
Recent Advances in the Synthesis of Borinic Acid Derivatives



Abstract
:1. Introduction
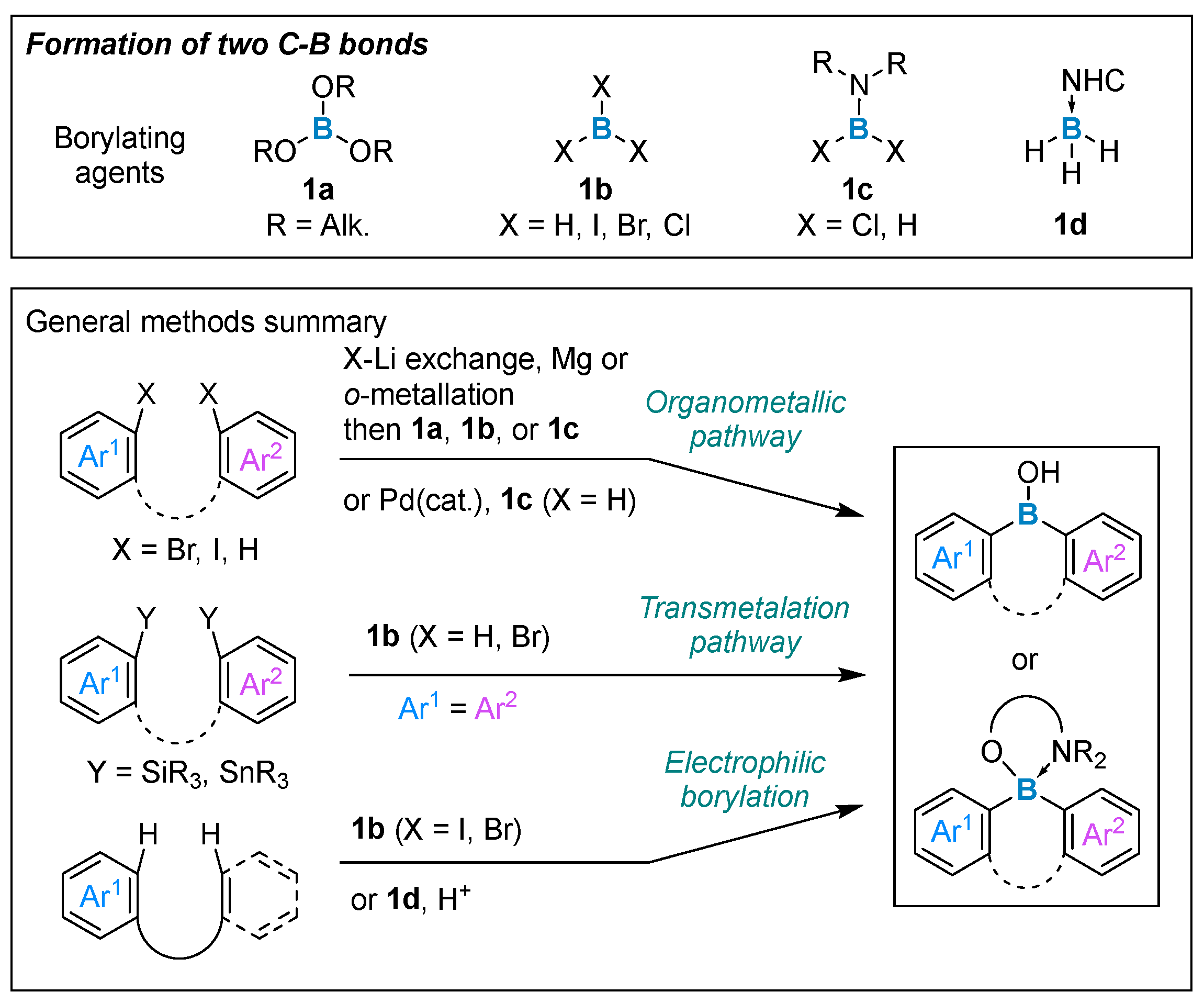
2. Formation of Two Carbon–Boron Bonds
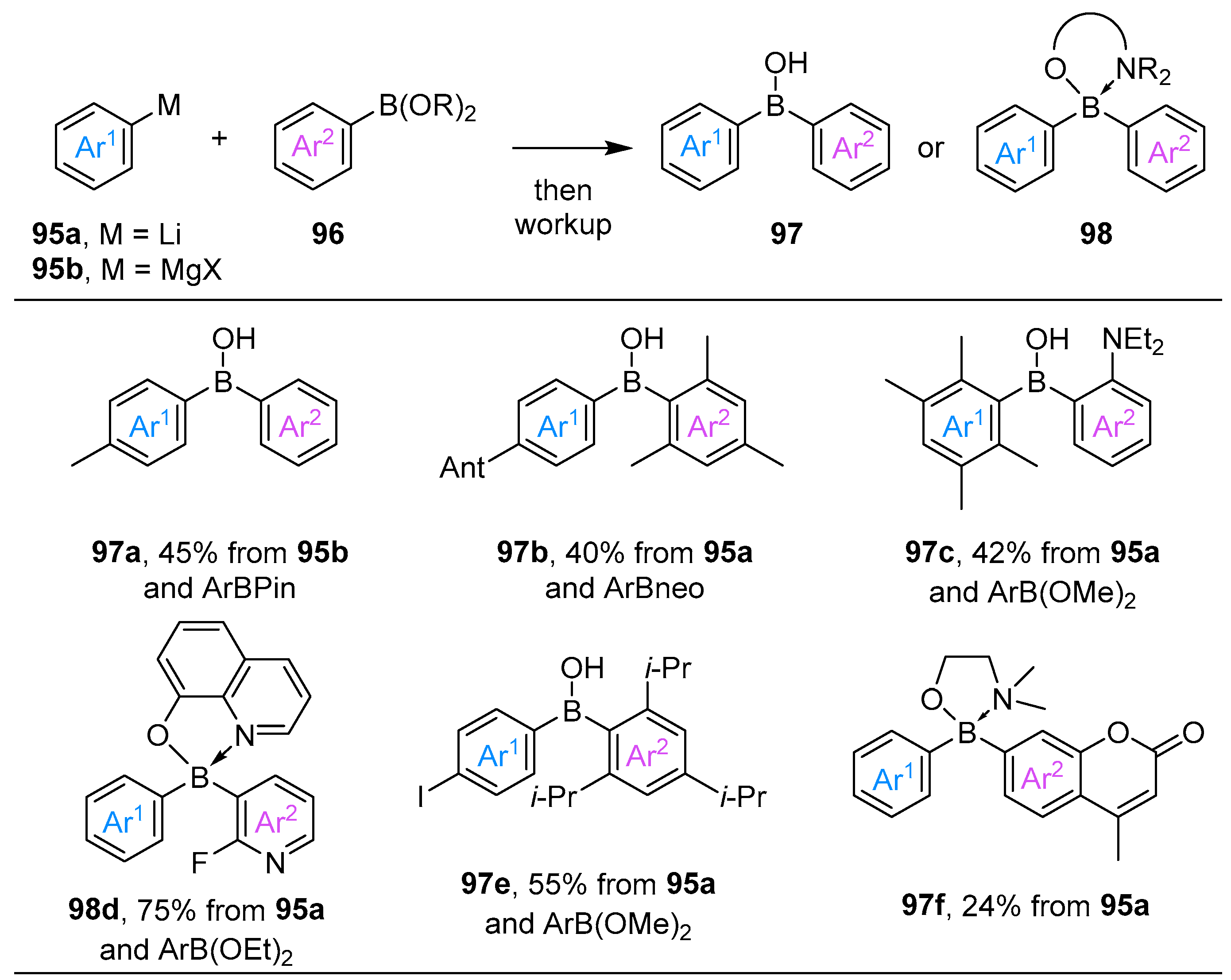
2.1. By Nucleophilic Addition of ArLi and ArMgX to Boron Reagents
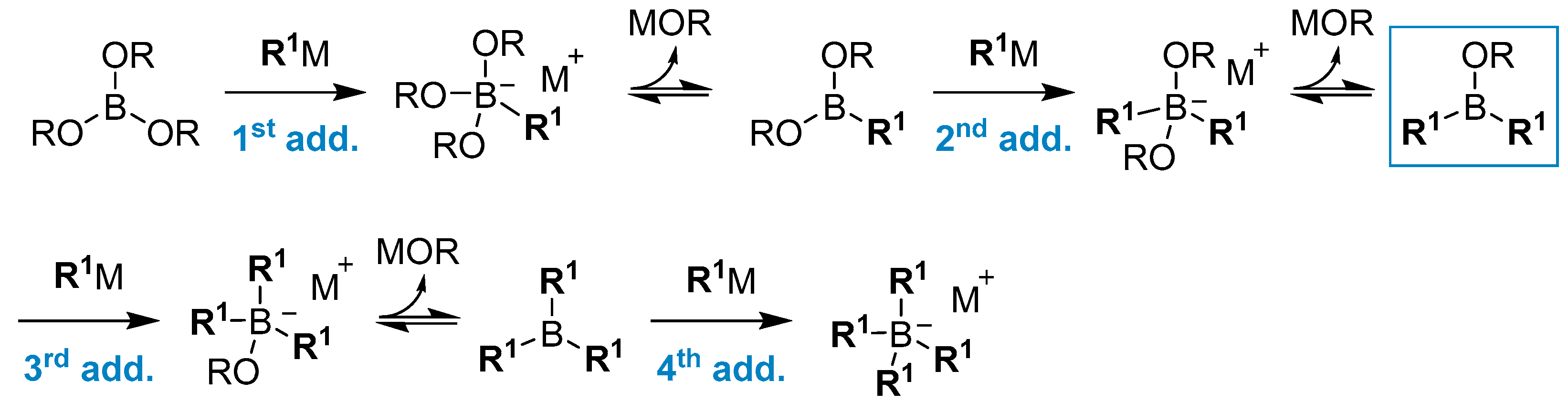
2.1.1. Use of Trialkoxyboranes
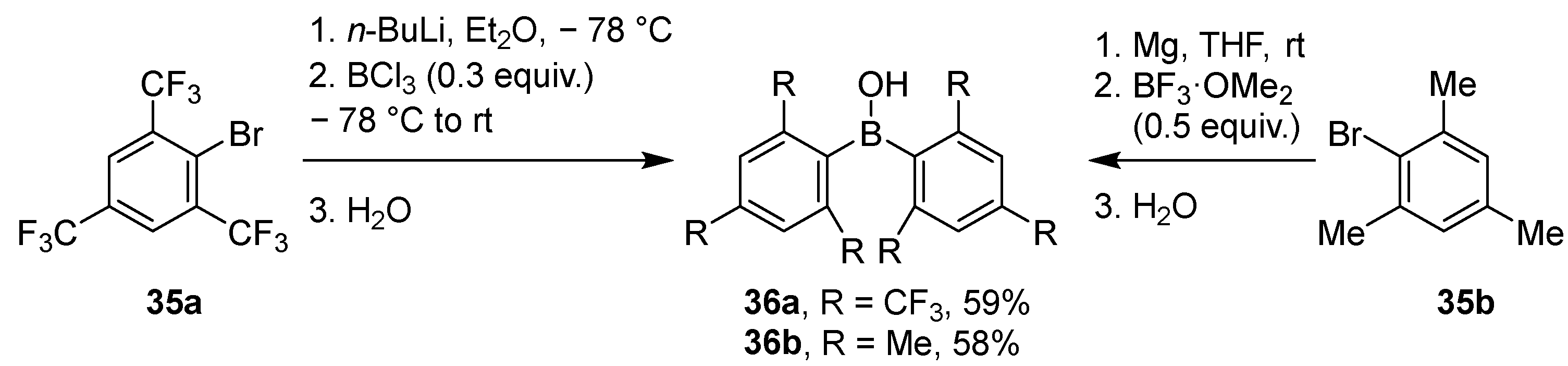
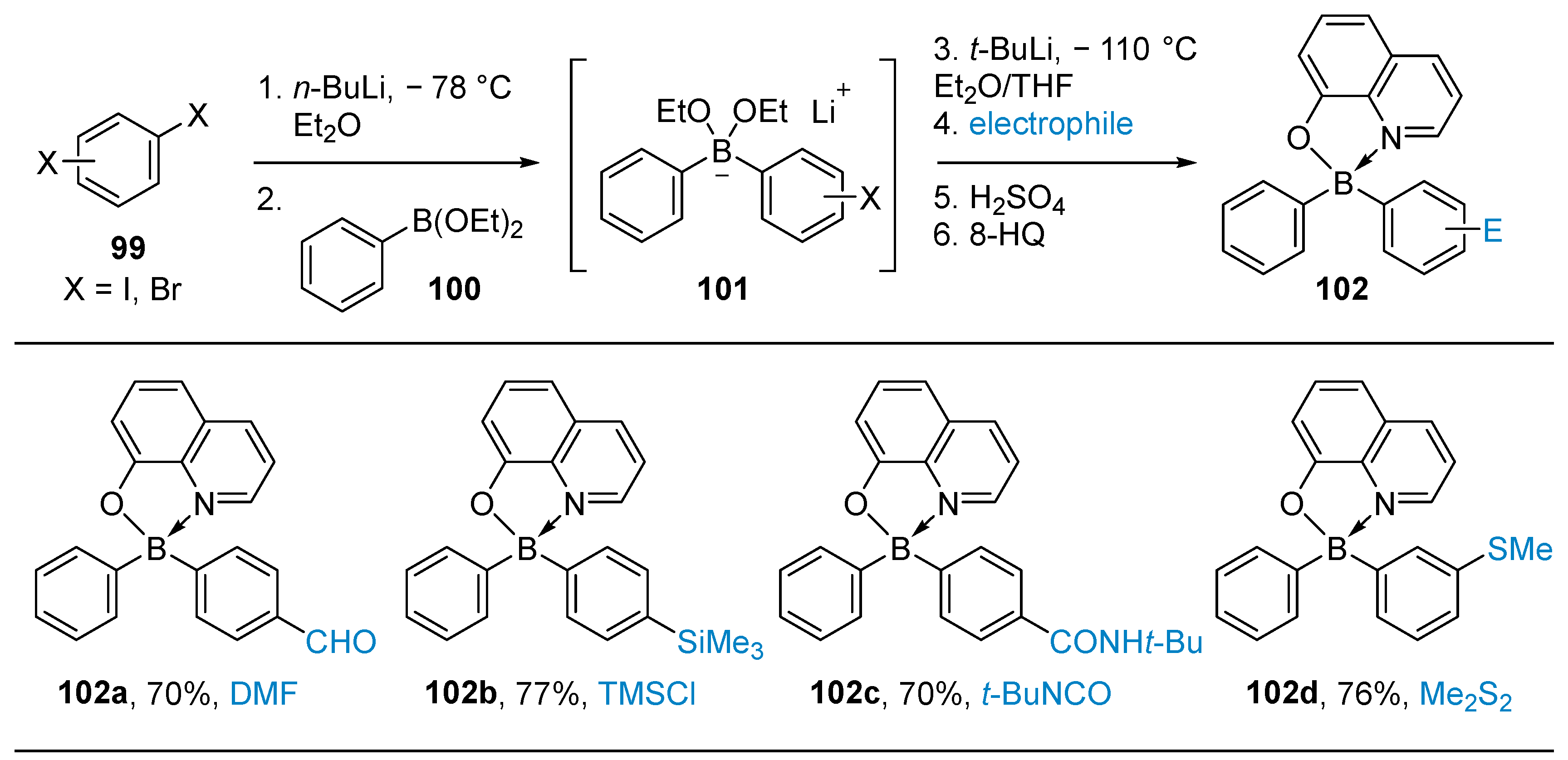
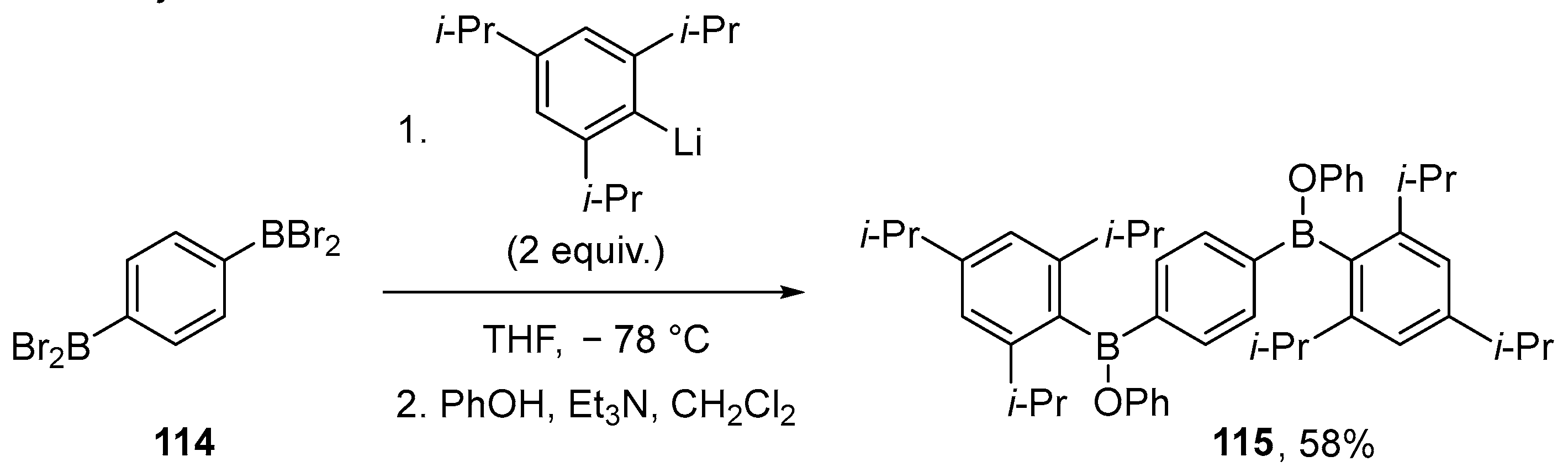
2.1.2. Use of Borontrihalides
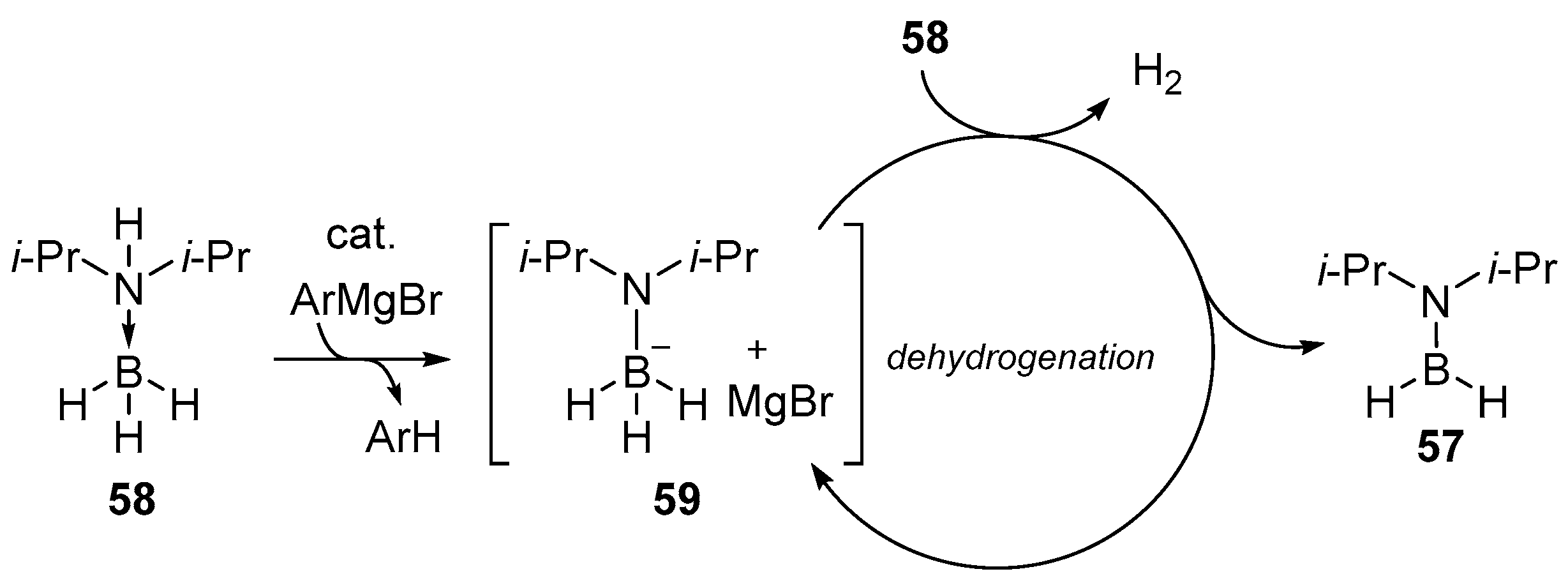
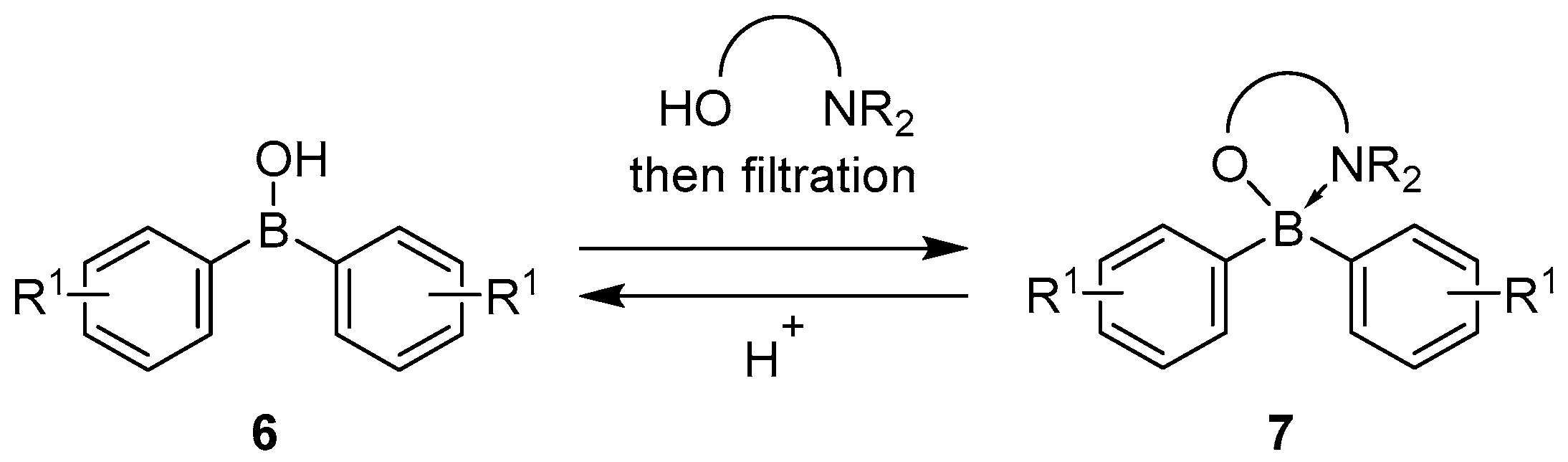
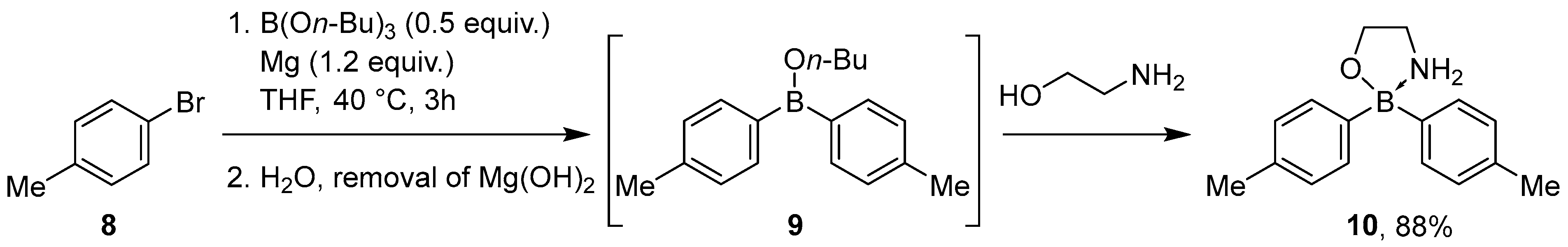
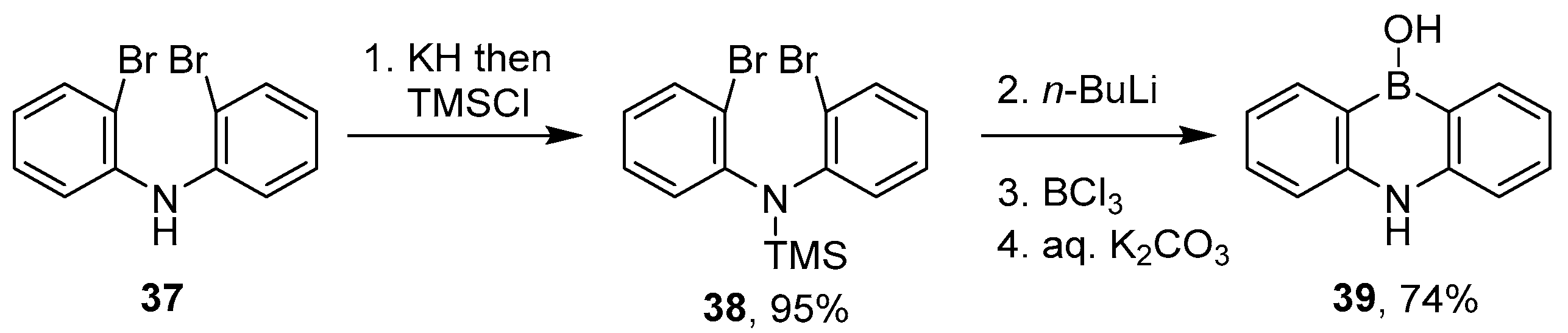
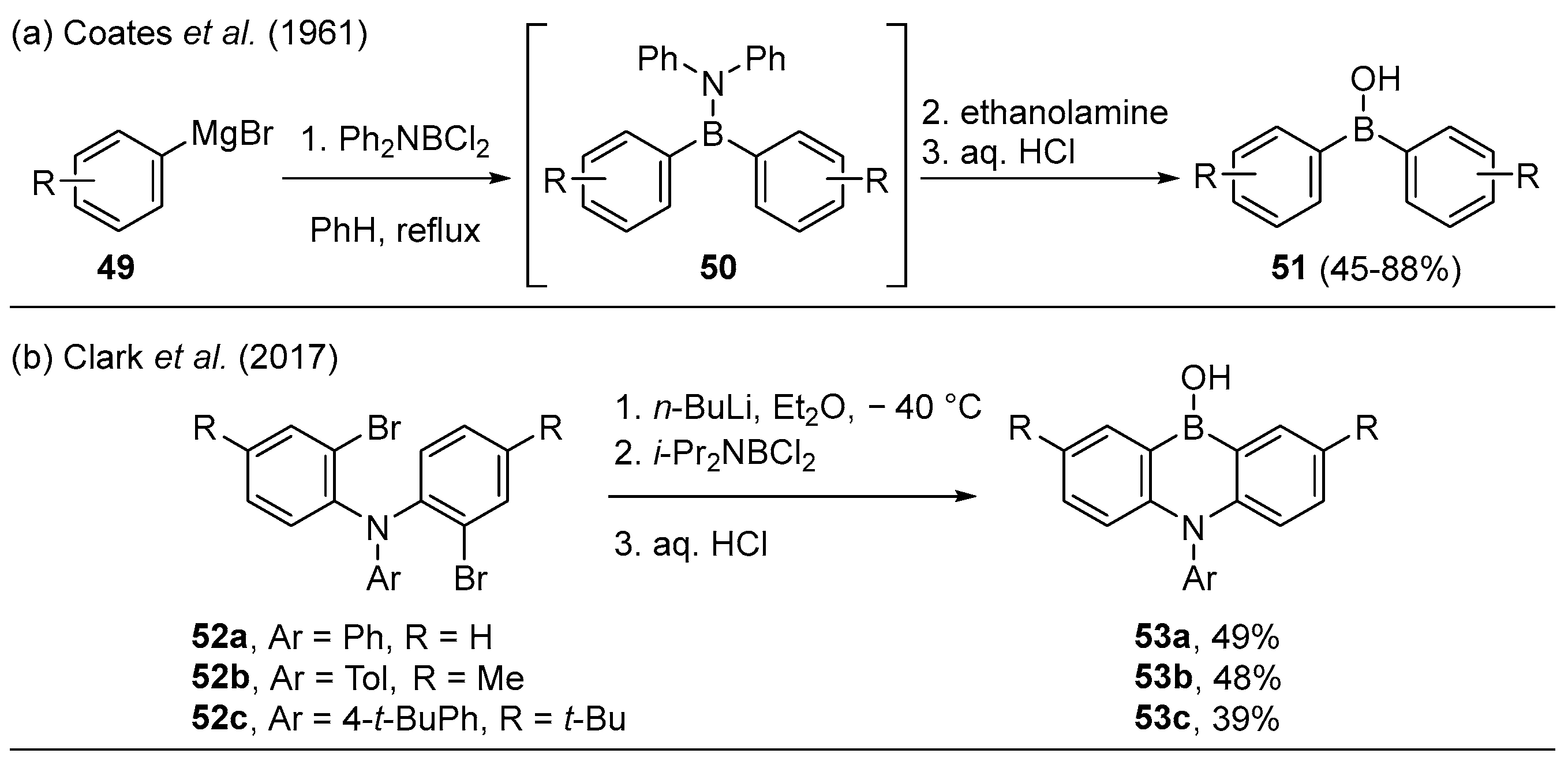
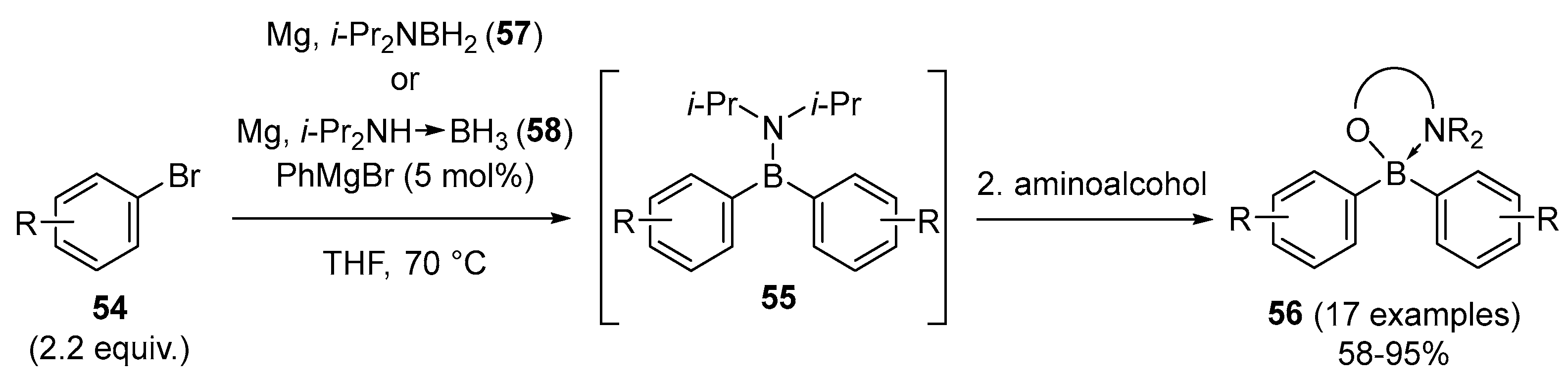
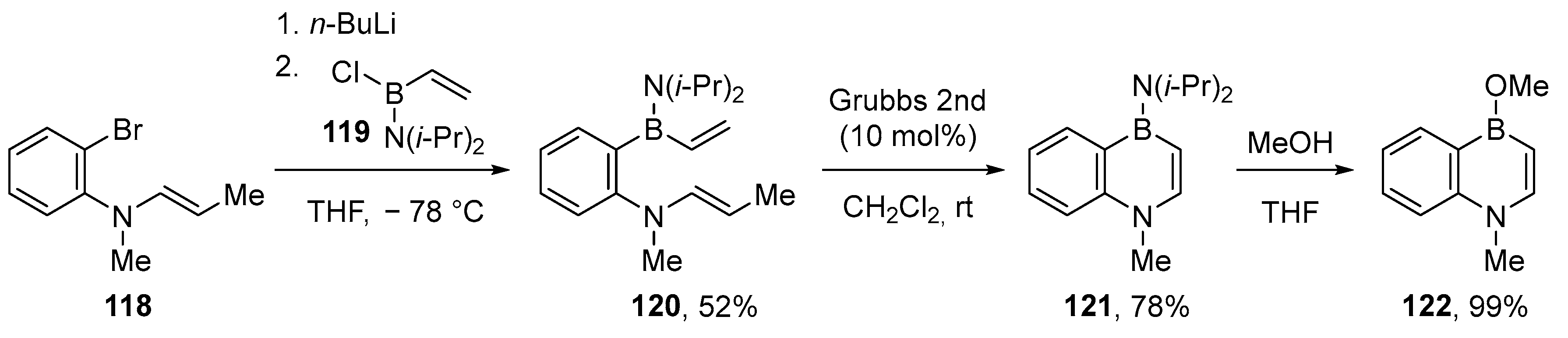
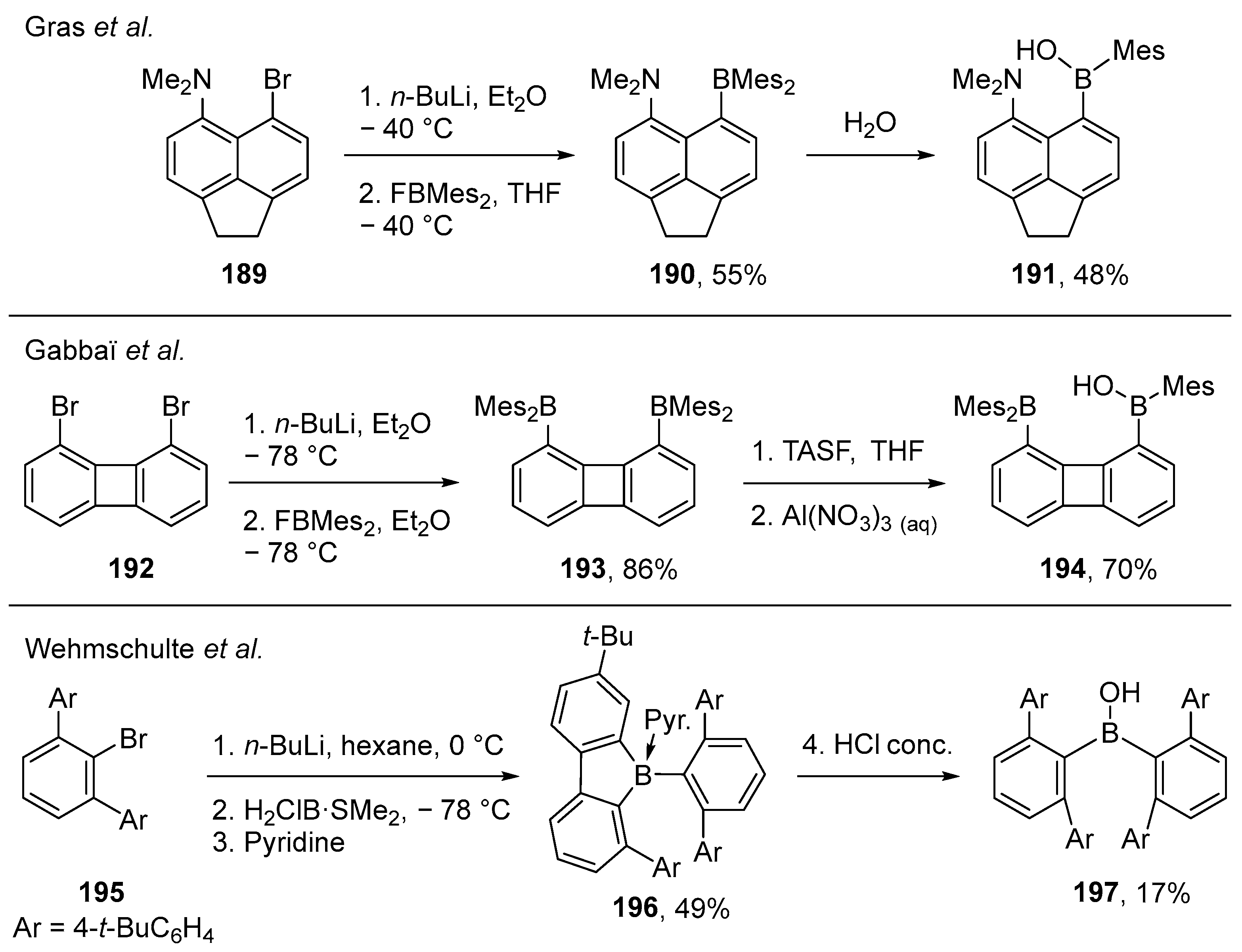
2.1.3. Use of Aminoboranes
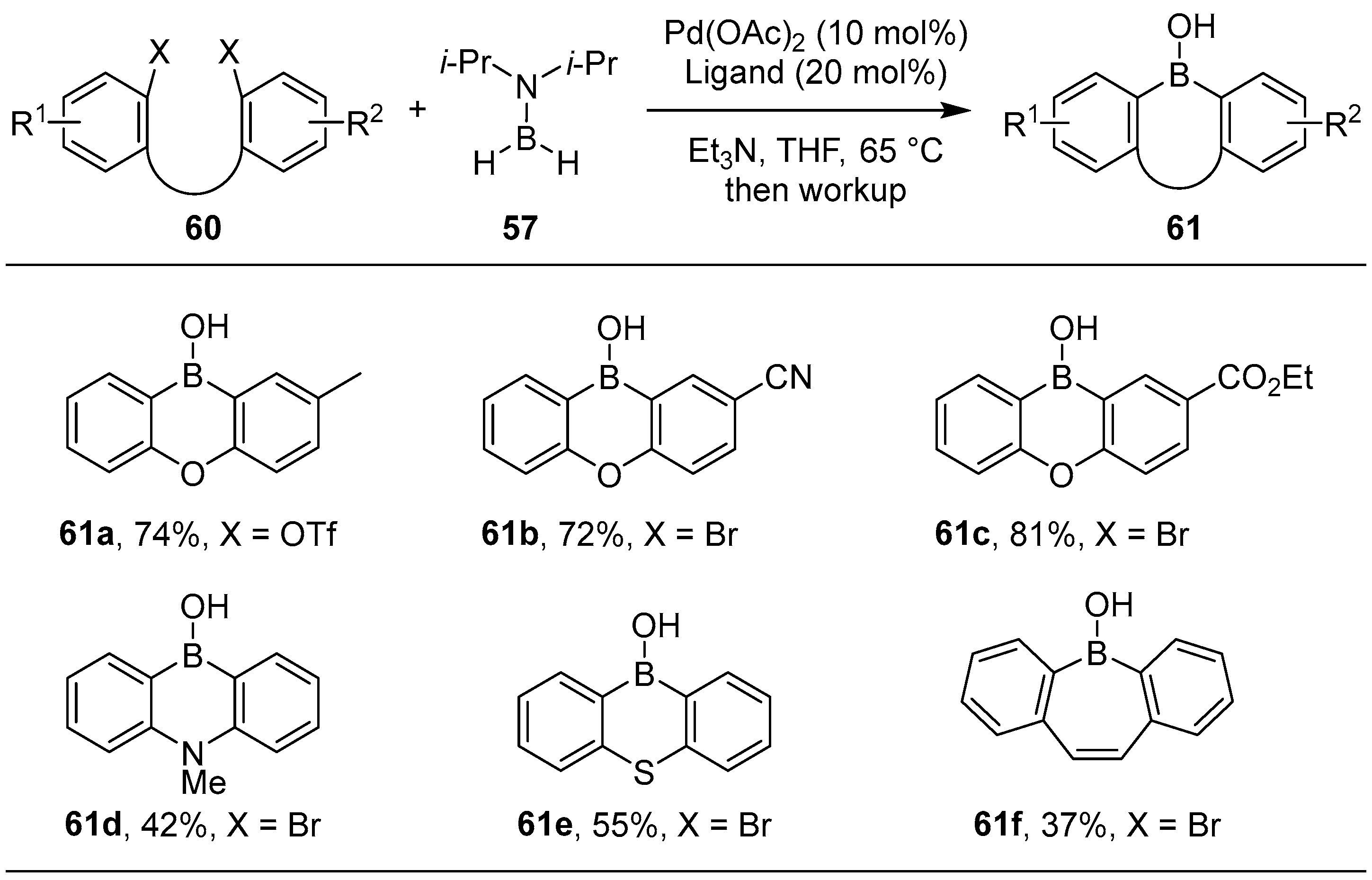
2.2. By Organometallic Catalysis with Boron Reagents
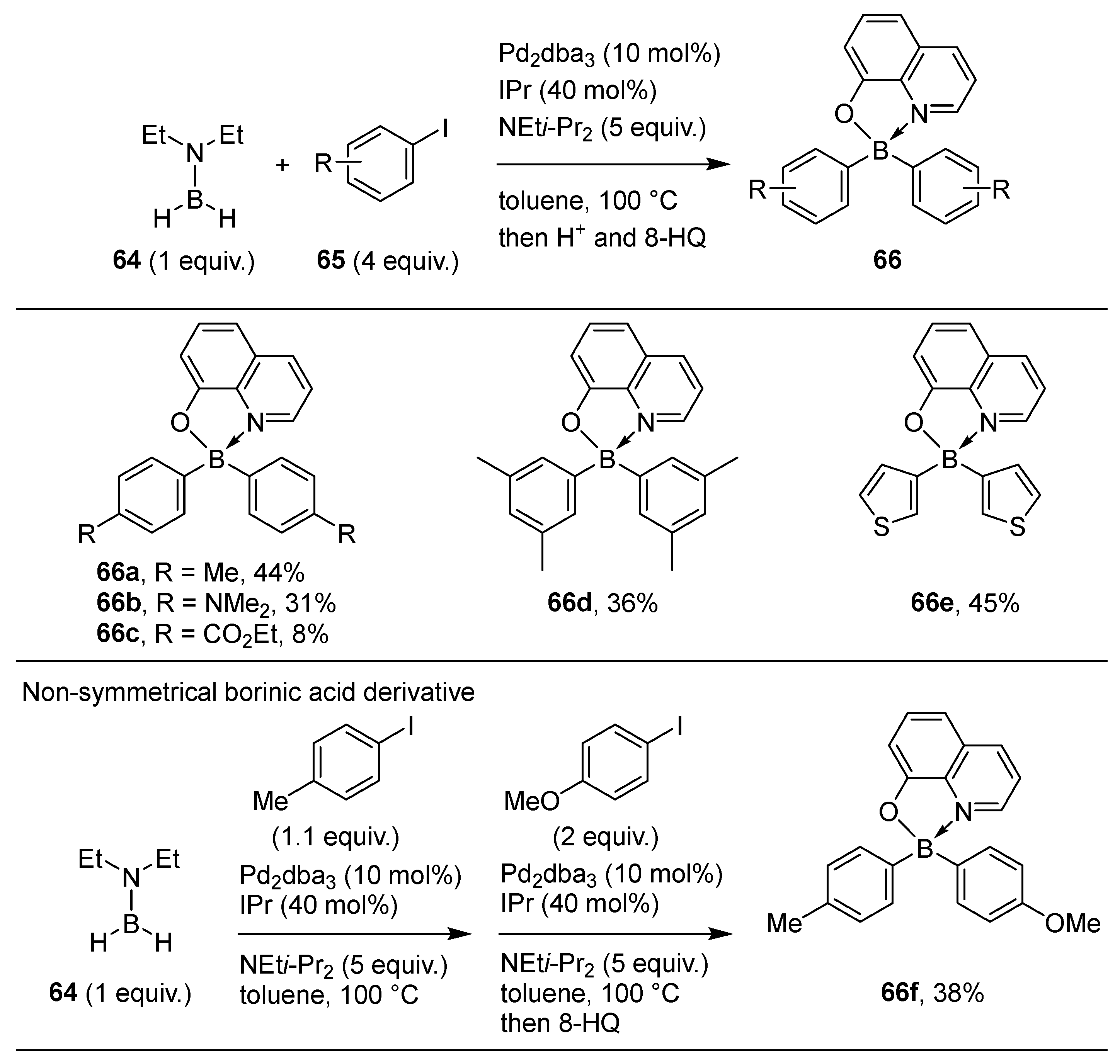
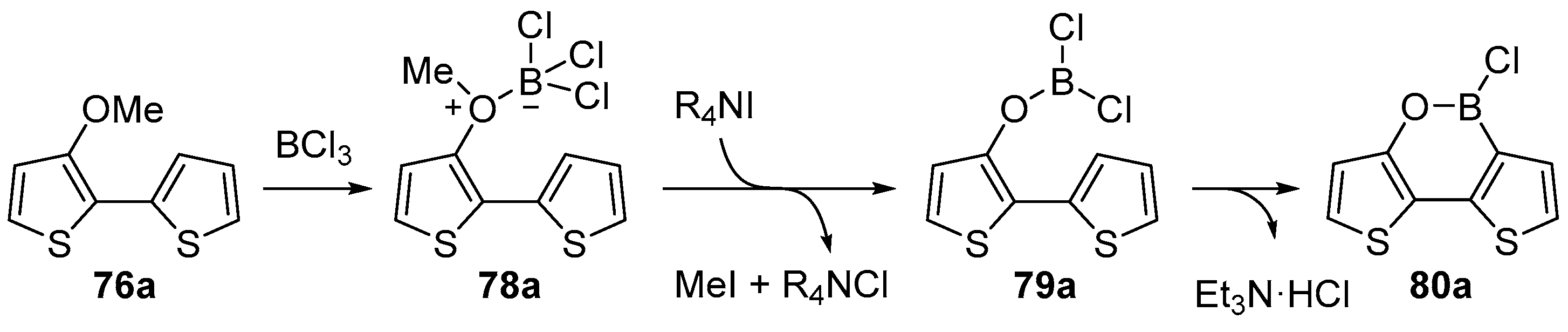
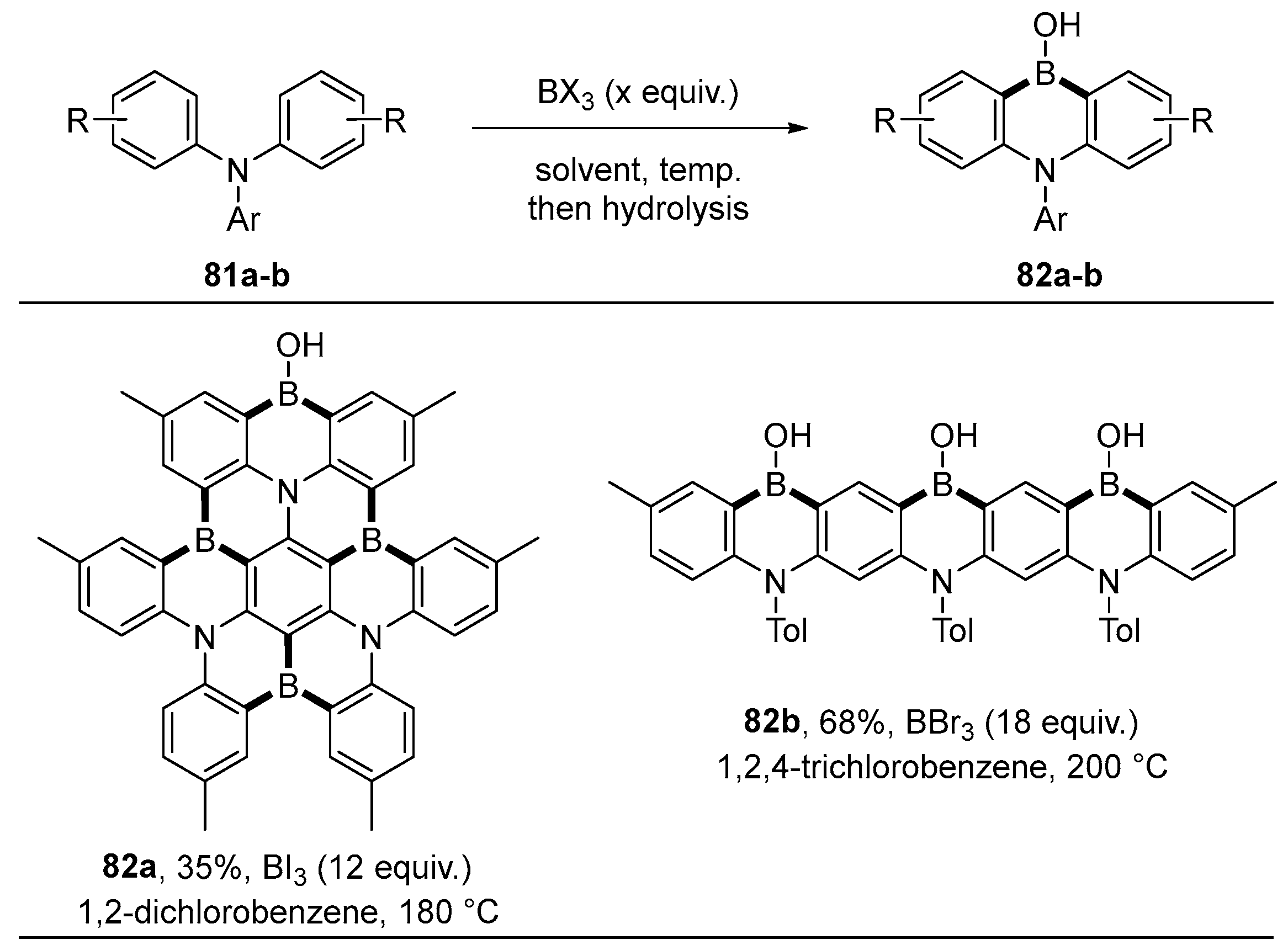
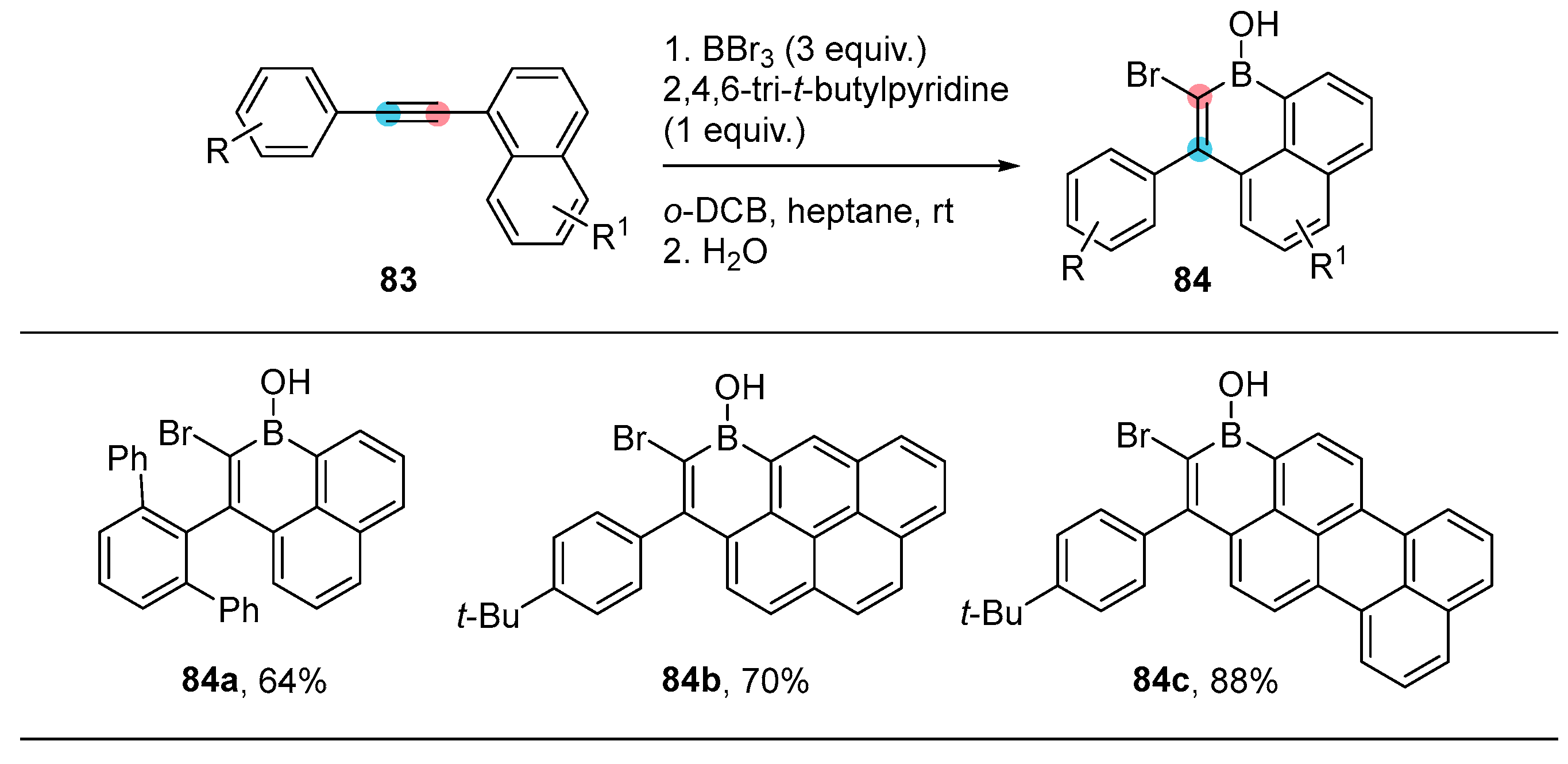
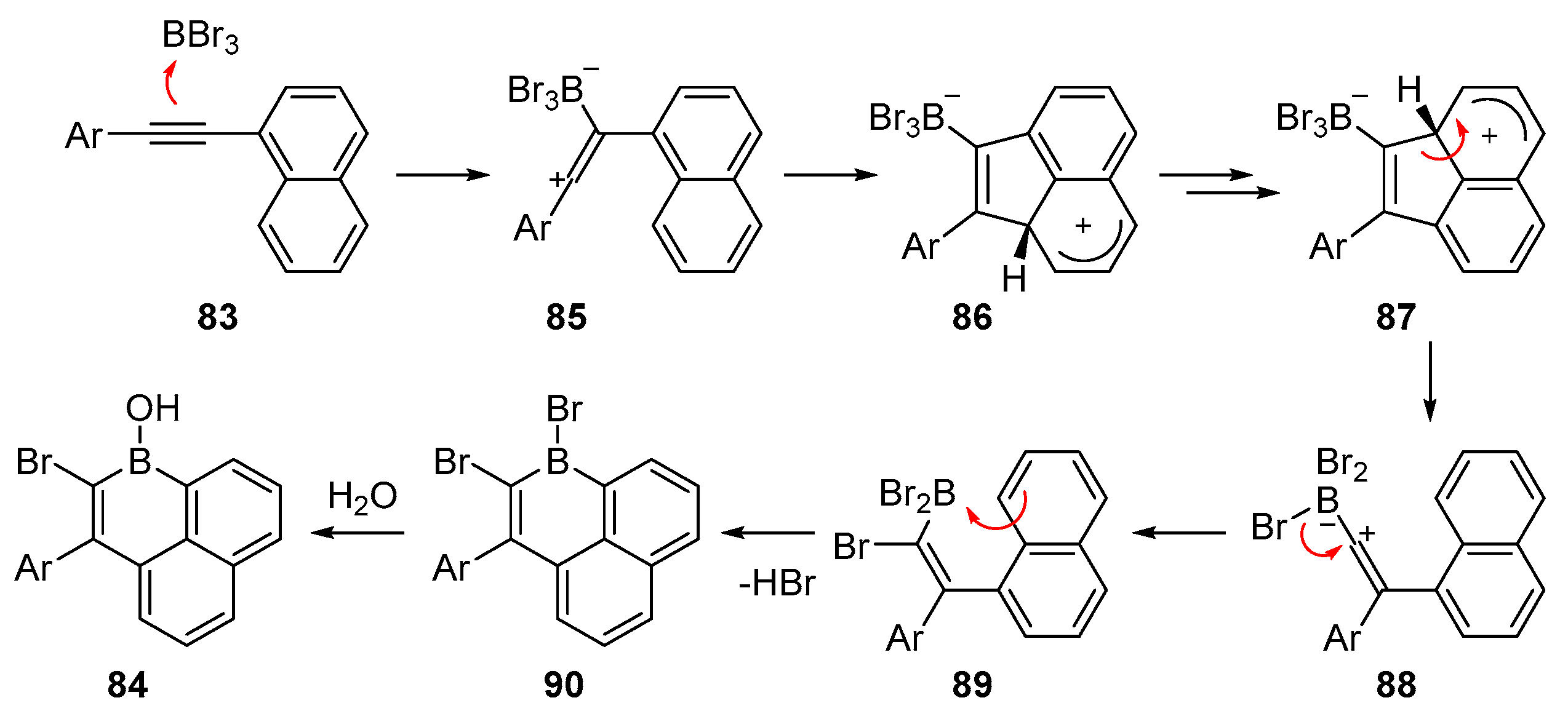
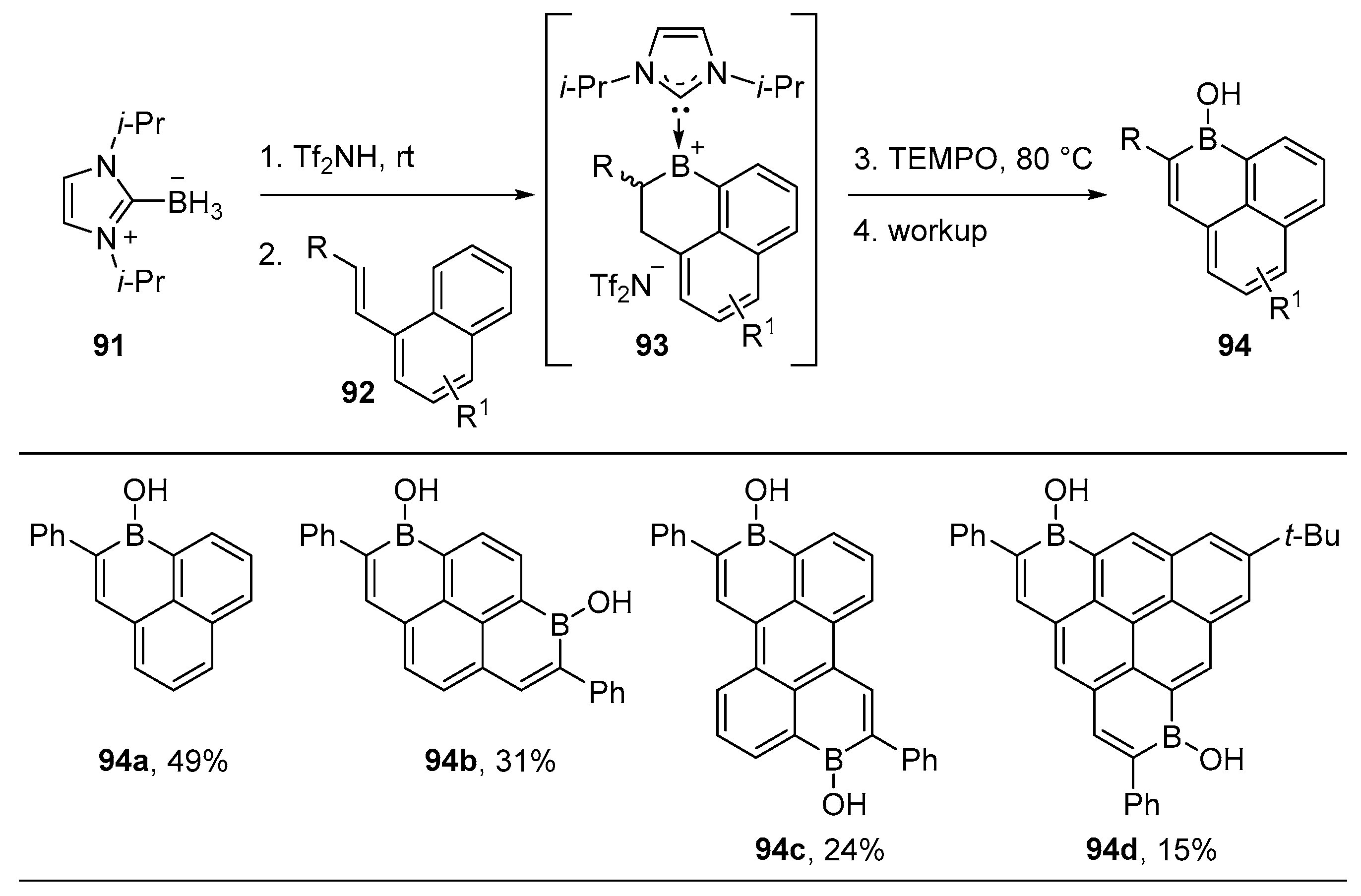
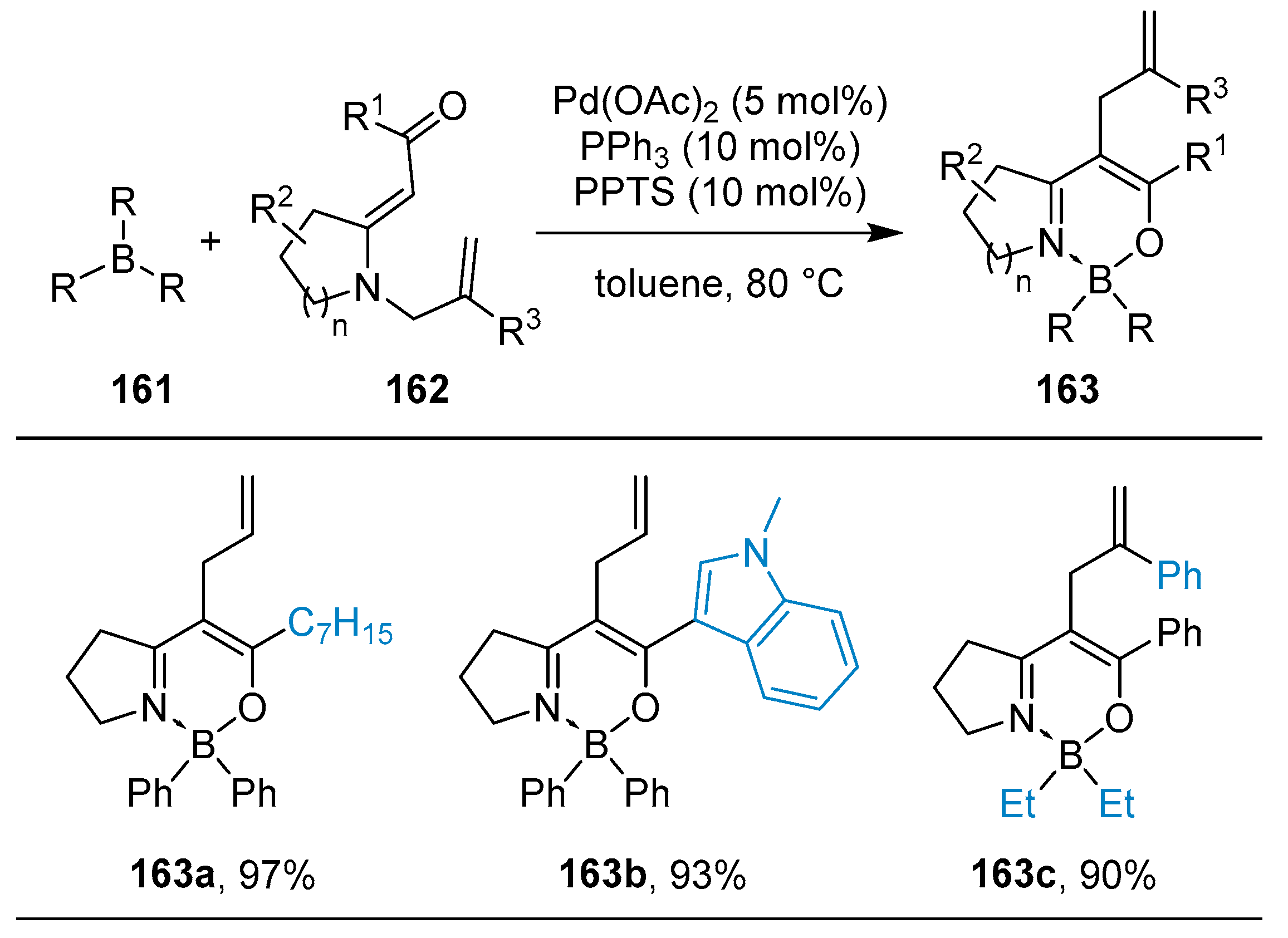
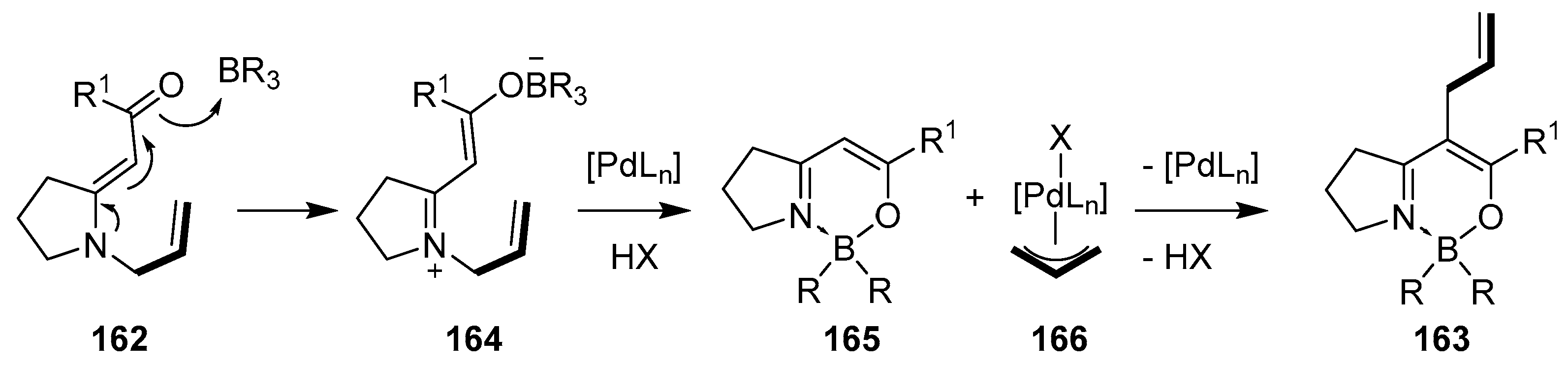
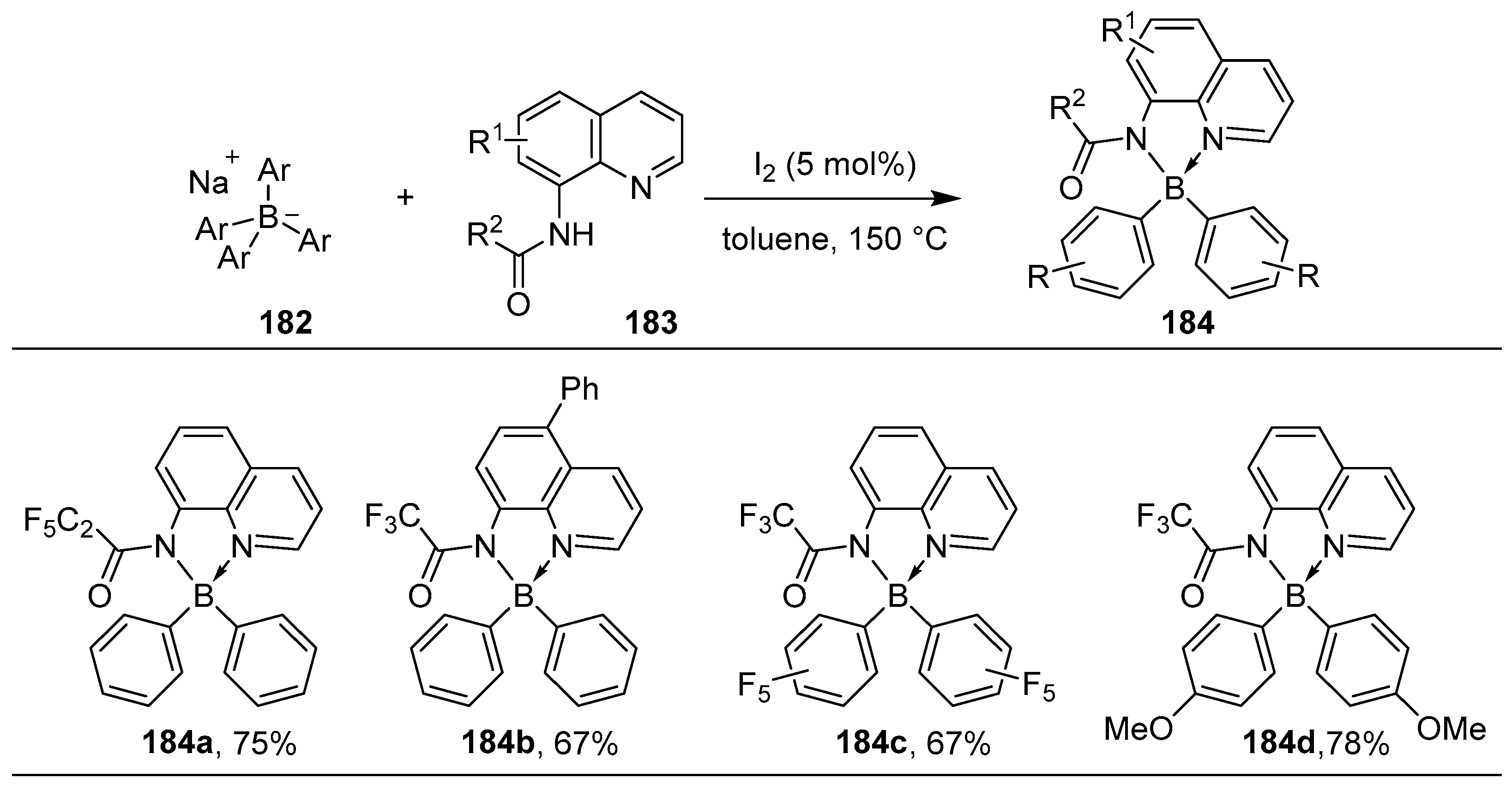
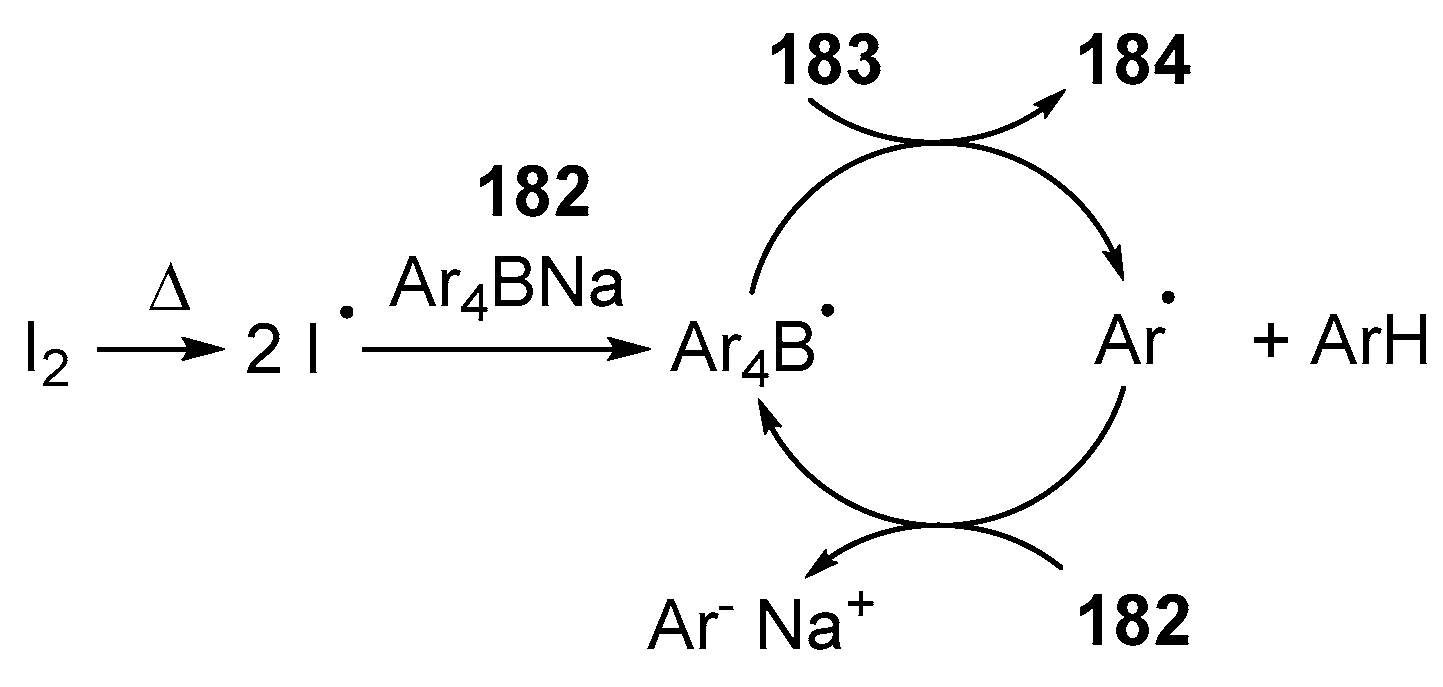
2.3. By Electrophilic Aromatic Substitution with Boron Reagents
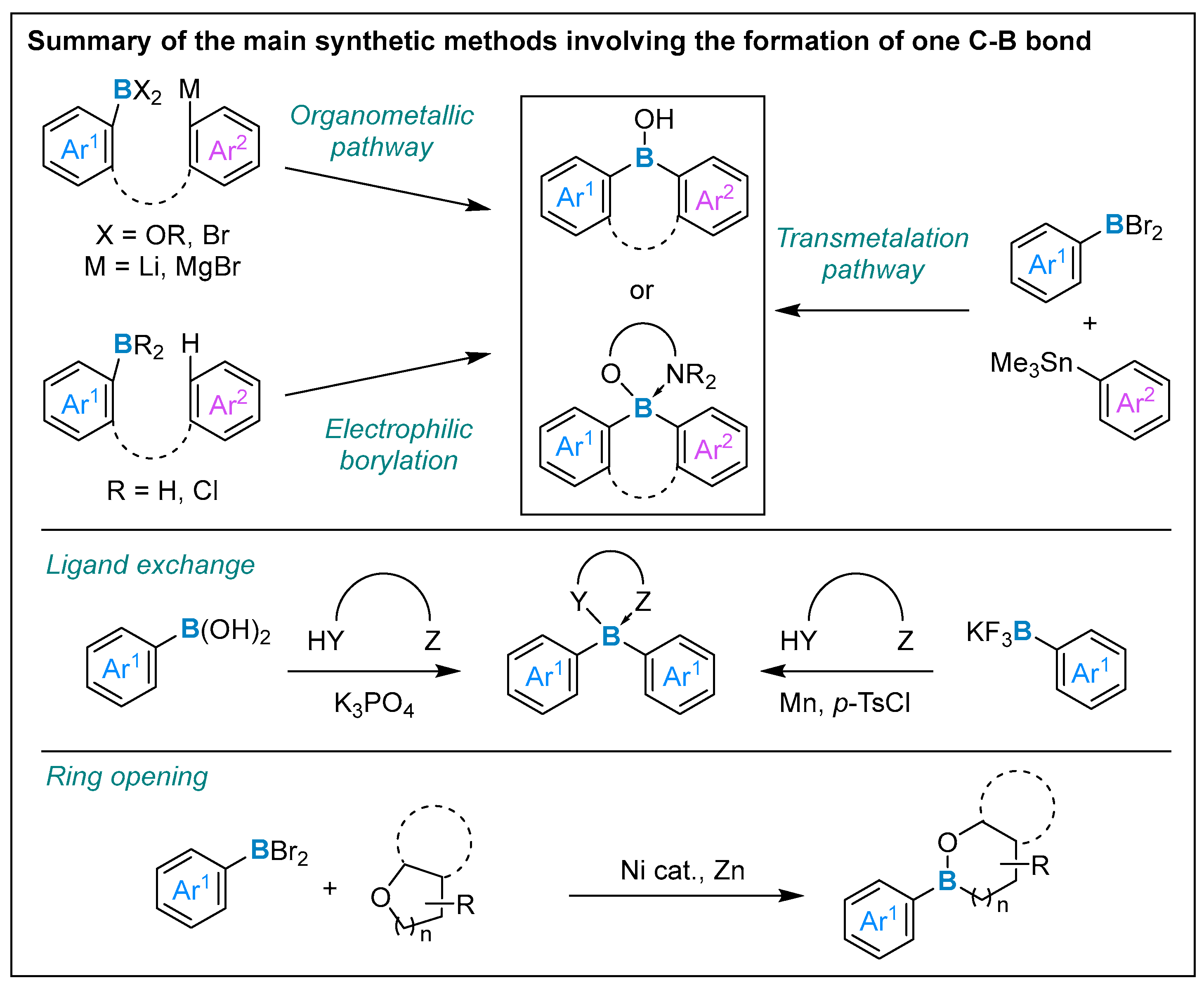
3. Formation of One Carbon–Boron Bond
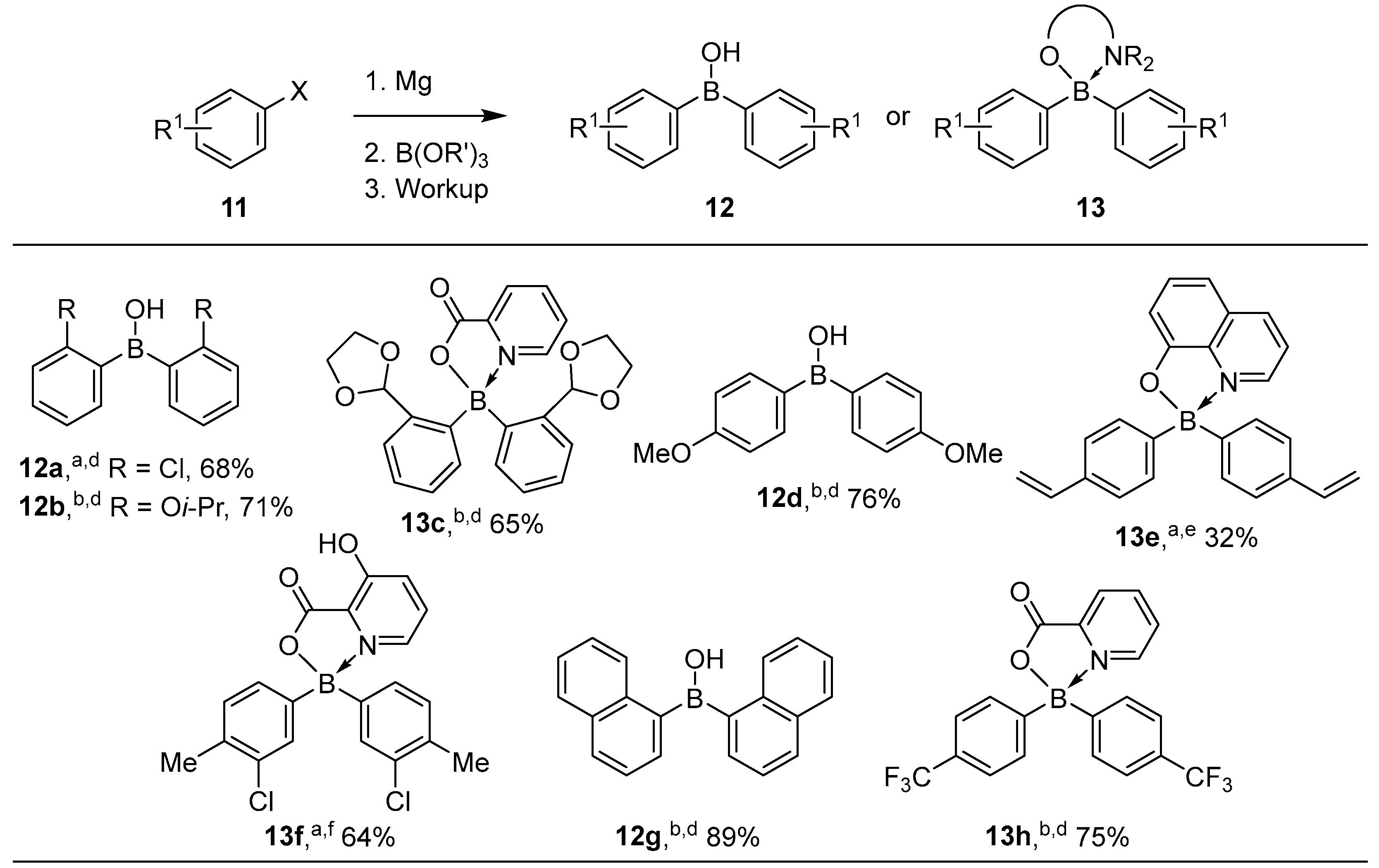
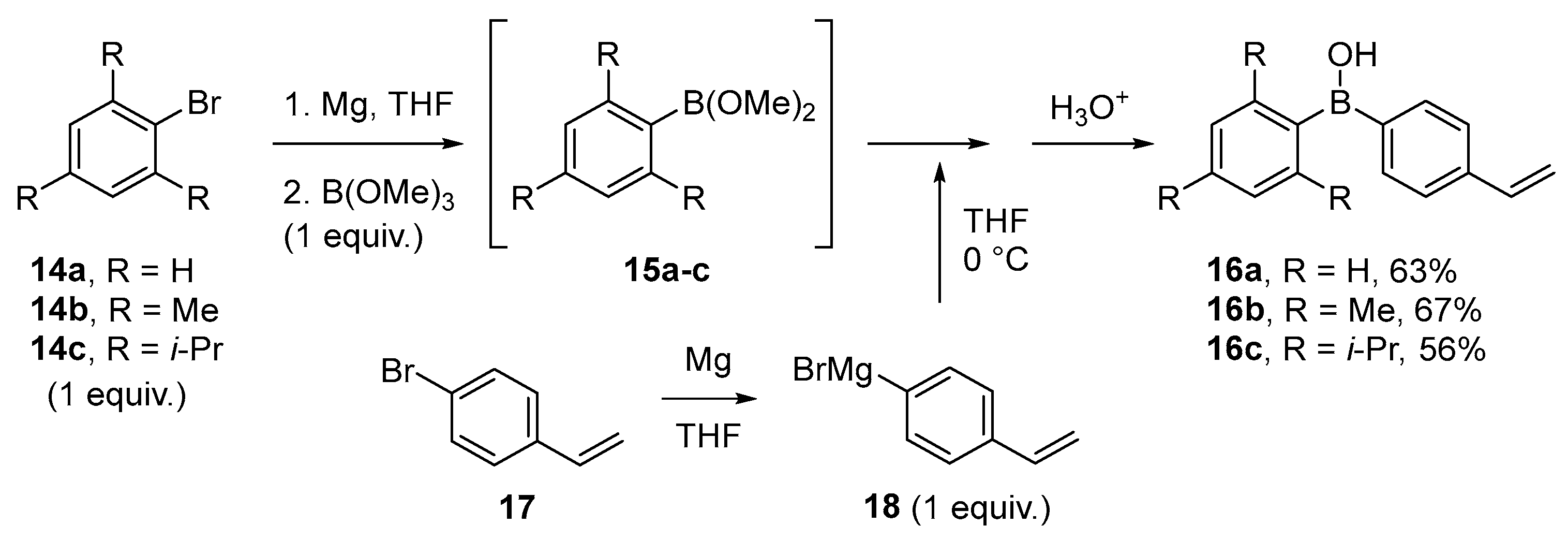
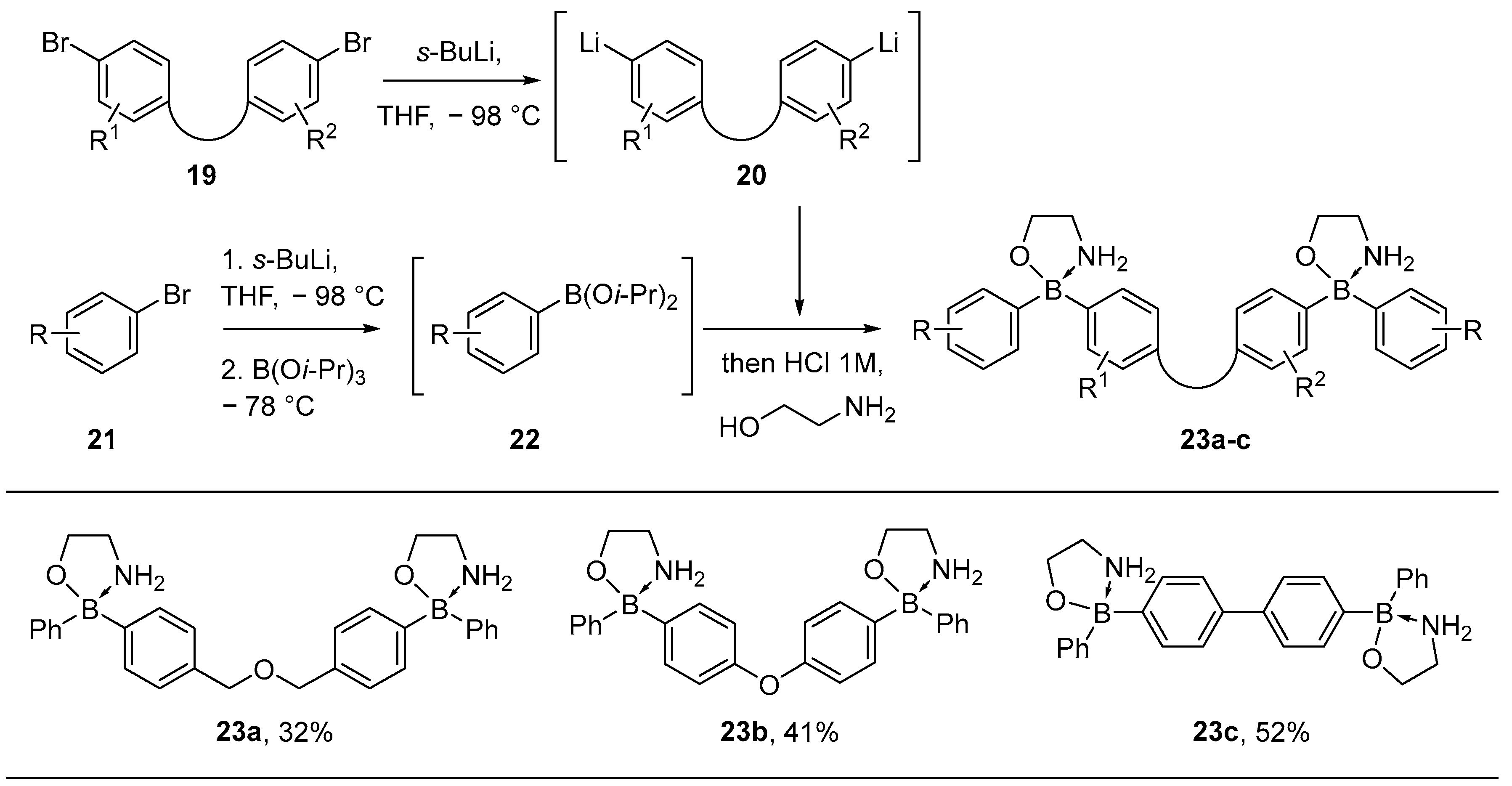
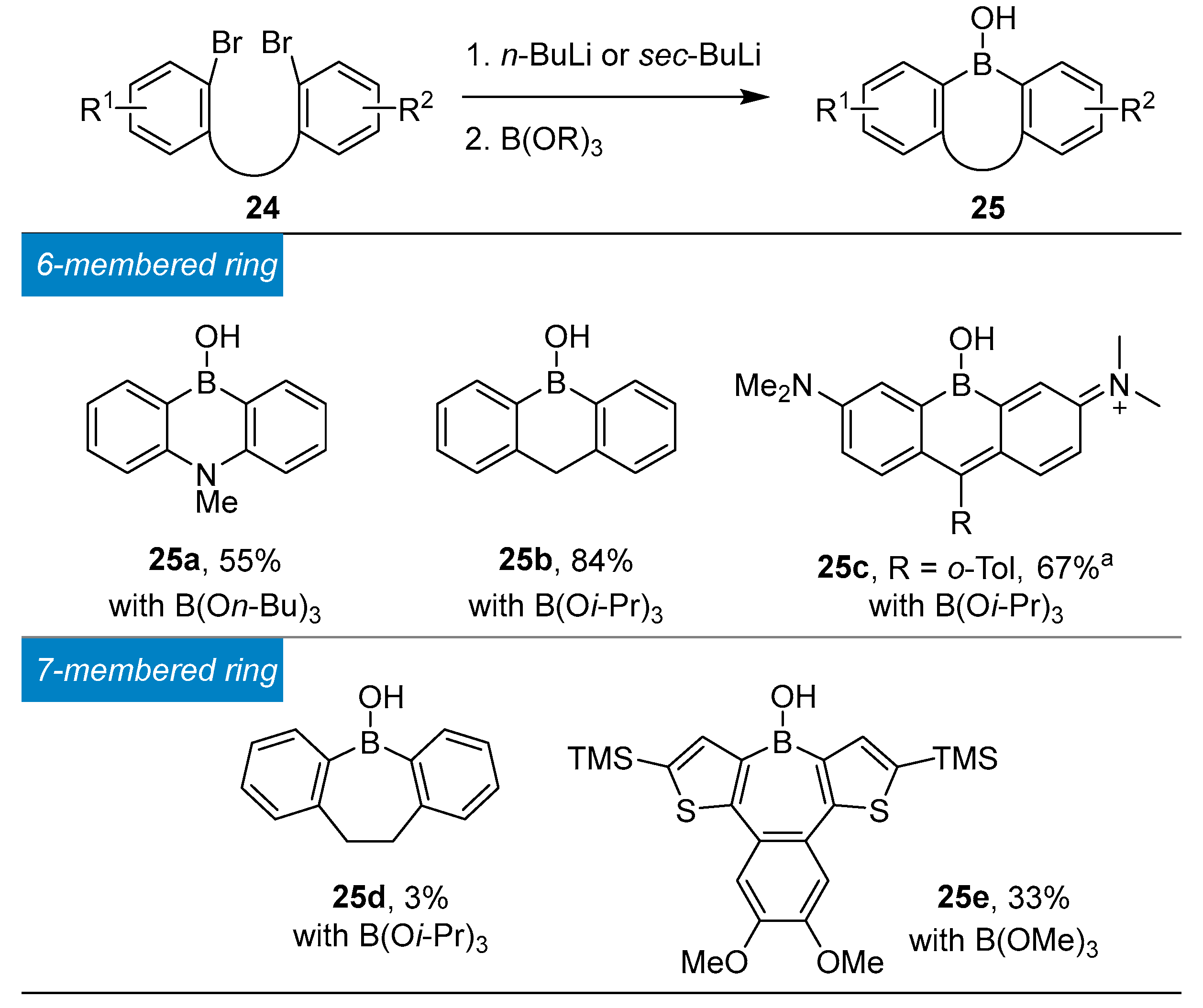
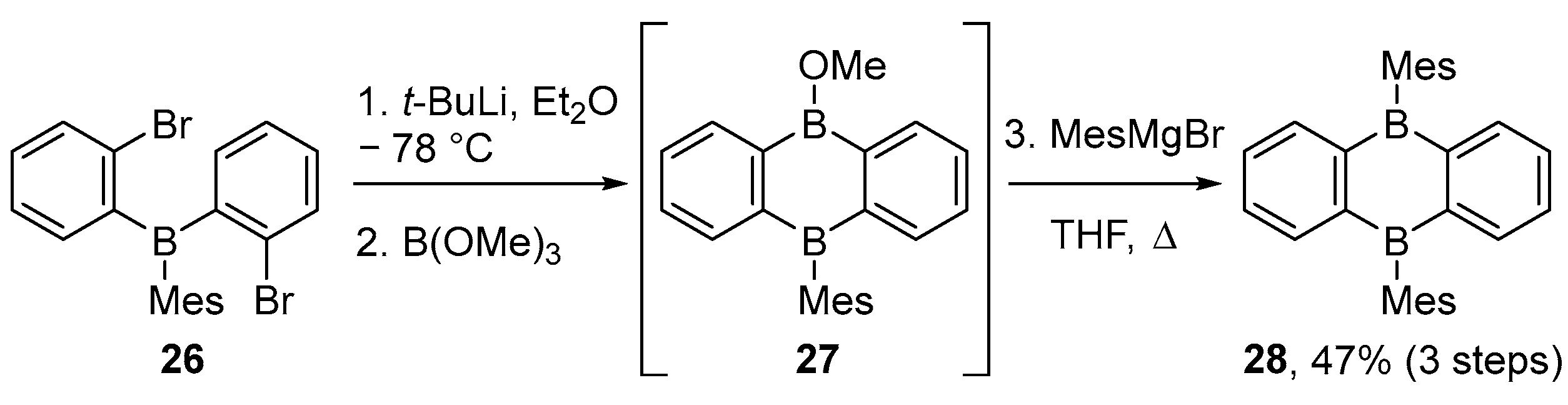
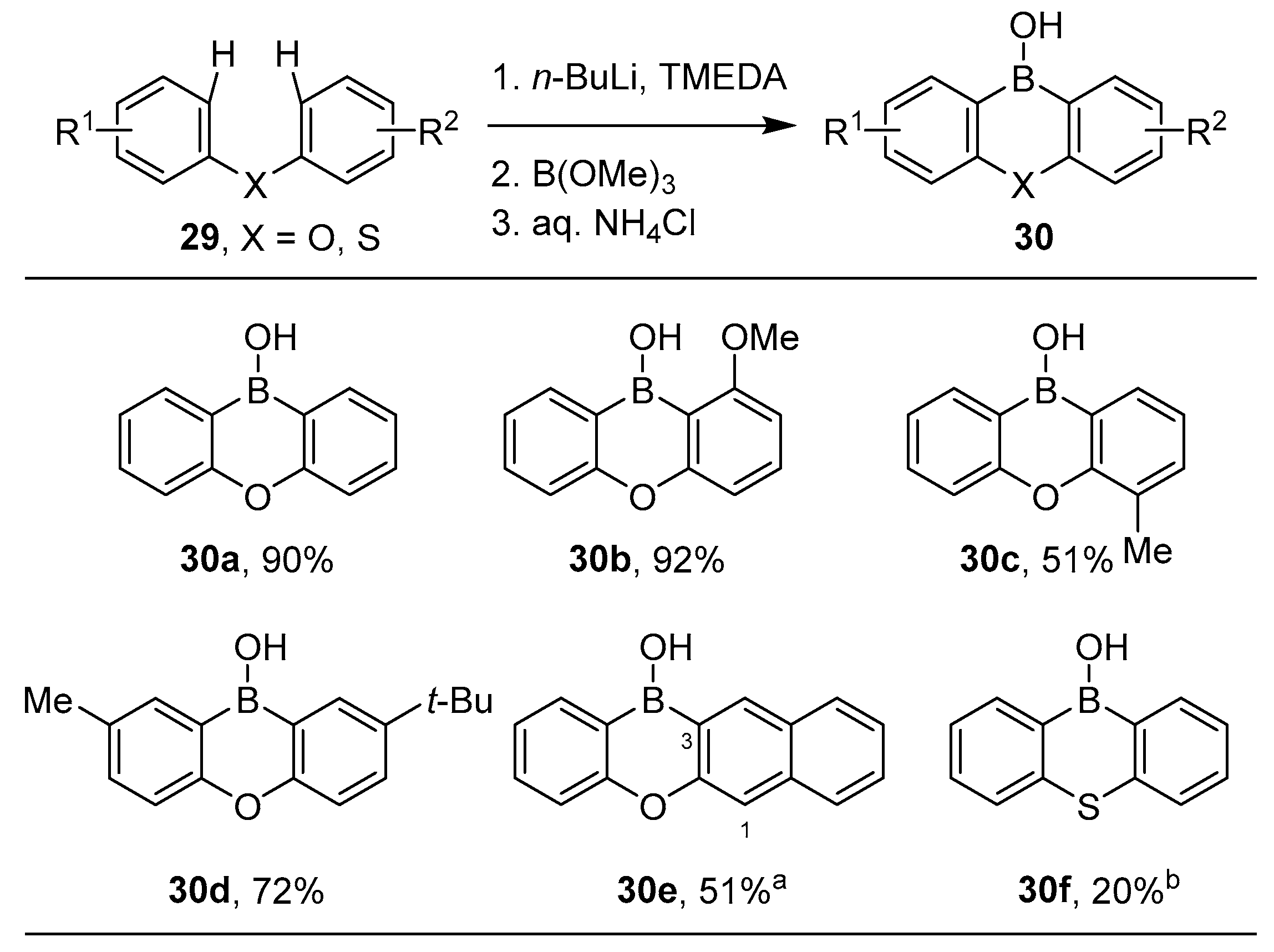
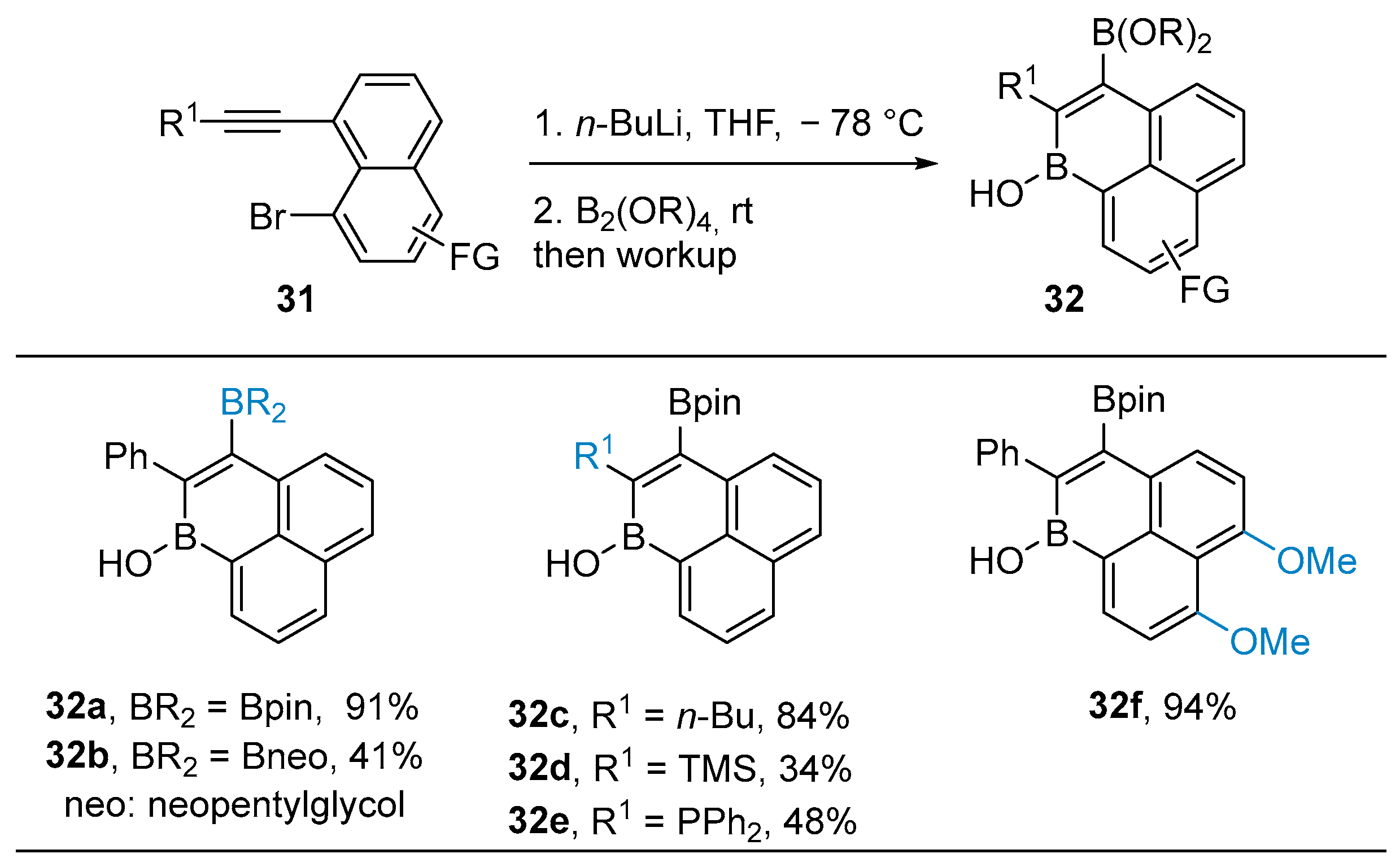
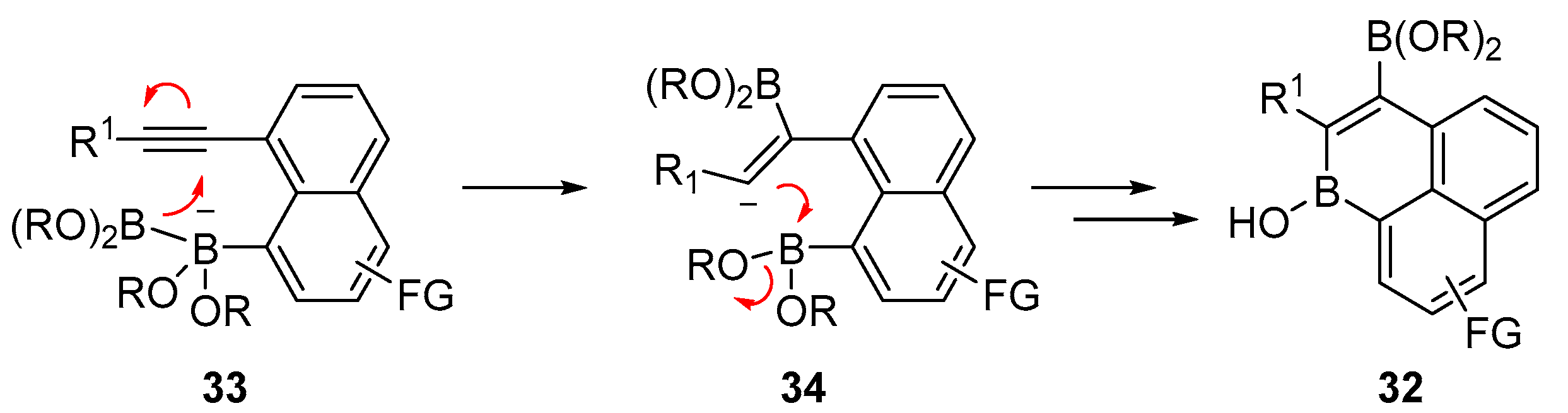
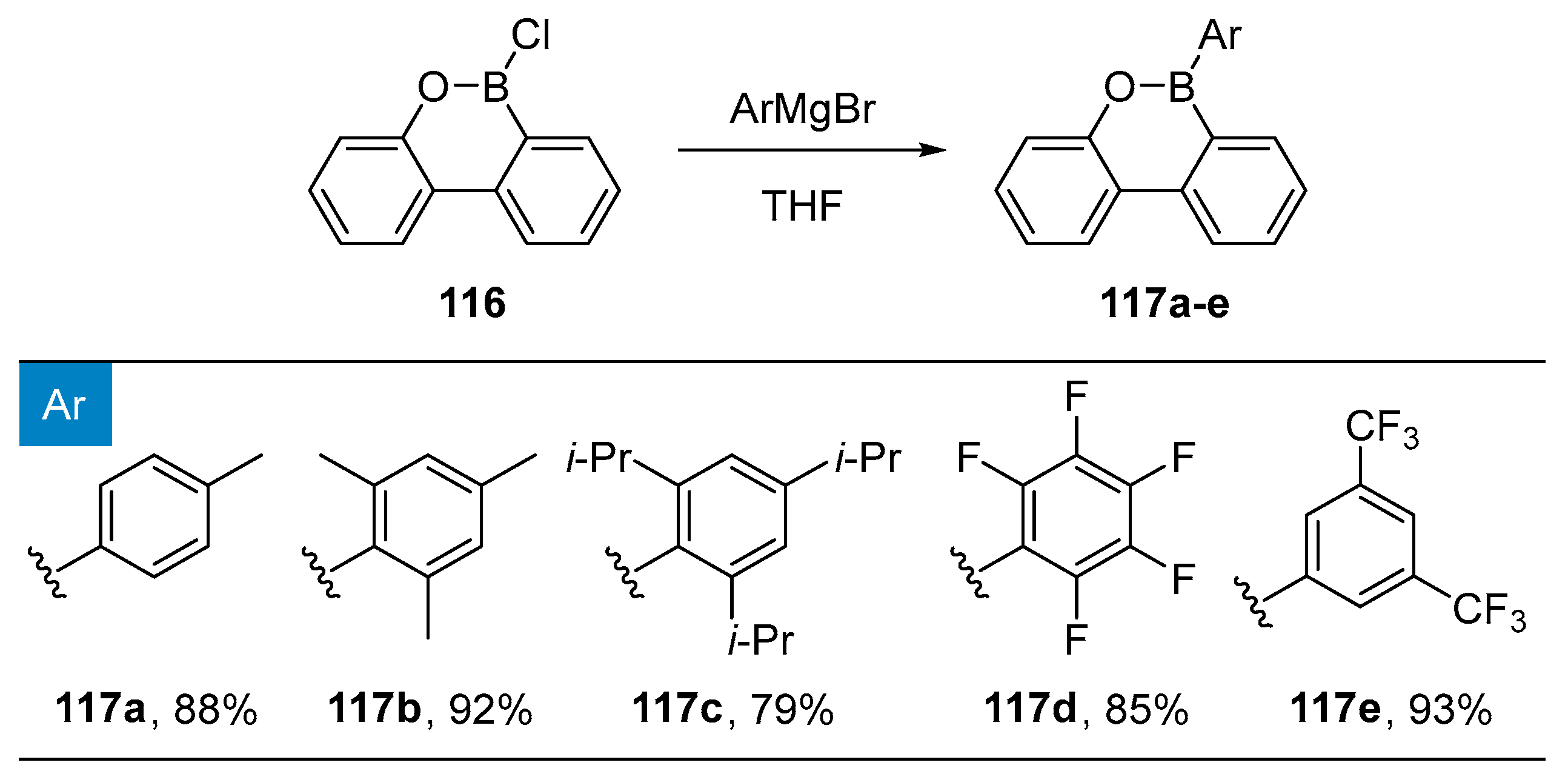
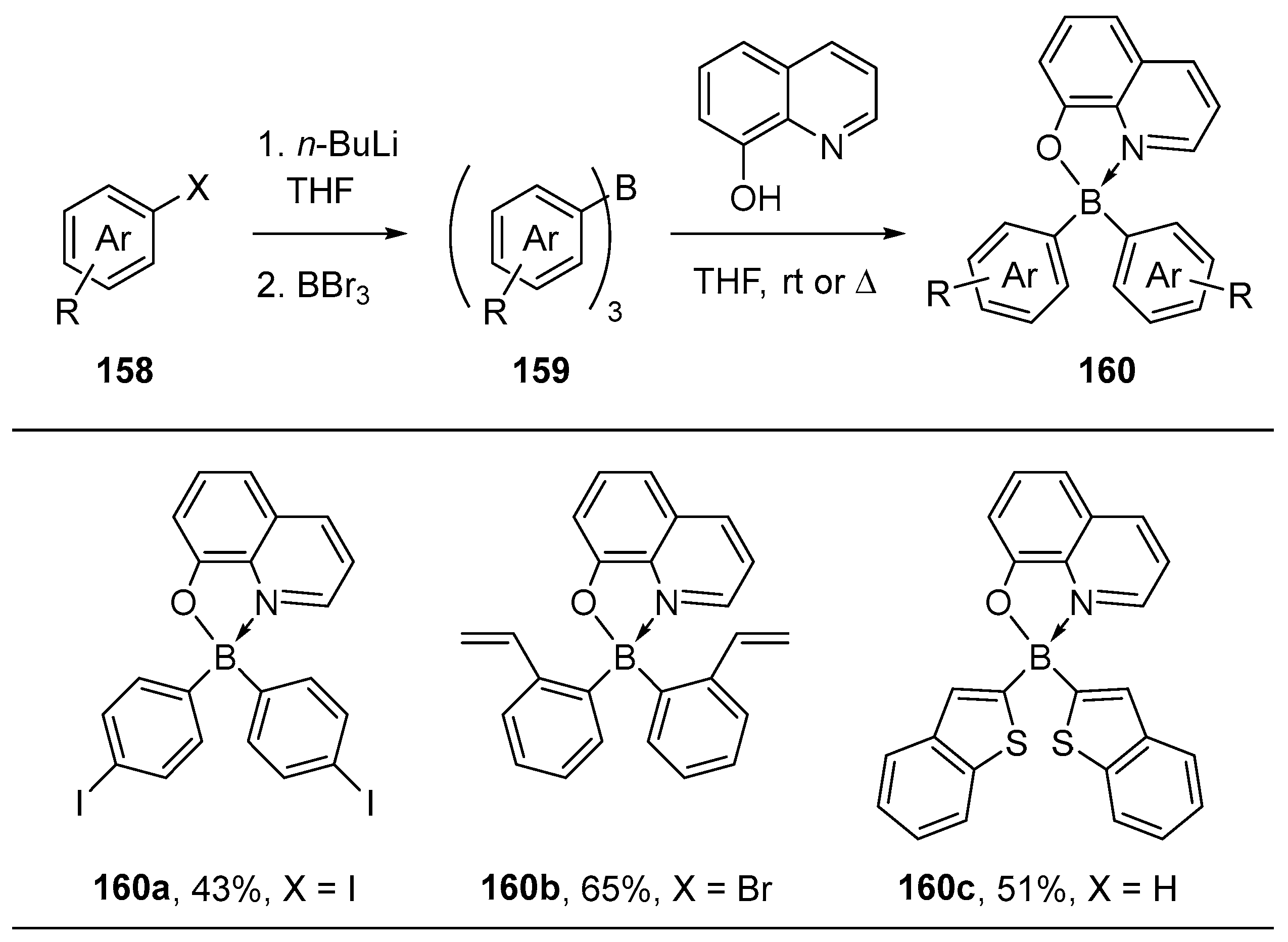
3.1. By Nucleophilic Addition of ArLi and ArMgX to Aryl Boron Reagents
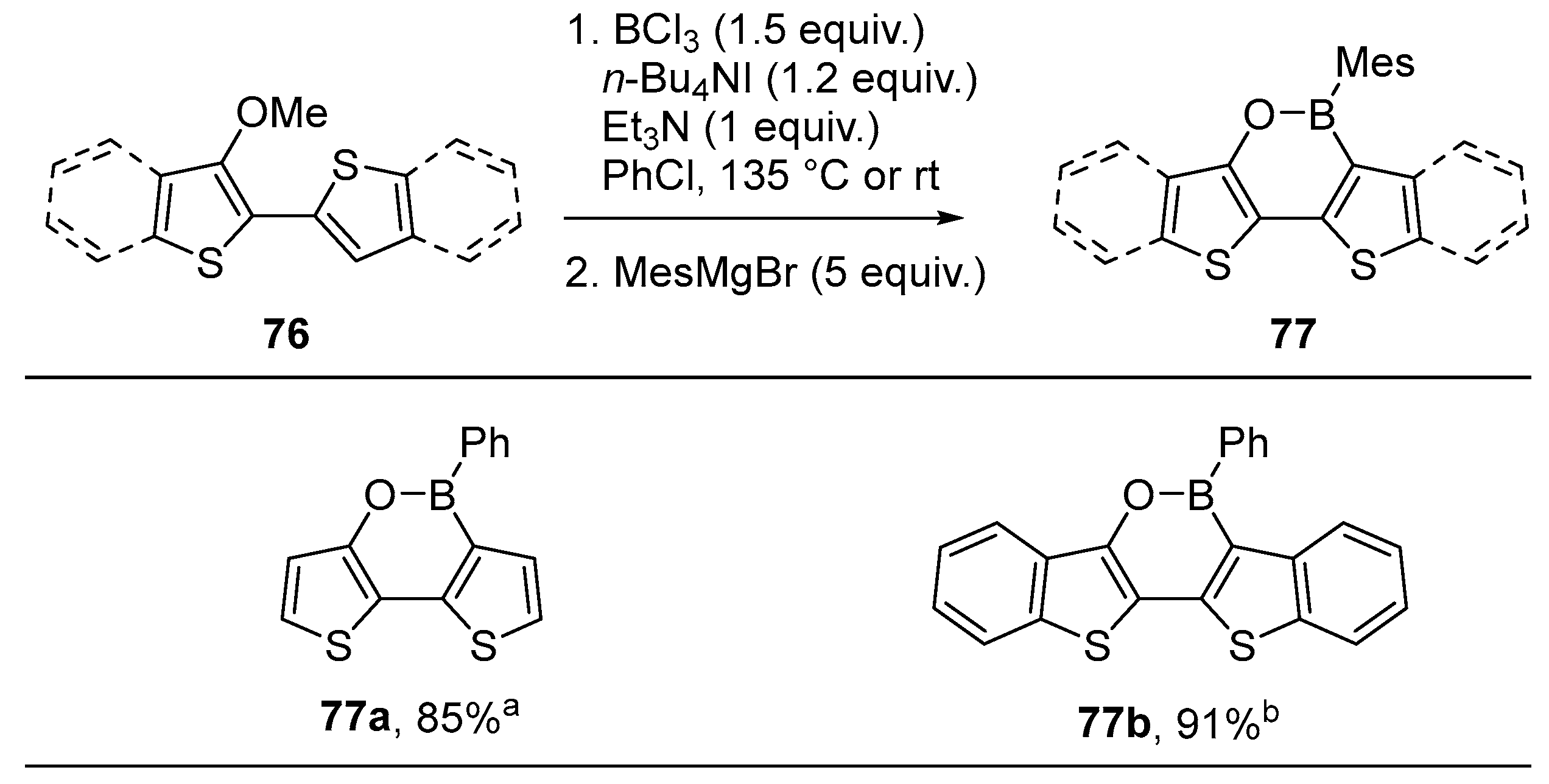
3.2. By Electrophilic Aromatic Substitution with Aryl Boron Reagents
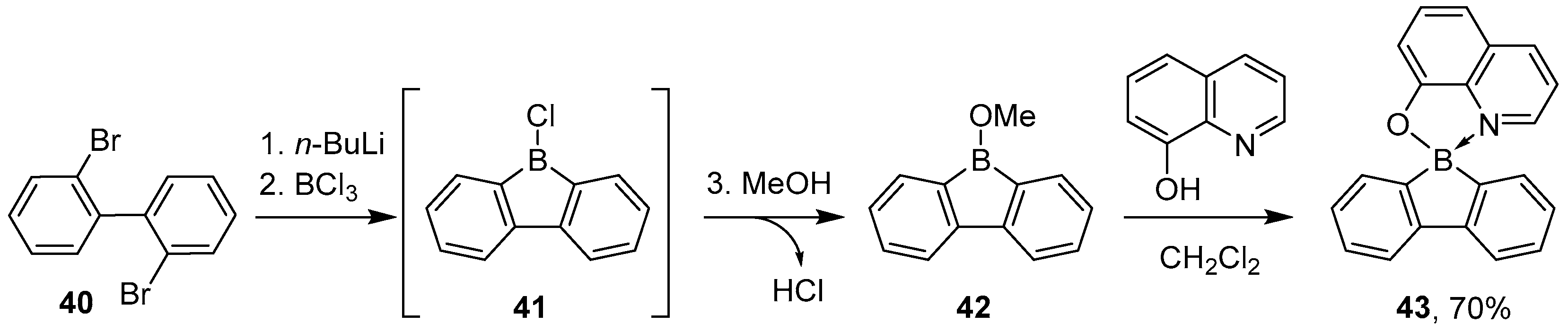
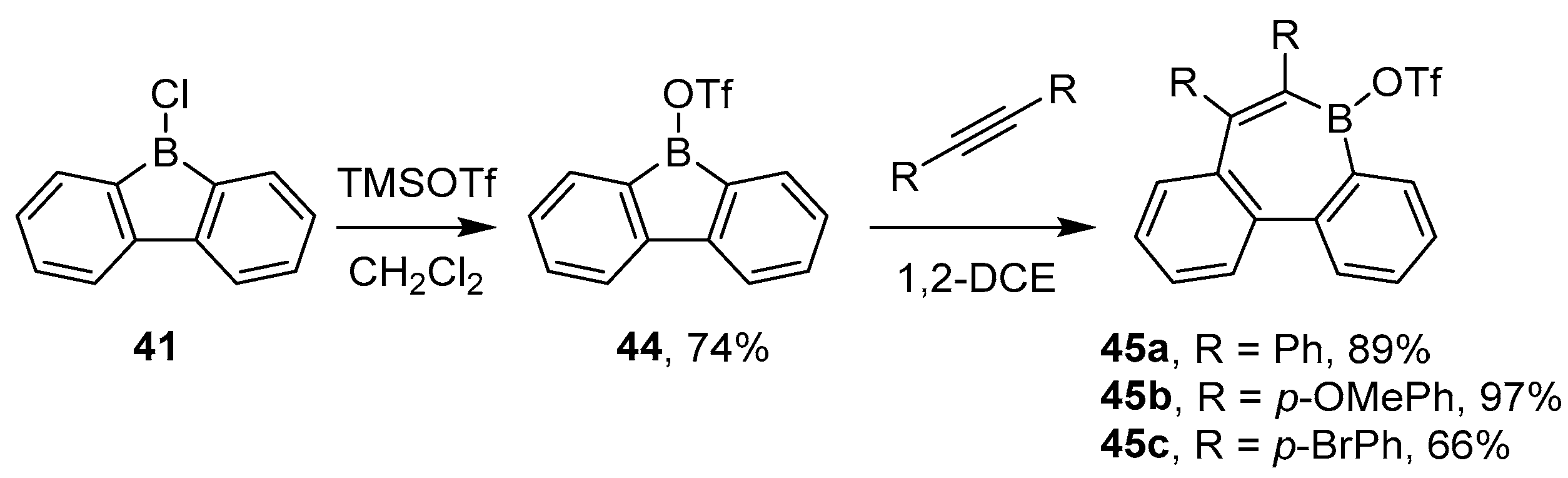
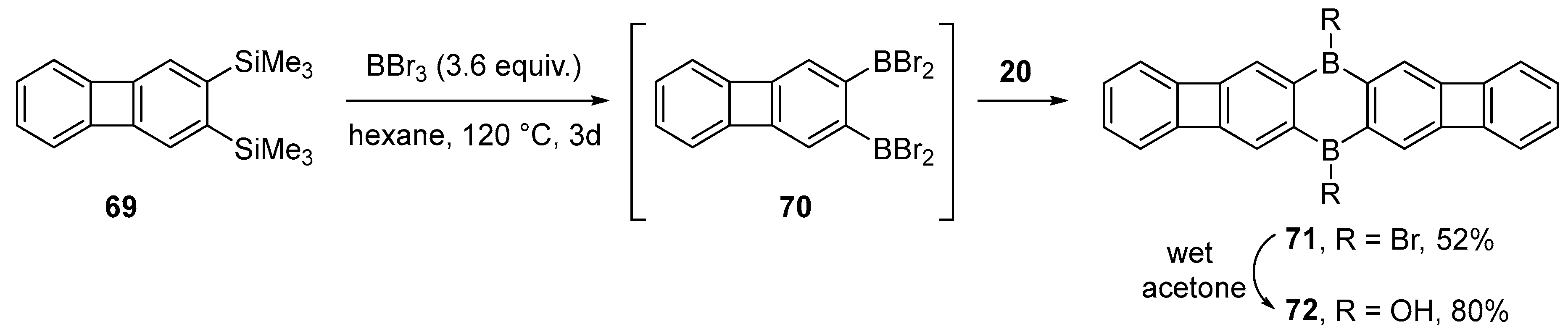
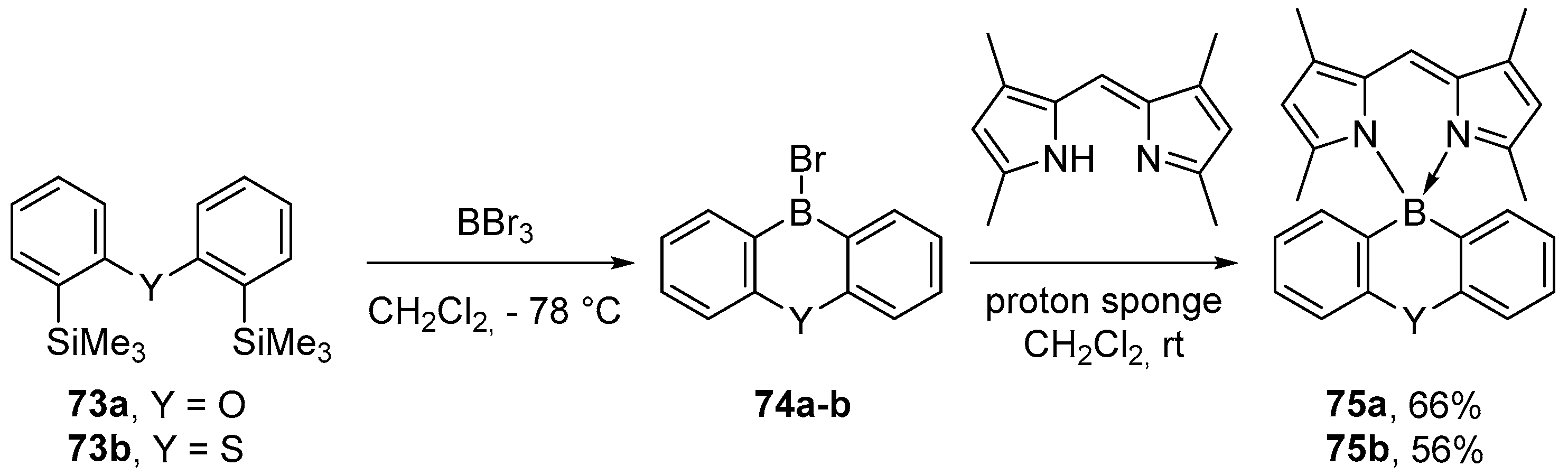
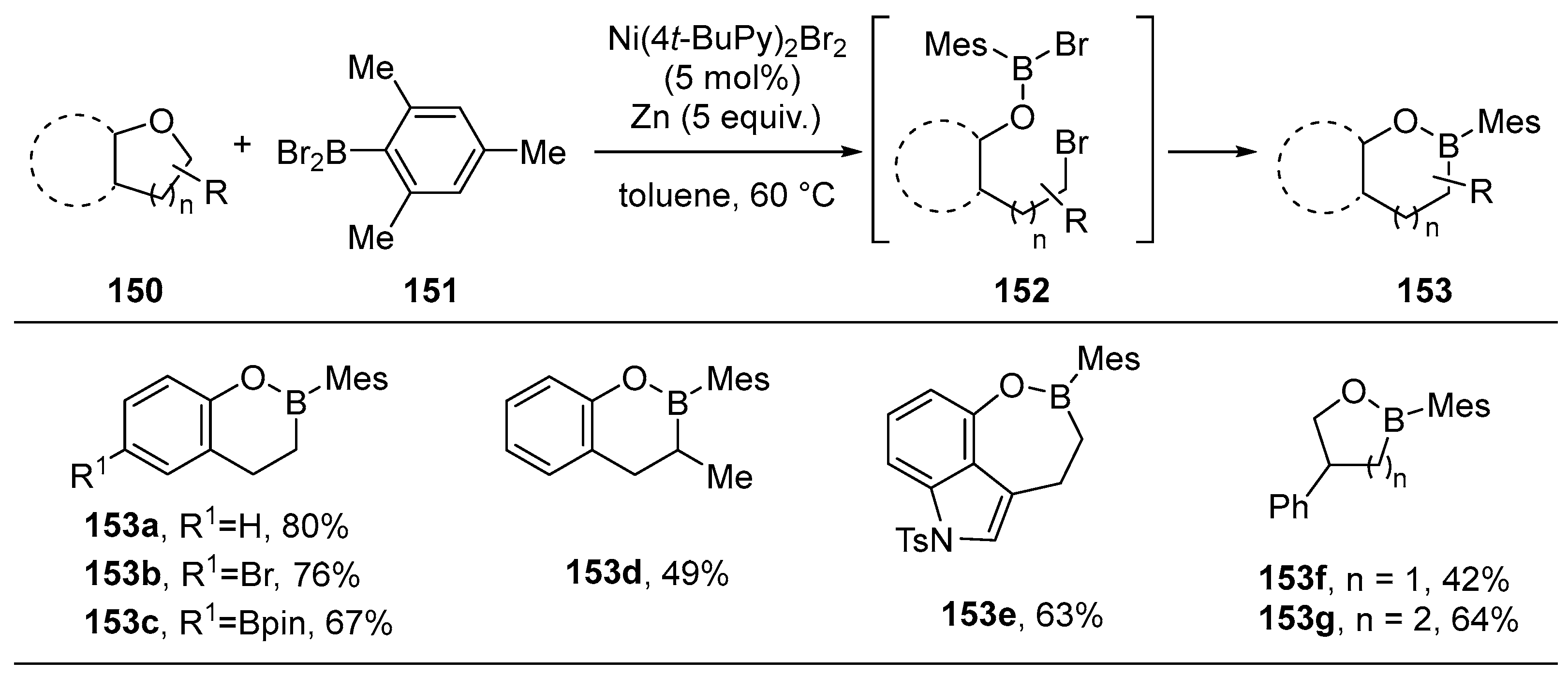
3.3. Other Methods
4. Cleavage of One C-B Bond
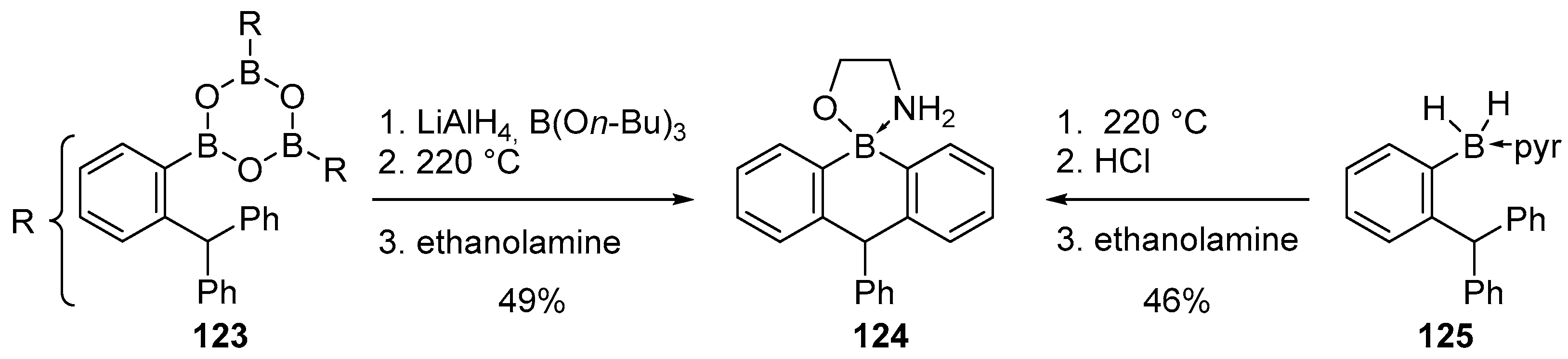
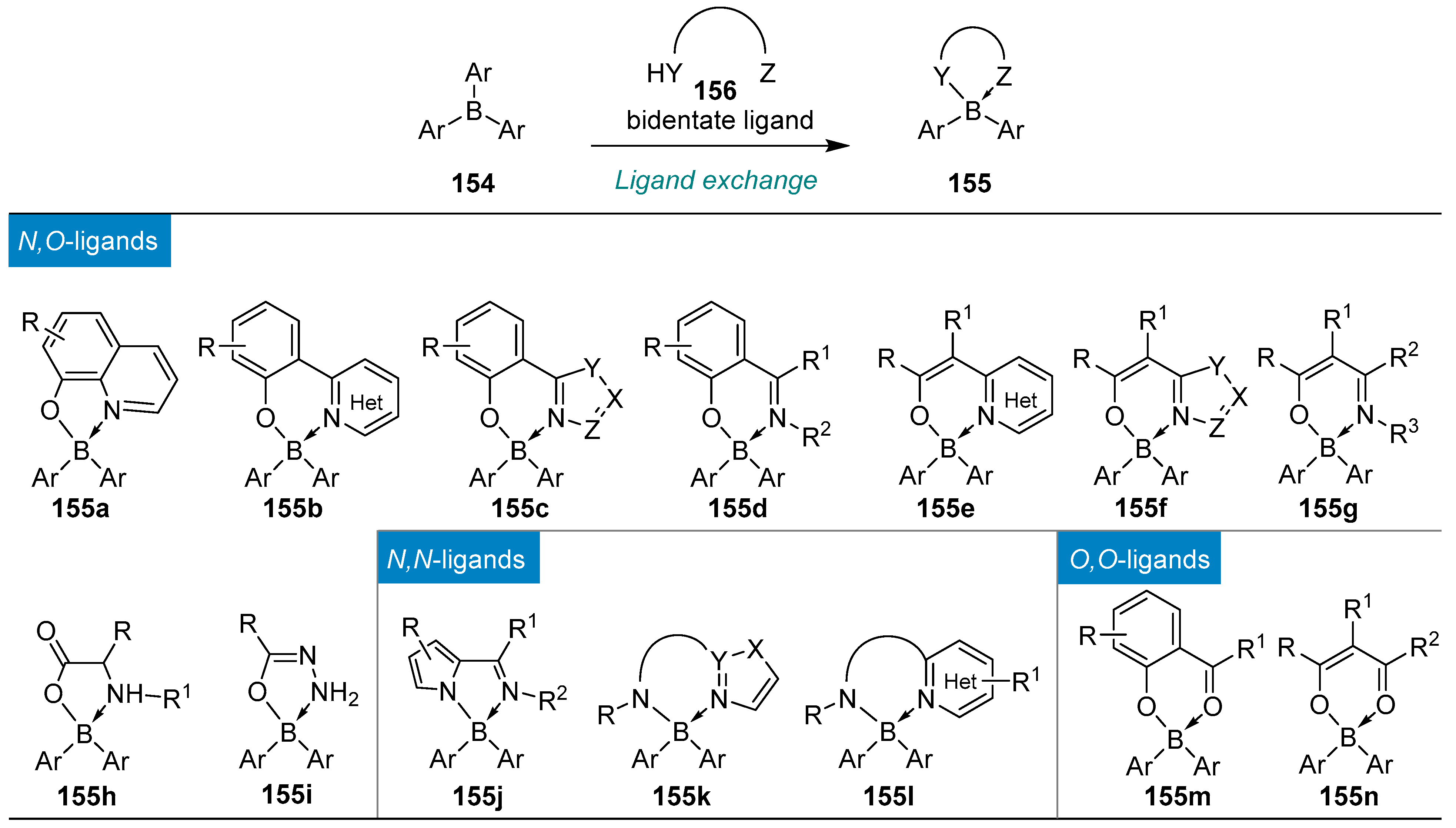
4.1. Synthesis of Four-Coordinated Borinic Acids from Triaryl Boranes
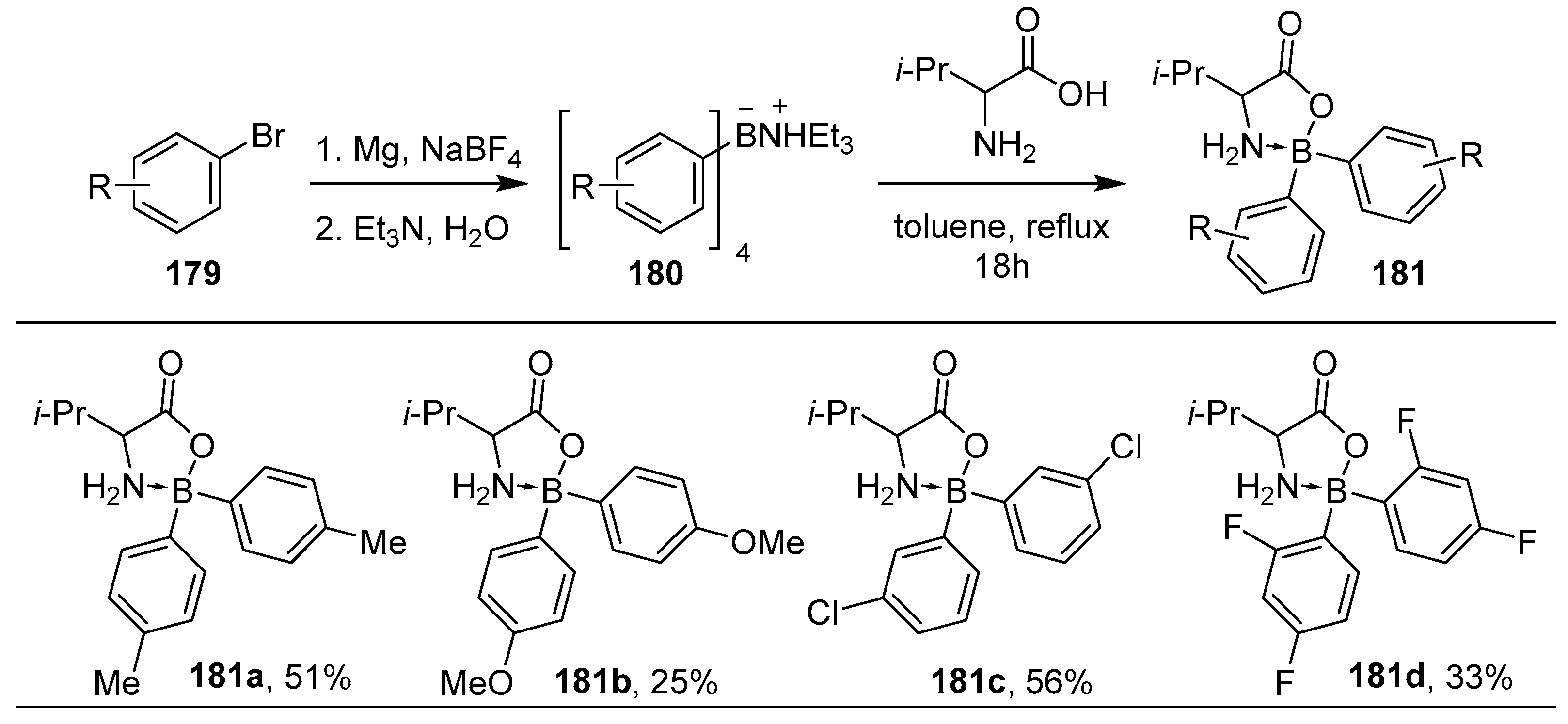
4.2. Synthesis of Four-Coordinated Borinic Acids from Tetraarylborate Salts
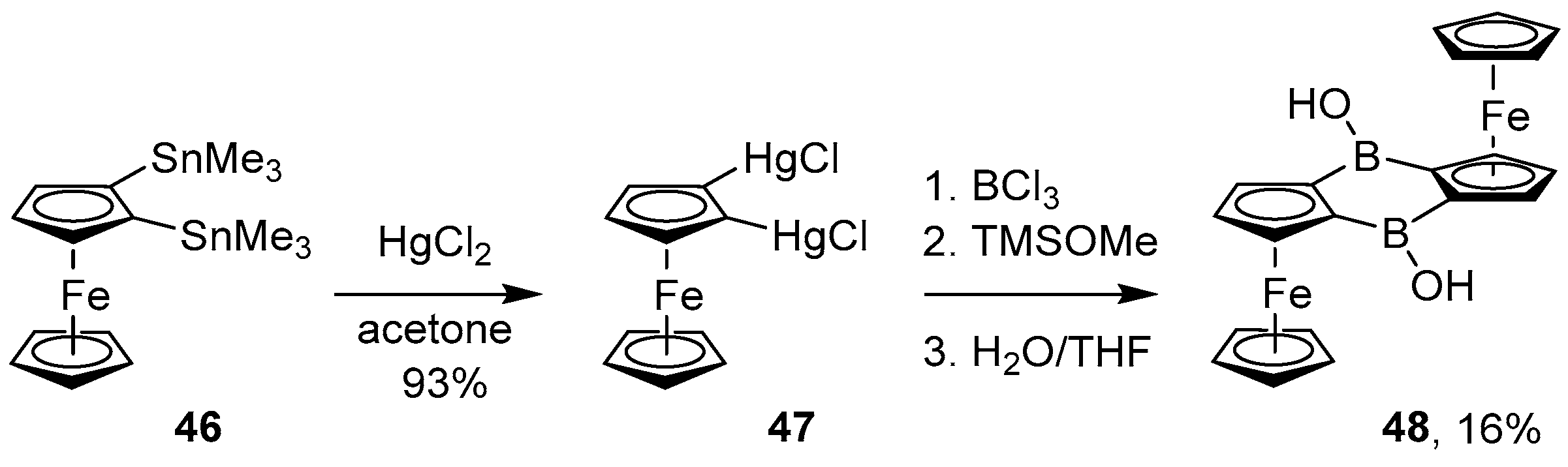
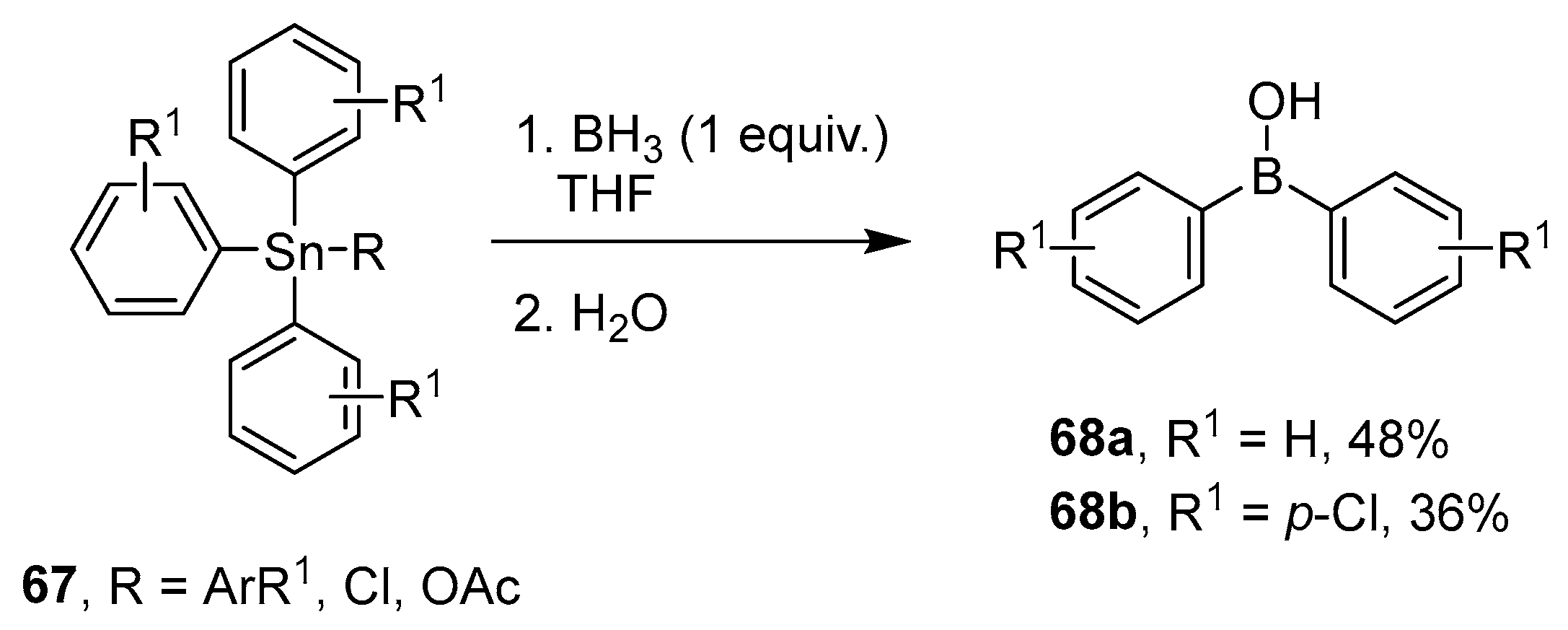
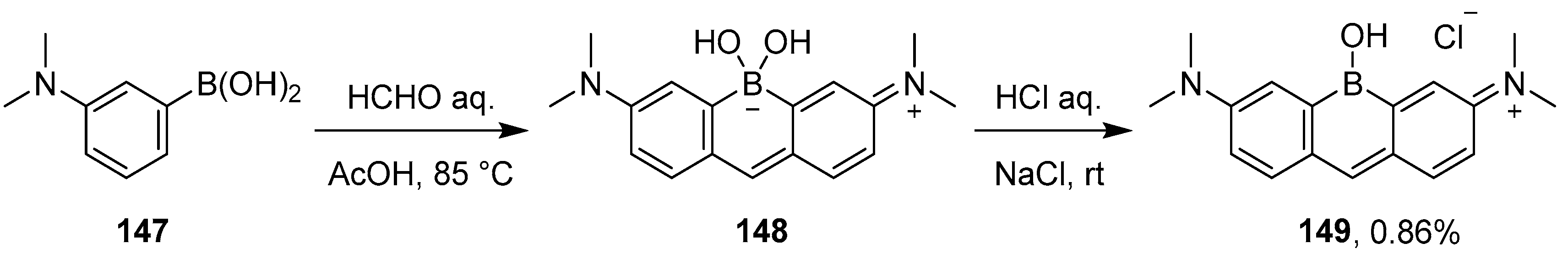
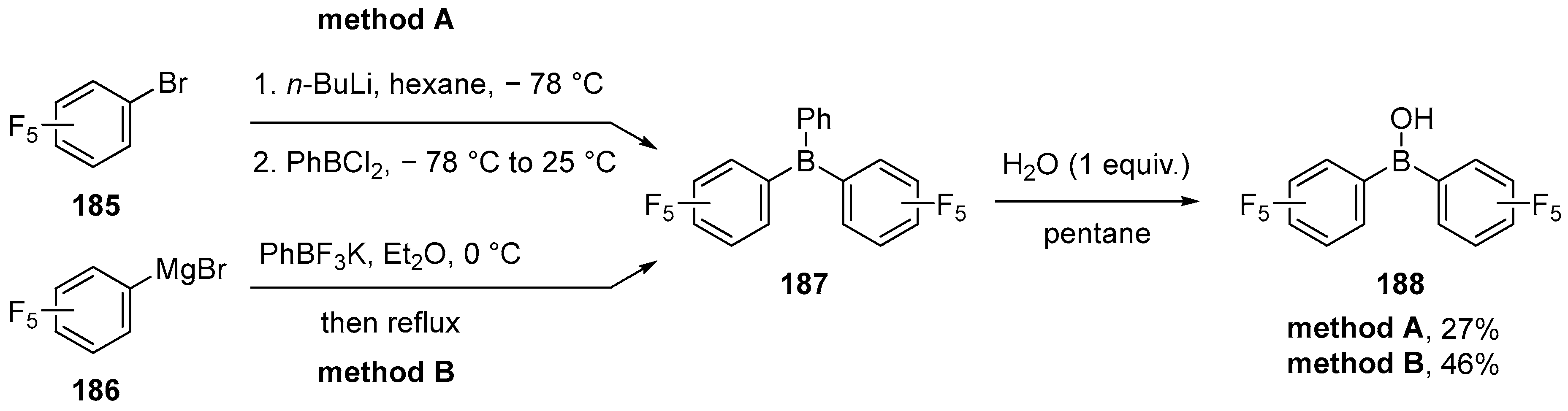
4.3. Synthesis of Borinic Acids by Hydrolysis or Oxidation of Triaryl Boranes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hiller, N.d.J.; do Amaral e Silva, N.A.; Tavares, T.A.; Faria, R.X.; Eberlin, M.N.; de Luna Martins, D. Arylboronic Acids and their Myriad of Applications Beyond Organic Synthesis. Eur. J. Org. Chem. 2020, 2020, 4841–4877. [Google Scholar] [CrossRef]
- Flores-Parra, A.; Contreras, R. Boron coordination compounds derived from organic molecules of biological interest. Coord. Chem. Rev. 2000, 196, 85–124. [Google Scholar] [CrossRef]
- Beringhelli, T.; D’Alfonso, G.; Donghi, D.; Maggioni, D.; Mercandelli, P.; Sironi, A. Aggregation and Ionization Equilibria of Bis(pentafluorophenyl)borinic Acid Driven by Hydrogen-Bonding with Tetrahydrofuran. Organometallics 2007, 26, 2088–2095. [Google Scholar] [CrossRef] [Green Version]
- Donghi, D.; Maggioni, D.; Beringhelli, T.; D’Alfonso, G.; Mercandelli, P.; Sironi, A. Hydrogen Bonding and Lewis Acid–Base Interactions in the System Bis(pentafluorophenyl)borinic Acid / Methanol. Eur. J. Inorg. Chem. 2008, 2008, 1645–1653. [Google Scholar] [CrossRef]
- Donghi, D.; Maggioni, D.; Beringhelli, T.; D’Alfonso, G. 19 F NMR Spectroscopic Investigation of the Reaction of Bis(pentafluorophenyl)borinic Acid with a “Proton Sponge”: Deprotonation, Trimerization and Stepwise Dearylation. Eur. J. Inorg. Chem. 2008, 2008, 3606–3613. [Google Scholar] [CrossRef]
- Chen, X.; Ke, H.; Zou, G. Nickel-Catalyzed Cross-Coupling of Diarylborinic Acids with Aryl Chlorides. ACS Catal. 2014, 4, 379–385. [Google Scholar] [CrossRef]
- Chen, X.; Ke, H.; Chen, Y.; Guan, C.; Zou, G. Cross-coupling of diarylborinic acids and anhydrides with arylhalides catalyzed by a phosphite/N-heterocyclic carbene co-supported palladium catalyst system. J. Org. Chem. 2012, 77, 7572–7578. [Google Scholar] [CrossRef]
- Hofer, A.; Kovacs, G.; Zappatini, A.; Leuenberger, M.; Hediger, M.A.; Lochner, M. Design, synthesis and pharmacological characterization of analogs of 2-aminoethyl diphenylborinate (2-APB), a known store-operated calcium channel blocker, for inhibition of TRPV6-mediated calcium transport. Bioorg. Med. Chem. 2013, 21, 3202–3213. [Google Scholar] [CrossRef]
- Dellis, O.; Mercier, P.; Chomienne, C. The boron-oxygen core of borinate esters is responsible for the store-operated calcium entry potentiation ability. BMC Pharmacol. 2011, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.Z.; Ozaki, S.; Goto, J.; Mikoshiba, K. Synthesis of bisboron compounds and their strong inhibitory activity on store-operated calcium entry. Bioorg. Med. Chem. Lett. 2010, 20, 1395–1398. [Google Scholar] [CrossRef]
- Haque, A.; Al-Balushi, R.A.; Raithby, P.R.; Khan, M.S. Recent Advances in π-Conjugated N^C-Chelate Organoboron Materials. Molecules 2020, 25, 2645. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S. Catalysis Based on Reversible Covalent Interactions of Organoboron Compounds. Acc. Chem. Res. 2014, 48, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Takemoto, Y. Regio- and stereoselective glycosylation of 1,2-O-unprotected sugars using organoboron catalysts. Tetrahedron 2020, 76, 131328. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Wang, Y. Four-coordinate organoboron compounds for organic light-emitting diodes (OLEDs). Chem. Soc. Rev. 2013, 42, 8416–8433. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Huang, L.; Ren, C.; Zou, G. Development of a Telescoped Process for Preparation of N,O-Chelated Diarylborinates. Org. Process Res. Dev. 2018, 22, 824–828. [Google Scholar] [CrossRef]
