at 6 mg mL
−1 was observed. More importantly, the chromatographic performance was also strongly reduced, as shown in
Figure 4C.
Indeed, the resolution between the two free chelator species was equal to 3.22 for injection #1, while this value drops to only 1.59 for injection #30. One of the reasons for this critical behaviour could be that impurities were analysed under RPLC conditions at only 20 °C in order to achieve sufficient retention for the early eluted impurities (i.e., succinimide and
N-hydroxysuccinimide). However, it has been reported that antibody-based drugs such as monoclonal antibodies (mAbs) and antibody–drug conjugates (ADCs) can be strongly adsorbed at the surface of the stationary phase (mostly through ionic interactions and hydrogen bonding) at such a low temperature [
33,
34]. Since the ACC is an antibody-based drug, the same behaviour can be expected. Therefore, it can be stated that the accumulation of ACC (from the injections at high concentration) at the surface of the stationary phase can cause both a chemical modification of the Synergi Polar RP material (responsible for distorted peaks and modification of retention) and physical clogging of the stationary phase pores (responsible for pressure increase).
Unfortunately, it was not possible to solve this issue by simply increasing the column temperature in the range 70–90 °C, as the column is only stable up to 60 °C and the retention of succinimide and
N-hydroxysuccinimide would be unacceptably low at the elevated temperature. An alternative solution that was tested in this work was ACC removal through filtration. Spin filters (Vivaspin) with a cut-off of 50 kDa were therefore used before the analysis by performing a centrifugation step at 10,000×
g for 10 min at 20 °C. As shown in
Figure 4B, an increase in column backpressure lower than 10 bar after 30 injections was observed after removal of the ACC.
Figure 4D shows the separation of the two free chelator species at injections #1 and #30. As highlighted, the peak shapes, retention times, and chromatographic resolution of these two species were not affected after ACC removal.
2.4. Issue #4: Improvement of Recovery of Impurities
Being chelating agents, the 3,2-HOPO chelator and its by-product TOP-succinimide might easily capture or interact with metal ions,
e.g., iron(III), ubiquitously present in the analytical system [
35,
36]. Such interactions can cause deleterious effects ranging from peak tailing and generation of additional peaks to a complete loss of the analyte signal. To mitigate such interactions, we added the competitive chelator EDTA to the sample diluent and saturated the chelation sites by adding Th-232 (with a half-life of 14 billion years) in excess. As an additional care, we also systematically used polypropylene vials to limit the contact with metal ions, such as Na
+ or K
+. In our opinion, the addition of Th-232 to the samples was probably the most critical step to limit adsorption of 3,2-HOPO chelator and 3,2-HOPO TOP succinimide.
Besides the chelating properties of some impurities, the spin filter procedure for removal of large species (previously discussed) can also be a potential source of sample loss. In the present case, we used the classical polyethersulfone membrane recommended by the provider. By performing a semiquantitative evaluation of the method, which was obviously not intended to be a full method validation, we tried to understand whether impurities were lost during this step of the process and how to circumvent this issue.
In a first instance, the method linearity and LOD/LOQ were evaluated for each impurity and compared with the ATP. The sample preparation protocol included the spiking of selected impurities in the ACC sample and then the filtration step performed with the Vivaspin. The corresponding LOD and LOQ corresponding to S/N ratio equal to 3 and 10, respectively, were summarized in
Table S3.
Figure 5 shows the chromatograms of 3,2-HOPO chelator and its by-product TOP succinimide at various concentrations ranging from 1 to 0.05 µg mL
−1, in addition to a blank injection (grey trace). As shown from the blank injection, the specificity of the method was found to be given for these two compounds, and the compounds were easily detected at 0.05 µg mL
−1, with resolution higher than 1.5. In addition, the regression lines were constructed. A linear model provides an
R2 higher than 0.999 in both cases, with RSD values on peak area (n = 3) for the various concentrations ranging from 0.32–1.57% for 3,2-HOPO chelator and 0.43–1.36% for its by-product TOP succinimide, confirming the good linearity of the method in the investigated concentration range. Some injections of succinimide and
N-hydroxysuccinimide at various concentrations comprised between 4 to 70 µg mL
−1 were also carried out, and the corresponding data were reported in
Figure S1. As illustrated, the linearity was higher than 0.998 for these two impurities and LOQ was well below the target level of 4 µg mL
−1. Moreover, RSD values on area (n = 3) ranging from 0.18–0.49% and 0.08–0.42% were obtained for succinimide and
N-hydroxysuccinimide, respectively. Last but not least, EDC-urea was injected at concentrations varying from 0.1 to 4 µg mL
−1 and detected with MS detector operating in SIR mode (
Figure S2). Here again, the method linearity was found to be excellent (R
2 > 0.999), RSD values on area (n = 3) ranging from 0.64–1.56% were determined for the various concentrations, and the lowest concentration could be easily detected. All the LOD/LOQ and linearity were found to be in line with the ATP criteria.
Next, the recovery and precision of the method was checked for a sample containing all the impurities. For this purpose, the sample preparation protocol included the spiking of the five impurities in the ACC sample. Results obtained with and without the filtration step were first compared, and data were summarized in
Table 1. In terms of precision (RSD on peak areas for three injections) for the sample without filtration, the values ranged from 0.22 to 0.89% for the four impurities detected in UV, while the RSD value was equal to 4.13% for the EDC-urea detected in MS. This behaviour is logical, as ESI-MS is known to produce much larger signal variability than UV detection. In the case where the sample was filtrated, the RSD values were slightly higher, comprised between 0.42 and 1.83%, while the RSD of EDC-urea was equal to 5.46%. From these results, it can be concluded that filtration does not alter the method precision, as comparable values were obtained with and without the filtration step. Thus, the precision of the method after filtration was found to be acceptable for a routine application of the method (<2% with UV detection and <6% with MS detection, in line with the ATP criteria). On the other hand, the average filtration recoveries (calculated by dividing the peak area without filtration by the peak area with filtration) were comprised between 89.3 and 91.3% for the first eluting peaks (
N-hydroxysuccinimide, succinimide, and EDC-urea), while the filtration recoveries were equal to 59.7% and 68.4% for the 3,2-HOPO chelator and its by-product TOP succinimide, respectively. These two last molecules are the most retained in the chromatographic method (eluted with pure MeOH) and are therefore the most lipophilic ones. The sample loss could be due to the quite hydrophobic composition of the membrane (polyethersulfone membrane), which could generate some chemical interactions with the two chelator species [
37]. Unfortunately, due to the large polarity range of the different impurities, the use of a more hydrophilic membrane does not seem to be a way to solve the problem for the lipophilic compounds, as it would most certainly result in a consequent loss of the more hydrophilic species.
As the 3,2-HOPO chelator and its by-product TOP succinimide were the most critical impurities for the filtration step, the method variability between six different injections from a single filtration and six injections from six independent filtrations were compared. The corresponding data were reported in
Table 2.
As shown, the variability was slightly higher when performing independent filtrations, but remains acceptable, lower than 5%. Finally, the results reported in
Table 1 and
Table 2 prove that a non-negligible proportion of 3,2-HOPO chelator and its by-product TOP succinimide were lost during the filtration procedure (mostly due to the hydrophobic nature of the membrane). However, since the observed loss was constant between different filtration procedures (RSD values around 3%), it is recommended to apply the same strategy to the external calibrants and the unknown samples (namely adding a filtration step) to achieve suitable method trueness. In other words, using such a procedure, the developed method could be applied in a semiquantitative way to support in-process control activities.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}