- Hessel, V.; Hofmann, C.; Löwe, H.; Meudt, A.; Scherer, S.; Schönfeld, F.; Werner, B. Selectivity Gains and Energy Savings for the Industrial Phenyl Boronic Acid Process Using Micromixer/Tubular Reactors. Org. Process Res. Dev. 2004, 8, 511–523. [Google Scholar] [CrossRef]
- Baldwin, A.G.; Tapia, V.S.; Swanton, T.; White, C.S.; Beswick, J.A.; Brough, D.; Freeman, S. Design, Synthesis and Evaluation of Oxazaborine Inhibitors of the NLRP3 Inflammasome. ChemMedChem 2018, 13, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Nudelman, F.; Matthes, R.R.; Shaver, M.P. Frustrated Lewis Pair Polymers as Responsive Self-Healing Gels. J. Am. Chem. Soc. 2017, 139, 14232–14236. [Google Scholar] [CrossRef] [Green Version]
- Povlock, T.P.; Lippincott, W.T. The Reaction of Trimethoxyboroxine with Aromatic Grignard Reagents: A New Synthesis of Borinic Acids1. J. Am. Chem. Soc. 1958, 80, 5409–5411. [Google Scholar] [CrossRef]
- Murakami, S.; Suzuki, T. Method for Producing Borinic Acid Derivative, and Novel Borinic Acid Derivative. WO2014030600, 2014. [Google Scholar]
- Hawkins, V.F.; Wilkinson, M.C.; Whiting, M. Mild and Selective Synthesis of an Aryl Boronic Ester by Equilibration of Mixtures of Boronic and Borinic Acid Derivatives. Org. Process Res. Dev. 2008, 12, 1265–1268. [Google Scholar] [CrossRef]
- Bell, B.M.; Clark, T.P.; De Vries, T.S.; Lai, Y.; Laitar, D.S.; Gallagher, T.J.; Jeon, J.-H.; Kearns, K.L.; McIntire, T.; Mukhopadhyay, S.; et al. Boron-based TADF emitters with improved OLED device efficiency roll-off and long lifetime. Dyes Pigm. 2017, 141, 83–92. [Google Scholar] [CrossRef]
- Davidson, J.M.; French, C.M. The synthesis and structure of aromatic boron compounds. J. Chem. Soc. 1960, 191–195. [Google Scholar] [CrossRef]
- El Dine, T.M.; Rouden, J.; Blanchet, J. Borinic acid catalysed peptide synthesis. Chem. Commun. 2015, 51, 16084–16087. [Google Scholar] [CrossRef]
- Baker, S.J.; Akama, T.; Zhang, Y.K.; Sauro, V.; Pandit, C.; Singh, R.; Kully, M.; Khan, J.; Plattner, J.J.; Benkovic, S.J.; et al. Identification of a novel boron-containing antibacterial agent (AN0128) with anti-inflammatory activity, for the potential treatment of cutaneous diseases. Bioorg. Med. Chem. Lett. 2006, 16, 5963–5967. [Google Scholar] [CrossRef]
- Ke, H.; Chen, X.; Zou, G. N-Heterocyclic Carbene-Assisted, Bis(phosphine)nickel-Catalyzed Cross-Couplings of Diarylborinic Acids with Aryl Chlorides, Tosylates, and Sulfamates. J. Org. Chem. 2014, 79, 7132–7140. [Google Scholar] [CrossRef] [PubMed]
- Torabi Kohlbouni, S.; Sarkar, A.; Zhang, J.; Li, X.; Borhan, B. Absolute stereochemical determination of 1,2-diols via complexation with dinaphthyl borinic acid. Chirality 2020, 32, 817–823. [Google Scholar] [CrossRef]
- Sengupta, A.; Doshi, A.; Jäkle, F.; Peetz, R.M. Segmented conjugated macromolecules containing silicon and boron: ADMET synthesis and luminescent properties. J. Polym. Sci. Pol. Chem. 2015, 53, 1707–1718. [Google Scholar] [CrossRef]
- Wan, W.-M.; Li, S.-S.; Liu, D.-M.; Lv, X.-H.; Sun, X.-L. Synthesis of Electron-Deficient Borinic Acid Polymers with Multiresponsive Properties and Their Application in the Fluorescence Detection of Alizarin Red S and Electron-Rich 8-Hydroxyquinoline and Fluoride Ion: Substituent Effects. Macromolecules 2017, 50, 6872–6879. [Google Scholar] [CrossRef]
- Lv, X.-H.; Li, S.-S.; Tian, C.-Y.; Yang, M.-M.; Li, C.; Zhou, Y.; Sun, X.-L.; Zhang, J.; Wan, W.-M. Borinic Acid Polymer: Simplified Synthesis and Enzymatic Biofuel Cell Application. Macromol. Rapid Commun. 2017, 38, 1600687. [Google Scholar] [CrossRef]
- Wang, G.; Taylor, M.S. Borinic Acid-Catalyzed Regioselective Ring-Opening of 3,4- and 2,3-Epoxy Alcohols with Halides. Adv. Synth. Catal. 2020, 362, 398–403. [Google Scholar] [CrossRef]
- Cheung, K.Y.; Miao, Q. A ketone-functionalized aromatic saddle as a potential building block for negatively curved carbon nanobelts. Chin. Chem. Lett. 2019, 30, 1506–1508. [Google Scholar] [CrossRef]
- Igarashi, T.; Tobisu, M.; Chatani, N. Catalytic Double Carbon-Boron Bond Formation for the Synthesis of Cyclic Diarylborinic Acids as Versatile Building Blocks for pi-Extended Heteroarenes. Angew. Chem. Int. Ed. 2017, 56, 2069–2073. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lesiak, L.; Lai, R.; Beck, J.R.; Zhao, J.; Elowsky, C.G.; Li, H.; Stains, C.I. Chemoselective Alteration of Fluorophore Scaffolds as a Strategy for the Development of Ratiometric Chemodosimeters. Angew. Chem. Int. Ed. 2017, 56, 4197–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Cha, I.; Lee, E.J.; Lee, Y.; Kim, Y.H.; Song, C. Annulated Borepin-1-ol: Coordinative Control of Aromaticity and Photophysical Properties. Chem. Lett. 2014, 43, 1432–1434. [Google Scholar] [CrossRef]
- Dimitrijević, E.; Taylor, M.S. 9-Hetero-10-boraanthracene-derived borinic acid catalysts for regioselective activation of polyols. Chem. Sci. 2013, 4, 3298–3303. [Google Scholar] [CrossRef]
- Dimitrijevic, E.; Cusimano, M.; Taylor, M.S. Synthesis of benzannulated heterocycles by twofold Suzuki-Miyaura couplings of cyclic diarylborinic acids. Org. Biomol. Chem. 2014, 12, 1391–1394. [Google Scholar] [CrossRef]
- Agou, T.; Sekine, M.; Kawashima, T. Stepwise synthesis and properties of a 9,10-dihydro-9,10-diboraanthracene. Tetrahedron Lett. 2010, 51, 5013–5015. [Google Scholar] [CrossRef]
- Niu, L.; Yang, H.; Jiang, Y.; Fu, H. Efficient Synthesis of Dibenzoxaborininols from Diaryl Ethers and Their Application to Dibenzofuran Synthesis. Adv. Synth. Catal. 2013, 355, 3625–3632. [Google Scholar] [CrossRef]
- Hirano, K.; Morimoto, K.; Fujioka, S.; Miyamoto, K.; Muranaka, A.; Uchiyama, M. Nucleophilic Diboration Strategy Targeting Diversified 1-Boraphenarene Architectures. Angew. Chem. Int. Ed. 2020, 59, 21448–21453. [Google Scholar] [CrossRef]
- Gibson, V.C.; Redshaw, C.; Clegg, W.; Elsegood, M.R.J. Synthesis and structural characterization of some novel metalloboroxides bearing boron-bound mesityl and fluoromesityl substituents: The molecular structure of the first metallaboroxane complex. Polyhedron 1997, 16, 2637–2641. [Google Scholar] [CrossRef]
- Cornet, S.M.; Dillon, K.B.; Entwistle, C.D.; Fox, M.A.; Goeta, A.E.; Goodwin, H.P.; Marder, T.B.; Thompson, A.L. Synthesis and characterisation of some new boron compounds containing the 2,4,6-(CF3)3C6H2(fluoromes = Ar), 2,6-(CF3)2C6H3(fluoroxyl = Ar′), or 2,4-(CF3)2C6H3(Ar″) ligands. Dalton Trans. 2003, 23, 4395–4405. [Google Scholar] [CrossRef]
- Chazalette, C.; Riviere-Baudet, M.; Scozzafava, A.; Abbate, F.; Maarouf, Z.B.; Supuran, C.T. Carbonic Anhydrase Inhibitors, Interaction of Boron Derivatives with Isozymes I and II: A New Binding Site for Hydrophobic Inhibitors at the Entrance of the Active Site as shown by Docking Studies. J. Enzyme Inhib. 2010, 16, 125–133. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Suzuki, K.; Hayashi, K.; Nema, S.-y.; Yamashita, M. Chlorine-Substituted 9,10-Dihydro-9-aza-10-boraanthracene as a Precursor for Various Boron- and Nitrogen-Containing π-Conjugated Compounds. Org. Lett. 2019, 21, 1722–1725. [Google Scholar] [CrossRef]
- Urban, M.; Durka, K.; Górka, P.; Wiosna-Sałyga, G.; Nawara, K.; Jankowski, P.; Luliński, S. The effect of locking π-conjugation in organoboron moieties in the structures of luminescent tetracoordinate boron complexes. Dalton Trans. 2019, 48, 8642–8663. [Google Scholar] [CrossRef]
- Shoji, Y.; Shigeno, N.; Takenouchi, K.; Sugimoto, M.; Fukushima, T. Mechanistic Study of Highly Efficient Direct 1,2-Carboboration of Alkynes with 9-Borafluorenes. Chem. Eur. J. 2018, 24, 13223–13230. [Google Scholar] [CrossRef] [PubMed]
- Venkatasubbaiah, K.; Pakkirisamy, T.; Lalancette, R.A.; Jäkle, F. Tuning the electronic structure of diboradiferrocenes. Dalton Trans. 2008, 33, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Thilagar, P.; Murillo, D.; Chen, J.; Jakle, F. Synthesis and supramolecular assembly of the bifunctional borinic acid [1,2-fcB(OH)]2. Dalton Trans. 2013, 42, 665–670. [Google Scholar] [CrossRef]
- Coates, G.E.; Livingstone, J.G. 965. Aminoditolylborane and the preparation of diarylborinic acids. J. Chem. Soc. 1961, 4909–4911. [Google Scholar] [CrossRef]
- Marciasini, L.; Cacciuttolo, B.; Vaultier, M.; Pucheault, M. Synthesis of Borinic Acids and Borinate Adducts Using Diisopropylaminoborane. Org. Lett. 2015, 17, 3532–3535. [Google Scholar] [CrossRef]
- Qi, Y.; Xu, W.; Kang, R.; Ding, N.; Wang, Y.; He, G.; Fang, Y. Discrimination of saturated alkanes and relevant volatile compounds via the utilization of a conceptual fluorescent sensor array based on organoboron-containing polymers. Chem. Sci. 2018, 9, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Pucheault, M.; Pinet, S.; Richard, J.; Birepinte, M.; Charbonnier, J.; Liautard, V. Borinic Acids via Direct Arylation of Amine–Borane Complexes: An Air- and Water-Stable Boron Source. Synthesis 2017, 49, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Guerrand, H.D.S.; Marciasini, L.D.; Jousseaume, M.; Vaultier, M.; Pucheault, M. Borylation of Unactivated Aryl Chlorides under Mild Conditions by Using Diisopropylaminoborane as a Borylating Reagent. Chem. Eur. J. 2014, 20, 5573–5579. [Google Scholar] [CrossRef] [PubMed]
- Shimazumi, R.; Igarashi, T.; Tobisu, M. Palladium-catalyzed B-Diarylation of Diethylaminoborane for the Synthesis of Diarylborinic Acids. Chem. Lett. 2020, 49, 760–763. [Google Scholar] [CrossRef]
- Pickles, G.M.; Spencer, T.; Thorpe, F.G.; Chopa, A.B.; Podesta, J.C. The reactions of diborane with aryl-organotin compounds. J. Organomet. Chem. 1984, 260, 7–15. [Google Scholar] [CrossRef]
- Kirschner, S.; Mewes, J.-M.; Bolte, M.; Lerner, H.-W.; Dreuw, A.; Wagner, M. How Boron Doping Shapes the Optoelectronic Properties of Canonical and Phenylene-Containing Oligoacenes: A Combined Experimental and Theoretical Investigation. Chem. Eur. J. 2017, 23, 5104–5116. [Google Scholar] [CrossRef]
- Kirschner, S.; Bao, S.-S.; Fengel, M.K.; Bolte, M.; Lerner, H.-W.; Wagner, M. Aryl–aryl coupling in a polycyclic aromatic hydrocarbon with embedded tetracoordinate boron centre. Org. Biomol. Chem. 2019, 17, 5060–5065. [Google Scholar] [CrossRef]
- Yuan, K.; Wang, X.; Mellerup, S.K.; Kozin, I.; Wang, S. Spiro-BODIPYs with a Diaryl Chelate: Impact on Aggregation and Luminescence. J. Org. Chem. 2017, 82, 13481–13487. [Google Scholar] [CrossRef]
- Sole, S.; Gabbai, F.P. A bidentate borane as colorimetric fluoride ion sensor. Chem. Commun. 2004, 11, 1284–1285. [Google Scholar] [CrossRef]
- Shigemori, K.; Watanabe, M.; Kong, J.; Mitsudo, K.; Wakamiya, A.; Mandai, H.; Suga, S. Iodide-Mediated or Iodide-Catalyzed Demethylation and Friedel-Crafts C-H Borylative Cyclization Leading to Thiophene-Fused 1,2-Oxaborine Derivatives. Org. Lett. 2019, 21, 2171–2175. [Google Scholar] [CrossRef]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-Shot Multiple Borylation toward BN-Doped Nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef]
- Suresh, S.M.; Duda, E.; Hall, D.; Yao, Z.; Bagnich, S.; Slawin, A.M.Z.; Bassler, H.; Beljonne, D.; Buck, M.; Olivier, Y.; et al. A Deep Blue B,N-Doped Heptacene Emitter That Shows Both Thermally Activated Delayed Fluorescence and Delayed Fluorescence by Triplet-Triplet Annihilation. J. Am. Chem. Soc. 2020, 142, 6588–6599. [Google Scholar] [CrossRef]
- Kahan, R.J.; Crossley, D.L.; Cid, J.; Radcliffe, J.E.; Ingleson, M.J. Synthesis, Characterization, and Functionalization of 1-Boraphenalenes. Angew. Chem. Int. Ed. 2018, 57, 8084–8088. [Google Scholar] [CrossRef]
- Yuan, K.; Kahan, R.J.; Si, C.; Williams, A.; Kirschner, S.; Uzelac, M.; Zysman-Colman, E.; Ingleson, M.J. The synthesis of brominated-boron-doped PAHs by alkyne 1,1-bromoboration: Mechanistic and functionalisation studies. Chem. Sci. 2020, 11, 3258–3267. [Google Scholar] [CrossRef] [Green Version]
- Curran, D.P.; Solovyev, A.; Makhlouf Brahmi, M.; Fensterbank, L.; Malacria, M.; Lacôte, E. Synthesis and Reactions of N-Heterocyclic Carbene Boranes. Angew. Chem. Int. Ed. 2011, 50, 10294–10317. [Google Scholar] [CrossRef]
- Farrell, J.M.; Stephan, D.W. Planar N-Heterocyclic Carbene Diarylborenium Ions: Synthesis by Cationic Borylation and Reactivity with Lewis Bases. Angew. Chem. Int. Ed. 2015, 54, 5214–5217. [Google Scholar] [CrossRef]
- Schnitzlein, M.; Zhu, C.; Shoyama, K.; Würthner, F. π-Extended Pleiadienes by [5+2] Annulation of 1-Boraphenalenes and ortho-Dihaloarenes. Chem. Eur. J. 2022, 28, e202202053. [Google Scholar] [CrossRef]
- Farrell, J.M.; Schmidt, D.; Grande, V.; Wurthner, F. Synthesis of a Doubly Boron-Doped Perylene through NHC-Borenium Hydroboration/C-H Borylation/Dehydrogenation. Angew. Chem. Int. Ed. 2017, 56, 11846–11850. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.M.; Mutzel, C.; Bialas, D.; Rudolf, M.; Menekse, K.; Krause, A.M.; Stolte, M.; Wurthner, F. Tunable Low-LUMO Boron-Doped Polycyclic Aromatic Hydrocarbons by General One-Pot C-H Borylations. J. Am. Chem. Soc. 2019, 141, 9096–9104. [Google Scholar] [CrossRef] [PubMed]
- Schnitzlein, M.; Mützel, C.; Shoyama, K.; Farrell, J.M.; Würthner, F. PAHs Containing both Heptagon and Pentagon: Corannulene Extension by [5+2] Annulation. Eur. J. Org. Chem. 2021, 2022, e202101273. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, X.; Bai, Y.; Wang, X.; Ru, C.; Chen, W.; Lv, X.; Pan, X.; Wu, J. Diradical Anion of Potassium Aggregate: Reduction of Dimer Boroxide Complex. Inorg. Chem. 2018, 57, 13544–13551. [Google Scholar] [CrossRef]
- Albrecht, K.; Kaiser, V.; Boese, R.; Adams, J.; Kaufmann, D.E. Synthesis and properties of fluorescent organoboranes: Triarylmethane-type dyes. J. Chem. Soc. Perkin Trans. 2 2000, 10, 2153–2157. [Google Scholar] [CrossRef]
- Wesela-Bauman, G.; Ciecwierz, P.; Durka, K.; Lulinski, S.; Serwatowski, J.; Woźniak, K. Heteroleptic (2-fluoro-3-pyridyl)arylborinic 8-oxyquinolinates for the potential application in organic light-emitting devices. Inorg. Chem. 2013, 52, 10846–10859. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Durka, K.; Kasprzak, A.; Klis, T.; Monkman, A.P.; Piszcz, M.; Woźniak, K. Excited-state photodynamics of pyrene-containing boronated dyes. Dyes Pigm. 2022, 197, 109934. [Google Scholar] [CrossRef]
- Wan, W.-M.; Cheng, F.; Jäkle, F. A Borinic Acid Polymer with Fluoride Ion- and Thermo-responsive Properties that are Tunable over a Wide Temperature Range. Angew. Chem. Int. Ed. 2014, 53, 8934–8938. [Google Scholar] [CrossRef] [PubMed]
- Gatin-Fraudet, B.; Ottenwelter, R.; Le Saux, T.; Norsikian, S.; Pucher, M.; Lombès, T.; Baron, A.; Durand, P.; Doisneau, G.; Bourdreux, Y.; et al. Evaluation of borinic acids as new, fast hydrogen peroxide–responsive triggers. Proc. Natl. Acad. Sci. USA 2021, 118, e2107503118. [Google Scholar] [CrossRef]
- Wesela-Bauman, G.; Jastrzębski, L.; Kurach, P.; Luliński, S.; Serwatowski, J.; Woźniak, K. Synthesis of functionalized diarylborinic 8-oxyquinolates via bimetallic boron–lithium intermediates. J. Organomet. Chem. 2012, 711, 1–9. [Google Scholar] [CrossRef]
- Luliński, S.; Smętek, J.; Durka, K.; Serwatowski, J. Tandem Synthesis of 9,10-Dihydro-9,10-diboraanthracenes via Elusiveortho-Lithiated Phenylboronates. Eur. J. Org. Chem. 2013, 2013, 8315–8322. [Google Scholar] [CrossRef]
- Brend’amour, S.; Gilmer, J.; Bolte, M.; Lerner, H.W.; Wagner, M. C-Halogenated 9,10-Diboraanthracenes: How the Halogen Load and Distribution Influences Key Optoelectronic Properties. Chem. Eur. J. 2018, 24, 16910–16918. [Google Scholar] [CrossRef]
- Kaehler, T.; Bolte, M.; Lerner, H.W.; Wagner, M. Introducing Perylene as a New Member to the Azaborine Family. Angew. Chem. Int. Ed. 2019, 58, 11379–11384. [Google Scholar] [CrossRef]
- Körner, C.; Starkov, P.; Sheppard, T.D. An Alternative Approach to Aldol Reactions: Gold-Catalyzed Formation of Boron Enolates from Alkynes. J. Am. Chem. Soc. 2010, 132, 5968–5969. [Google Scholar] [CrossRef] [Green Version]
- Ouadoudi, O.; Kaehler, T.; Bolte, M.; Lerner, H.-W.; Wagner, M. One tool to bring them all: Au-catalyzed synthesis of B,O- and B,N-doped PAHs from boronic and borinic acids. Chem. Sci. 2021, 12, 5898–5909. [Google Scholar] [CrossRef] [PubMed]
- Brosge, F.; Lorenz, T.; Helten, H.; Bolm, C. BN- and BO-Doped Inorganic-Organic Hybrid Polymers with Sulfoximine Core Units. Chem. Eur. J. 2019, 25, 12708–12711. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, T.; Crumbach, M.; Eckert, T.; Lik, A.; Helten, H. Poly(p-phenylene iminoborane): A Boron-Nitrogen Analogue of Poly(p-phenylene vinylene). Angew. Chem. Int. Ed. 2017, 56, 2780–2784. [Google Scholar] [CrossRef] [PubMed]
- Budanow, A.; von Grotthuss, E.; Bolte, M.; Wagner, M.; Lerner, H.-W. 10,9-Oxaboraphenanthrenes as luminescent fluorophores. Tetrahedron 2016, 72, 1477–1484. [Google Scholar] [CrossRef]
- Xu, S.; Haeffner, F.; Li, B.; Zakharov, L.N.; Liu, S.Y. Monobenzofused 1,4-azaborines: Synthesis, characterization, and discovery of a unique coordination mode. Angew. Chem. Int. Ed. 2014, 53, 6795–6799. [Google Scholar] [CrossRef] [PubMed]
- van Veen, R.; Bickelhaupt, F. Synthesis of the 9-mesityl-10-phenyl-9-boraanthracene anion. J. Organomet. Chem. 1972, 43, 241–248. [Google Scholar] [CrossRef]
- Van Veen, R.; Bickelhaupt, F. Synthesis of 5-and 6-membered carbon-boron heterocycles by pyrolysis of pyridine-arylboranes. J. Organomet. Chem. 1973, 47, 33–38. [Google Scholar] [CrossRef]
- You, C.; Sakai, M.; Daniliuc, C.G.; Bergander, K.; Yamaguchi, S.; Studer, A. Regio- and Stereoselective 1,2-Carboboration of Ynamides with Aryldichloroboranes. Angew. Chem. Int. Ed. 2021, 60, 21697–21701. [Google Scholar] [CrossRef]
- Baraniak, M.K.; Lalancette, R.A.; Jakle, F. Electron-Deficient Borinic Acid Polymers: Synthesis, Supramolecular Assembly, and Examination as Catalysts in Amide Bond Formation. Chem. Eur. J. 2019, 25, 13799–13810. [Google Scholar] [CrossRef]
- Cheng, F.; Jakle, F. RAFT polymerization of luminescent boron quinolate monomers. Chem. Commun. 2010, 46, 3717–3719. [Google Scholar] [CrossRef]
- Qin, Y.; Kiburu, I.; Shah, S.; Jakle, F. Luminescence tuning of organoboron quinolates through substituent variation at the 5-position of the quinolato moiety. Org. Lett. 2006, 8, 5227–5230. [Google Scholar] [CrossRef] [PubMed]
- Sadu, V.S.; Bin, H.R.; Lee, D.M.; Lee, K.I. One-pot synthesis of four-coordinate boron(III) complexes by the ligand-promoted organic group migration between boronic acids. Sci. Rep. 2017, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Zu, W.; Day, C.; Wei, L.; Jia, X.; Xu, L. Dual aminoquinolate diarylboron and nickel catalysed metallaphotoredox platform for carbon-oxygen bond construction. Chem. Commun. 2020, 56, 8273–8276. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Kirschner, S.; Fengel, M.K.; Bolte, M.; Lerner, H.-W.; Wagner, M. Simultaneous expansion of 9,10 boron-doped anthracene in longitudinal and lateral directions. Dalton Trans. 2019, 48, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Seven, Ö.; Qu, Z.-W.; Zhu, H.; Bolte, M.; Lerner, H.-W.; Holthausen, M.C.; Wagner, M. Synthesis, Coupling, and Condensation Reactions of 1,2-Diborylated Benzenes: An Experimental and Quantum-Chemical Study. Chem. Eur. J. 2012, 18, 11284–11295. [Google Scholar] [CrossRef]
- Januszewski, E.; Lorbach, A.; Grewal, R.; Bolte, M.; Bats, J.W.; Lerner, H.W.; Wagner, M. Unsymmetrically substituted 9,10-dihydro-9,10-diboraanthracenes as versatile building blocks for boron-doped pi-conjugated systems. Chem. Eur. J. 2011, 17, 12696–12705. [Google Scholar] [CrossRef] [PubMed]
- Chinnapattu, M.; Sathiyanarayanan, K.I.; Iyer, P.S. Synthesis of benzofused 1,4-azaborinols via [4 + 2] annulation strategy and its application in indole synthesis. RSC Adv. 2015, 5, 37716–37720. [Google Scholar] [CrossRef]
- Shimomura, N.; Egawa, Y.; Miki, R.; Fujihara, T.; Ishimaru, Y.; Seki, T. A red fluorophore comprising a borinate-containing xanthene analogue as a polyol sensor. Org. Biomol. Chem. 2016, 14, 10031–10036. [Google Scholar] [CrossRef] [Green Version]
- Lyu, H.; Kevlishvili, I.; Yu, X.; Liu, P.; Dong, G. Boron insertion into alkyl ether bonds via zinc/nickel tandem catalysis. Science 2021, 372, 175–182. [Google Scholar] [CrossRef]
- Matsuda, T.; Sato, S. Synthesis of dibenzoheteropines of group 13-16 elements via ring-closing metathesis. J. Org. Chem. 2013, 78, 3329–3335. [Google Scholar] [CrossRef]
- Tokoro, Y.; Nagai, A.; Kokado, K.; Chujo, Y. Synthesis of Organoboron Quinoline-8-thiolate and Quinoline-8-selenolate Complexes and Their Incorporation into the π-Conjugated Polymer Main-Chain. Macromolecules 2009, 42, 2988–2993. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, Q.D.; Bai, D.R.; Jia, W.L.; Tao, Y.; Wang, S. Organoboron compounds with an 8-hydroxyquinolato chelate and its derivatives: Substituent effects on structures and luminescence. Inorg. Chem. 2005, 44, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Kappaun, S.; Rentenberger, S.; Pogantsch, A.; Zojer, E.; Mereiter, K.; Trimmel, G.; Saf, R.; Möller, K.C.; Stelzer, F.; Slugovc, C. Organoboron Quinolinolates with Extended Conjugated Chromophores: Synthesis, Structure, and Electronic and Electroluminescent Properties. Chem. Mater. 2006, 18, 3539–3547. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, S. Diboron and triboron compounds based on linear and star-shaped conjugated ligands with 8-hydroxyquinolate functionality: Impact of intermolecular interaction and boron coordination on luminescence. J. Org. Chem. 2006, 71, 6485–6496. [Google Scholar] [CrossRef]
- Kim, N.G.; Shin, C.H.; Lee, M.H.; Do, Y. Four-coordinate boron compounds derived from 2-(2-pyridyl)phenol ligand as novel hole-blocking materials for phosphorescent OLEDs. J. Organomet. Chem. 2009, 694, 1922–1928. [Google Scholar] [CrossRef]
- Yan, Y.; Feng, P.; Zheng, Q.Z.; Liang, Y.F.; Lu, J.F.; Cui, Y.; Jiao, N. PdCl2 and N-hydroxyphthalimide co-catalyzed C(sp2)-H hydroxylation by dioxygen activation. Angew. Chem. Int. Ed. 2013, 52, 5827–5831. [Google Scholar] [CrossRef]
- Tokoro, Y.; Nagai, A.; Chujo, Y. Synthesis of π-Conjugated Polymers Containing Organoboron Benzo[h]quinolate in the Main Chain. Macromolecules 2010, 43, 6229–6233. [Google Scholar] [CrossRef]
- Qiu, F.; Zhang, N.; Tang, R.; Zhou, M.; Wang, Y.; Wei, W.; Bi, S.; Han, S.; Zhang, F. Asymmetric Boron-Cored Aggregation-Induced Emission Luminogen with Multiple Functions Synthesized through Stepwise Conversion from a Symmetric Ligand. J. Org. Chem. 2018, 83, 12977–12984. [Google Scholar] [CrossRef]
- Pais, V.F.; Ramirez-Lopez, P.; Romero-Arenas, A.; Collado, D.; Najera, F.; Perez-Inestrosa, E.; Fernandez, R.; Lassaletta, J.M.; Ros, A.; Pischel, U. Red-Emitting Tetracoordinate Organoboron Chelates: Synthesis, Photophysical Properties, and Fluorescence Microscopy. J. Org. Chem. 2016, 81, 9605–9611. [Google Scholar] [CrossRef] [Green Version]
- Massue, J.; Retailleau, P.; Ulrich, G.; Ziessel, R. Synthesis of luminescent BPh2-coordinated 2-(2′-hydroxyphenyl)benzoxazole (HBO). N. J. Chem. 2013, 37, 1224–1230. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Jiao, C.; Ye, K.; Zhang, H.; Zhang, J.; Wang, Y. 2-(2-Hydroxyphenyl)benzimidazole-based four-coordinate boron-containing materials with highly efficient deep-blue photoluminescence and electroluminescence. Inorg. Chem. 2015, 54, 2652–2659. [Google Scholar] [CrossRef]
- Yagishita, F.; Kinouchi, T.; Hoshi, K.; Tezuka, Y.; Jibu, Y.; Karatsu, T.; Uemura, N.; Yoshida, Y.; Mino, T.; Sakamoto, M.; et al. Highly efficient blue emission from boron complexes of 1-(o-hydroxyphenyl)imidazo[1,5-a]pyridine. Tetrahedron 2018, 74, 3728–3733. [Google Scholar] [CrossRef]
- Li, D.; Wang, K.; Huang, S.; Qu, S.; Liu, X.; Zhu, Q.; Zhang, H.; Wang, Y. Brightly fluorescent red organic solids bearing boron-bridged π–conjugated skeletons. J. Mater. Chem. 2011, 21, 15298–15304. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Wang, C.; Huang, S.; Guo, J.; Wang, Y. Construction of full-color-tunable and strongly emissive materials by functionalizing a boron-chelate four-ring-fused π-conjugated core. J. Mater. Chem. 2012, 22, 4319–4328. [Google Scholar] [CrossRef]
- Benelhadj, K.; Massue, J.; Ulrich, G. 2,4 and 2,5-bis(benzooxazol-2′-yl)hydroquinone (DHBO) and their borate complexes: Synthesis and optical properties. New J. Chem. 2016, 40, 5877–5884. [Google Scholar] [CrossRef]
- Frath, D.; Azizi, S.; Ulrich, G.; Retailleau, P.; Ziessel, R. Facile synthesis of highly fluorescent Boranil complexes. Org. Lett. 2011, 13, 3414–3417. [Google Scholar] [CrossRef] [PubMed]
- Dobkowski, J.; Wnuk, P.; Buczynska, J.; Pszona, M.; Orzanowska, G.; Frath, D.; Ulrich, G.; Massue, J.; Mosquera-Vazquez, S.; Vauthey, E.; et al. Substituent and solvent effects on the excited state deactivation channels in anils and boranils. Chem. Eur. J. 2015, 21, 1312–1327. [Google Scholar] [CrossRef]
- Frath, D.; Benelhadj, K.; Munch, M.; Massue, J.; Ulrich, G. Polyanils and Polyboranils: Synthesis, Optical Properties, and Aggregation-Induced Emission. J. Org. Chem. 2016, 81, 9658–9668. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Z.; Liu, Q.; Wang, X.; Yan, H.; Gong, S.; Liu, Z.; He, W. High solid-state luminescence in propeller-shaped AIE-active pyridine-ketoiminate-boron complexes. Org. Biomol. Chem. 2015, 13, 5775–5782. [Google Scholar] [CrossRef]
- Huang, J.; Wang, Y.; Van Hecke, K.; Pereshivko, O.P.; Peshkov, V.A. Studies on Functionalization of N,O-Chelated Isoquinoline-Enol Boron Complexes. Eur. J. Org. Chem. 2019, 2019, 2490–2497. [Google Scholar] [CrossRef]
- Zhang, H.; Hong, X.; Ba, X.; Yu, B.; Wen, X.; Wang, S.; Wang, X.; Liu, L.; Xiao, J. Synthesis, Physical Properties, and Photocurrent Behavior of Strongly Emissive Boron-Chelate Heterochrysene Derivatives. Asian J. Org. Chem. 2014, 3, 1168–1172. [Google Scholar] [CrossRef]
- Kubota, Y.; Kasatani, K.; Niwa, T.; Sato, H.; Funabiki, K.; Matsui, M. Synthesis and Fluorescence Properties of Pyrimidine-Based Diboron Complexes with Donor-pi-Acceptor Structures. Chem. Eur. J. 2016, 22, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Kasatani, K.; Takai, H.; Funabiki, K.; Matsui, M. Strategy to enhance solid-state fluorescence and aggregation-induced emission enhancement effect in pyrimidine boron complexes. Dalton Trans. 2015, 44, 3326–3341. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Sakuma, Y.; Funabiki, K.; Matsui, M. Solvatochromic fluorescence properties of pyrazine-boron complex bearing a beta-iminoenolate ligand. J. Phys. Chem. A 2014, 118, 8717–8729. [Google Scholar] [CrossRef]
- Kubota, Y.; Hara, H.; Tanaka, S.; Funabiki, K.; Matsui, M. Synthesis and fluorescence properties of novel pyrazine-boron complexes bearing a beta-iminoketone ligand. Org. Lett. 2011, 13, 6544–6547. [Google Scholar] [CrossRef]
- Kubota, Y.; Tanaka, S.; Funabiki, K.; Matsui, M. Synthesis and fluorescence properties of thiazole-boron complexes bearing a beta-ketoiminate ligand. Org. Lett. 2012, 14, 4682–4685. [Google Scholar] [CrossRef]
- Gong, S.; Liu, Q.; Wang, X.; Xia, B.; Liu, Z.; He, W. AIE-active organoboron complexes with highly efficient solid-state luminescence and their application as gas sensitive materials. Dalton Trans. 2015, 44, 14063–14070. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xu, D.; Gao, H.; Han, A.; Yang, Y.; Zhang, C.; Liu, X.; Zhao, F. Effects of cyano groups on the properties of thiazole-based β-ketoiminate boron complexes: Aggregation-induced emission and mechanofluorochromism. RSC Adv. 2016, 6, 69560–69568. [Google Scholar] [CrossRef]
- Kubota, Y.; Niwa, T.; Jin, J.; Funabiki, K.; Matsui, M. Synthesis, Absorption, and Electrochemical Properties of Quinoid-Type Bisboron Complexes with Highly Symmetrical Structures. Org. Lett. 2015, 17, 3174–3177. [Google Scholar] [CrossRef]
- Mikysek, T.; Kvapilová, H.; Doušová, H.; Josefík, F.; Šimůnek, P.; Růžičková, Z.; Ludvík, J. Synthesis, electrochemical, structural and theoretical study of new derivatives of OBN and OBO heterocycles. Inorg. Chim. Acta 2017, 455, 465–472. [Google Scholar] [CrossRef]
- Kunick, C.; Tolle, N.; Dunkel, U.; Oehninger, L.; Ott, I.; Preu, L.; Haase, T.; Behrends, S.; Jones, P.; Totzke, F.; et al. Synthesis and Structure of Fluorescent Chelate Boron Complexes of 4-Anilinomethylidene-1-benzazepine-2,5-dione Ligands. Synthesis 2011, 2011, 2848–2858. [Google Scholar] [CrossRef]
- Doušová, H.; Šimůnek, P.; Almonasy, N.; Růžičková, Z. Synthesis, NMR, X-ray and UV/Vis characterization and preliminary luminescence study of some boron β-iminoenolates having 6-aminocoumarin moiety. J. Organomet. Chem. 2016, 802, 60–71. [Google Scholar] [CrossRef]
- Pešková, M.; Šimůnek, P.; Bertolasi, V.; Macháček, V.; Lyčka, A. Novel 5-(4-Substituted-phenyldiazenyl)-1,3,2λ4-oxazaborines and Their Rearrangement to 1,2,4,3λ4-Triazaborines. Organometallics 2006, 25, 2025–2030. [Google Scholar] [CrossRef]
- González, A.; Granell, J.; Pinlella, J.F.; Alvarez-Larena, A. Boron complexes of L-cysteine. Tetrahedron 1998, 54, 13313–13322. [Google Scholar] [CrossRef]
- Gazis, T.A.; Dasgupta, A.; Hill, M.S.; Rawson, J.M.; Wirth, T.; Melen, R.L. Reactions of hydrazones and hydrazides with Lewis acidic boranes. Dalton Trans. 2019, 48, 12391–12395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.I.; Figueira, C.A.; Gomes, C.S.B.; Suresh, D.; Ferreira, B.; Di Paolo, R.E.; Pereira, D.S.; Dias, F.B.; Calhorda, M.J.; Morgado, J.; et al. Boron complexes of aromatic 5-substituted iminopyrrolyl ligands: Synthesis, structure, and luminescence properties. Dalton Trans. 2019, 48, 13337–13352. [Google Scholar] [CrossRef]
- Krishnamoorthy, P.; Ferreira, B.; Gomes, C.S.B.; Vila-Viçosa, D.; Charas, A.; Morgado, J.; Calhorda, M.J.; Maçanita, A.L.; Gomes, P.T. Violet-blue emitting 2-(N-alkylimino)pyrrolyl organoboranes: Synthesis, structure and luminescent properties. Dyes Pigm. 2017, 140, 520–532. [Google Scholar] [CrossRef]
- Suresh, D.; Lopes, P.S.; Ferreira, B.; Figueira, C.A.; Gomes, C.S.; Gomes, P.T.; Di Paolo, R.E.; Macanita, A.L.; Duarte, M.T.; Charas, A.; et al. Tunable fluorophores based on 2-(N-arylimino)pyrrolyl chelates of diphenylboron: Synthesis, structure, photophysical characterization, and application in OLEDs. Chem. Eur. J. 2014, 20, 4126–4140. [Google Scholar] [CrossRef]
- Suresh, D.; Gomes, C.S.; Lopes, P.S.; Figueira, C.A.; Ferreira, B.; Gomes, P.T.; Di Paolo, R.E.; Macanita, A.L.; Duarte, M.T.; Charas, A.; et al. Luminescent Di- and Trinuclear Boron Complexes Based on Aromatic Iminopyrrolyl Spacer Ligands: Synthesis, Characterization, and Application in OLEDs. Chem. Eur. J. 2015, 21, 9133–9149. [Google Scholar] [CrossRef] [PubMed]
- Suresh, D.; Gomes, C.S.; Gomes, P.T.; Di Paolo, R.E.; Macanita, A.L.; Calhorda, M.J.; Charas, A.; Morgado, J.; Duarte, M.T. Syntheses and photophysical properties of new iminopyrrolyl boron complexes and their application in efficient single-layer non-doped OLEDs prepared by spin coating. Dalton Trans. 2012, 41, 8502–8505. [Google Scholar] [CrossRef]
- Suresh, D.; Ferreira, B.; Lopes, P.S.; Gomes, C.S.; Krishnamoorthy, P.; Charas, A.; Vila-Vicosa, D.; Morgado, J.; Calhorda, M.J.; Macanita, A.L.; et al. Boron complexes of aromatic ring fused iminopyrrolyl ligands: Synthesis, structure, and luminescence properties. Dalton Trans. 2016, 45, 15603–15620. [Google Scholar] [CrossRef] [PubMed]
- Gai, L.; Xu, J.; Wu, Y.; Lu, H.; Shen, Z. Synthesis and spectroscopic properties of novel N–N linked bis-(diphenylboron) complexes. N. J. Chem. 2016, 40, 5752–5757. [Google Scholar] [CrossRef]
- Maeda, C.; Nagahata, K.; Ema, T. Carbazole-based BODIPYs with ethynyl substituents at the boron center: Solid-state excimer fluorescence in the VIS/NIR region. Org. Biomol. Chem. 2017, 15, 7783–7788. [Google Scholar] [CrossRef]
- Mas-Montoya, M.; Usea, L.; Espinosa Ferao, A.; Montenegro, M.F.; Ramirez de Arellano, C.; Tarraga, A.; Rodriguez-Lopez, J.N.; Curiel, D. Single Heteroatom Fine-Tuning of the Emissive Properties in Organoboron Complexes with 7-(Azaheteroaryl)indole Systems. J. Org. Chem. 2016, 81, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Qiu, F.; Zhang, F.; Mai, Y.; Wang, Y.; Fu, S.; Tang, R.; Zhuang, X.; Feng, X. A dual-boron-cored luminogen capable of sensing and imaging. Chem. Commun. 2015, 51, 5298–5301. [Google Scholar] [CrossRef] [Green Version]
- Liddle, B.J.; Silva, R.M.; Morin, T.J.; Macedo, F.P.; Shukla, R.; Lindeman, S.V.; Gardinier, J.R. BORAZANs: Tunable fluorophores based on 2-(pyrazolyl)aniline chelates of diphenylboron. J. Org. Chem. 2007, 72, 5637–5646. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Murayama, A.; Haruyama, T.; Iino, T.; Mori, S.; Furuta, H.; Kobayashi, N. Benzo[c,d]indole-Containing Aza-BODIPY Dyes: Asymmetrization-Induced Solid-State Emission and Aggregation-Induced Emission Enhancement as New Properties of a Well-Known Chromophore. Chem. Eur. J. 2015, 21, 12996–13003. [Google Scholar] [CrossRef]
- Crandall, L.A.; Rhoda, H.M.; Nemykin, V.N.; Ziegler, C.J. Boron templated synthesis of a BODIPY analogue from a phthalocyanine precursor. N. J. Chem. 2016, 40, 5675–5678. [Google Scholar] [CrossRef]
- Dijkstra, P.; Angelone, D.; Talnishnikh, E.; Wortche, H.J.; Otten, E.; Browne, W.R. Pyridyl-1,2,4-triazole diphenyl boron complexes as efficient tuneable blue emitters. Dalton Trans. 2014, 43, 17740–17745. [Google Scholar] [CrossRef]
- Chen, H.Y.; Chi, Y.; Liu, C.S.; Yu, J.K.; Cheng, Y.M.; Chen, K.S.; Chou, P.T.; Peng, S.M.; Lee, G.H.; Carty, A.J.; et al. Rational Color Tuning and Luminescent Properties of Functionalized Boron-Containing 2-Pyridyl Pyrrolide Complexes. Adv. Funct. Mater. 2005, 15, 567–574. [Google Scholar] [CrossRef]
- Chen, T.-R.; Chien, R.-H.; Jan, M.-S.; Yeh, A.; Chen, J.-D. Syntheses and structures of new luminescent B(III) complexes: BPh2(2-(2-pyridyl)naphtho[b]imidazole) and BF2(2-(2-pyridyl)naphtho[b]imidazole). J. Organomet. Chem. 2006, 691, 799–804. [Google Scholar] [CrossRef]
- Baker, S.J.; Zhang, Y.K.; Akama, T.; Lau, A.; Zhou, H.; Hernandez, V.; Mao, W.; Alley, M.R.; Sanders, V.; Plattner, J.J. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J. Med. Chem. 2006, 49, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-G.; Ranganathan, R.; Lin, J.-A.; Huang, C.-Y.; Ho, M.-L.; Chi, Y.; Chou, P.-T. Ratiometric Tuning of Luminescence: Interplay between the Locally Excited and Interligand Charge-Transfer States in Pyrazolate-Based Boron Compounds. J. Phys. Chem. C 2019, 123, 4022–4028. [Google Scholar] [CrossRef]
- Ho, M.L.; Hwang, F.M.; Chen, P.N.; Hu, Y.H.; Cheng, Y.M.; Chen, K.S.; Lee, G.H.; Chi, Y.; Chou, P.T. Design and synthesis of iridium(III) azacrown complex: Application as a highly sensitive metal cation phosphorescence sensor. Org. Biomol. Chem. 2006, 4, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagata, T.; Kuwabara, J.; Kanbara, T. Optical properties of highly planar diketopyrrolopyrrole derivatives fixed by coordinate bonds. Tetrahedron 2014, 70, 1451–1457. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.D.; Mudadu, M.S.; Thummel, R.; Tao, Y.; Wang, S. From Blue to Red: Syntheses, Structures, Electronic and Electroluminescent Properties of Tunable Luminescent N,N Chelate Boron Complexes. Adv. Funct. Mater. 2005, 15, 143–154. [Google Scholar] [CrossRef]
- Curiel, D.; Mas-Montoya, M.; Usea, L.; Espinosa, A.; Orenes, R.A.; Molina, P. Indolocarbazole-based ligands for ladder-type four-coordinate boron complexes. Org. Lett. 2012, 14, 3360–3363. [Google Scholar] [CrossRef]
- Mula, S.; Leclerc, N.; Leveque, P.; Retailleau, P.; Ulrich, G. Synthesis of Indolo[3,2- b]carbazole-Based Boron Complexes with Tunable Photophysical and Electrochemical Properties. J. Org. Chem. 2018, 83, 14406–14418. [Google Scholar] [CrossRef]
- Nakamura, T.; Furukawa, S.; Nakamura, E. Benzodipyrrole-based Donor-Acceptor-type Boron Complexes as Tunable Near-infrared-Absorbing Materials. Chem. Asian J. 2016, 11, 2016–2020. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, H.; Wang, S.; Li, Z.; Shen, Z. Asymmetric boron-complexes containing keto-isoindolinyl and pyridyl groups: Solvatochromic fluorescence, efficient solid-state emission and DFT calculations. J. Mater. Chem. C 2015, 3, 12281–12289. [Google Scholar] [CrossRef]
- Liu, F.; Ding, Z.; Liu, J.; Wang, L. An organoboron compound with a wide absorption spectrum for solar cell applications. Chem. Commun. 2017, 53, 12213–12216. [Google Scholar] [CrossRef]
- Wang, T.; Dou, C.; Liu, J.; Wang, L. Effects of the Substituents of Boron Atoms on Conjugated Polymers Containing B<--N Units. Chem. Eur. J. 2018, 24, 13043–13048. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, J.; Wang, L. Effect of fluorine substitution in organoboron electron acceptors for photovoltaic application. Org. Chem. Front. 2019, 6, 1996–2003. [Google Scholar] [CrossRef]
- Qiu, F.; Zhang, F.; Tang, R.; Fu, Y.; Wang, X.; Han, S.; Zhuang, X.; Feng, X. Triple Boron-Cored Chromophores Bearing Discotic 5,11,17-Triazatrinaphthylene-Based Ligands. Org. Lett. 2016, 18, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Tokoro, Y.; Nagai, A.; Chujo, Y. Synthesis of highly luminescent organoboron polymers connected by bifunctional 8-aminoquinolate linkers. J. Polym. Sci. Pol. Chem. 2010, 48, 3693–3701. [Google Scholar] [CrossRef]
- Nagata, Y.; Chujo, Y. Main-Chain-TypeN,N′-Chelate Organoboron Aminoquinolate Polymers: Synthesis, Luminescence, and Energy Transfer Behavior. Macromolecules 2008, 41, 3488–3492. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Liu, Q.; Li, Z.; Yan, H.; Ji, C.; Duan, J.; Liu, Z. Aggregation-induced emission (AIE) of pyridyl-enamido-based organoboron luminophores. Chem. Commun. 2015, 51, 784–787. [Google Scholar] [CrossRef]
- Cheng, X.; Li, D.; Zhang, Z.; Zhang, H.; Wang, Y. Organoboron compounds with morphology-dependent NIR emissions and dual-channel fluorescent ON/OFF switching. Org. Lett. 2014, 16, 880–883. [Google Scholar] [CrossRef]
- Nagai, A.; Kokado, K.; Nagata, Y.; Arita, M.; Chujo, Y. Highly intense fluorescent diarylboron diketonate. J. Org. Chem. 2008, 73, 8605–8607. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Cheng, X.; Ye, K.; Li, F.; Wang, Y.; Zhang, H. Red emissive diarylboron diketonate crystals: Aggregation-induced color change and amplified spontaneous emission. J. Mater. Chem. C 2015, 3, 499–505. [Google Scholar] [CrossRef]
- Hu, J.; He, Z.; Wang, Z.; Li, X.; You, J.; Gao, G. A simple approach to aggregation-induced emission in difluoroboron dibenzoylmethane derivatives. Tetrahedron Lett. 2013, 54, 4167–4170. [Google Scholar] [CrossRef]
- Terashima, Y.; Takayama, M.; Isozaki, K.; Maeda, H. Ion-based materials of boron-modified dipyrrolyldiketones as anion receptors. Chem. Commun. 2013, 49, 2506–2508. [Google Scholar] [CrossRef]
- Poon, C.T.; Lam, W.H.; Yam, V.W. Synthesis, photochromic, and computational studies of dithienylethene-containing beta-diketonate derivatives and their near-infrared photochromic behavior upon coordination of a boron(III) center. Chem. Eur. J. 2013, 19, 3467–3476. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.T.; Lam, W.H.; Wong, H.L.; Yam, V.W. A versatile photochromic dithienylethene-containing beta-diketonate ligand: Near-infrared photochromic behavior and photoswitchable luminescence properties upon incorporation of a boron(III) center. J. Am. Chem. Soc. 2010, 132, 13992–13993. [Google Scholar] [CrossRef] [PubMed]
- Kersten, L.; Roesner, S.; Hilt, G. Synthesis and characterization of polycarbonyl compounds via their BF2-adducts. Org. Lett. 2010, 12, 4920–4923. [Google Scholar] [CrossRef] [PubMed]
- Hugelshofer, C.L.; Palani, V.; Sarpong, R. Oxazaborinines from Vinylogous N-Allylic Amides: Reactivities of Underexplored Heterocyclic Building Blocks. Org. Lett. 2018, 20, 2649–2653. [Google Scholar] [CrossRef] [PubMed]
- Koeritz, M.T.; Banovetz, H.K.; Prell, S.A.; Stanley, L.M. Synthesis of oxaboranes via nickel-catalyzed dearylative cyclocondensation. Chem. Sci. 2022, 13, 7790–7795. [Google Scholar] [CrossRef]
- Gazis, T.A.; Mohajeri Thaker, B.A.J.; Willcox, D.; Ould, D.M.C.; Wenz, J.; Rawson, J.M.; Hill, M.S.; Wirth, T.; Melen, R.L. 1,3-Carboboration of iodonium ylides. Chem. Commun. 2020, 56, 3345–3348. [Google Scholar] [CrossRef] [Green Version]
- Josefík, F.; Svobodová, M.; Bertolasi, V.; Šimůnek, P.; Macháček, V.; Almonasy, N.; Černošková, E. A new bicyclic oxazaborines with a bridged nitrogen atom, their thermic rearrangement and fluorescence properties. J. Organomet. Chem. 2012, 699, 75–81. [Google Scholar] [CrossRef]
- Josefík, F.; Mikysek, T.; Svobodová, M.; Šimůnek, P.; Kvapilová, H.; Ludvík, J. New Triazaborine Chromophores: Their Synthesis via Oxazaborines and Electrochemical and DFT Study of Their Fundamental Properties. Organometallics 2014, 33, 4931–4939. [Google Scholar] [CrossRef]
- Svobodová, M.; Bárta, J.; Šimůnek, P.; Bertolasi, V.; Macháček, V. Straightforward access to oxazaborines, diazaborinones and triazaborines by reactions of β-enaminoamides with 4-methylbenzenediazonium tetraphenylborate. J. Organomet. Chem. 2009, 694, 63–71. [Google Scholar] [CrossRef]
- Svobodová, M.; Šimůnek, P.; Macháček, V.; Štruncová, L.; Růžička, A. Four-coordinate organoboron compounds from β-enaminonitriles and diazonium salts. Tetrahedron 2012, 68, 2052–2060. [Google Scholar] [CrossRef]
- Raunio, J.; Mannoja, J.; Nguyen, T.; Ahmad, N.; Kemppainen, N.M.; Franzén, R.G.; Kandhavelu, M.; Candeias, N.R. Base catalysed N-functionalisation of boroxazolidones. RSC Adv. 2017, 7, 20620–20627. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Cao, X.; Zhang, X.-X.; Ou, Y.; Au, C.-T.; Yin, S.-F.; Qiu, R. Iodine-Catalyzed Synthesis of N,N′-Chelate Organoboron Aminoquinolate. J. Org. Chem. 2020, 85, 12430–12443. [Google Scholar] [CrossRef]
- Zhang, J.; Park, S.; Chang, S. Selective C−O Bond Cleavage of Sugars with Hydrosilanes Catalyzed by Piers’ Borane Generated In Situ. Angew. Chem. Int. Ed. 2017, 56, 13757–13761. [Google Scholar] [CrossRef]
- Pla, D.; Sadek, O.; Cadet, S.; Mestre-Voegtlé, B.; Gras, E. Naphthylaminoborane: From structural switches to frustrated Lewis pair reactivity. Dalton Trans. 2015, 44, 18340–18346. [Google Scholar] [CrossRef]
- Chen, C.H.; Gabbai, F.P. Exploiting the Strong Hydrogen Bond Donor Properties of a Borinic Acid Functionality for Fluoride Anion Recognition. Angew. Chem. Int. Ed. 2018, 57, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Wehmschulte, R.J.; Diaz, A.A.; Khan, M.A. Unsymmetrical 9-Borafluorenes via Low-Temperature C−H Activation ofm-Terphenylboranes. Organometallics 2003, 22, 83–92. [Google Scholar] [CrossRef]
- Hoffend, C.; Schickedanz, K.; Bolte, M.; Lerner, H.-W.; Wagner, M. Functionalization of boron-doped tri(9,10-anthrylene)s. Tetrahedron 2013, 69, 7073–7081. [Google Scholar] [CrossRef]
- Lai, Y.-Y.; Bornand, M.; Chen, P. Homogeneous Model Complexes for Supported Rhenia Metathesis Catalysts. Organometallics 2012, 31, 7558–7565. [Google Scholar] [CrossRef]

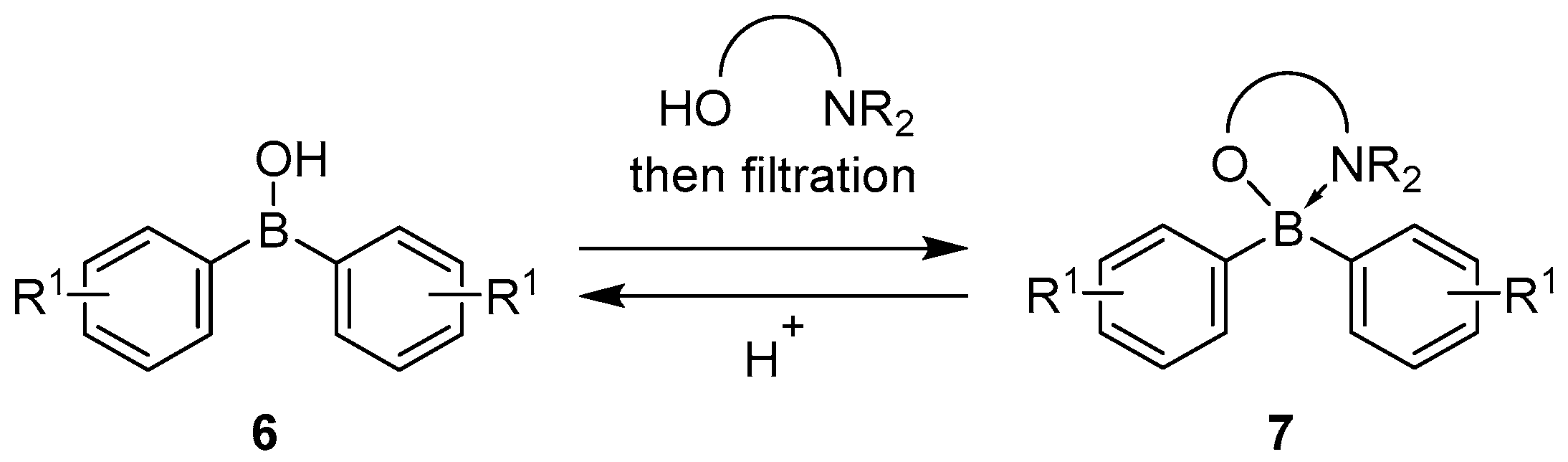



































































{kind=link}
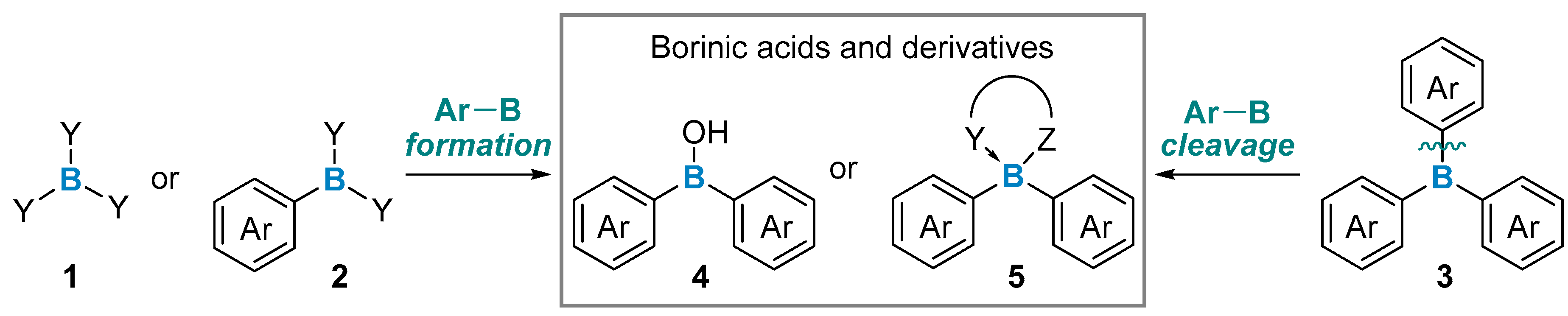
{kind=link}
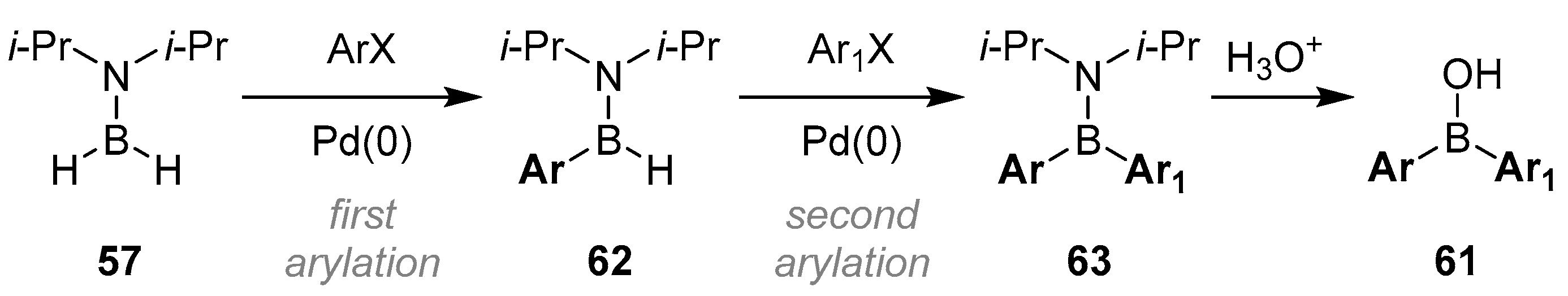{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
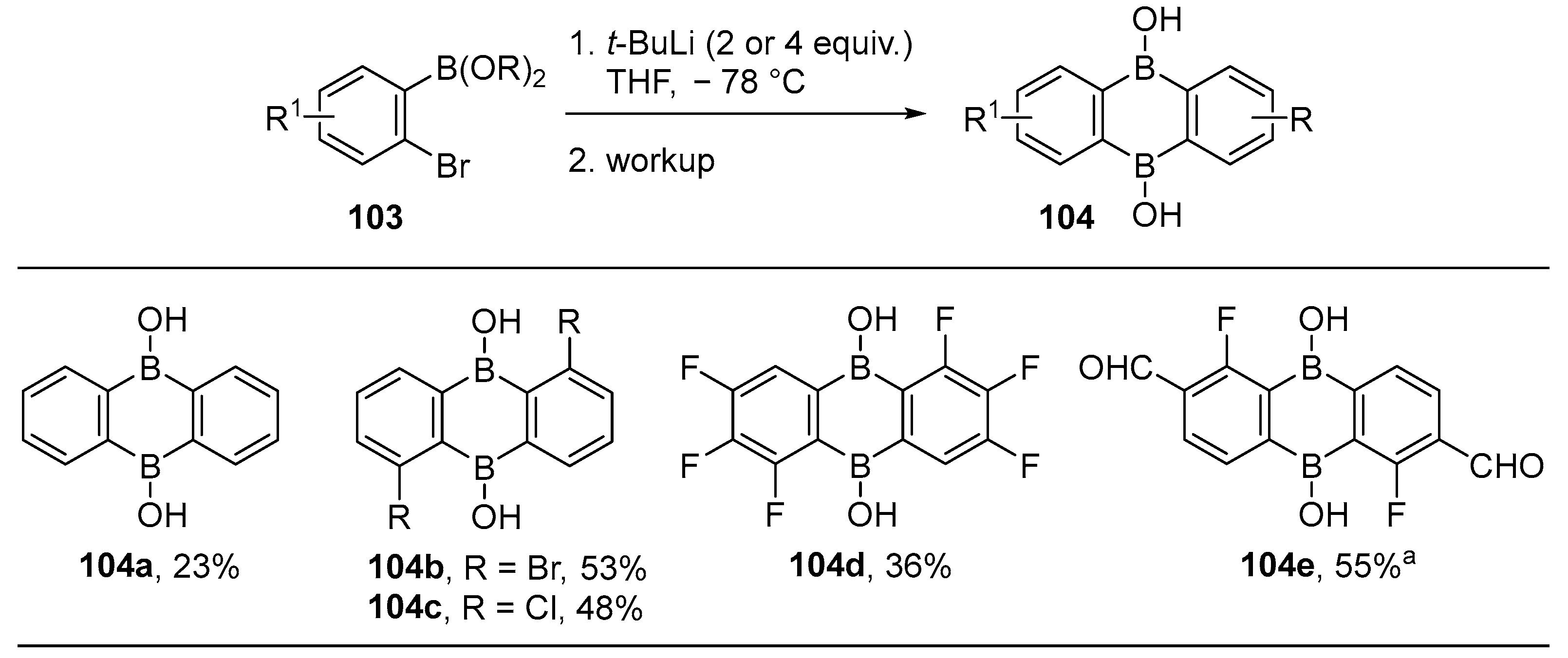{kind=link}
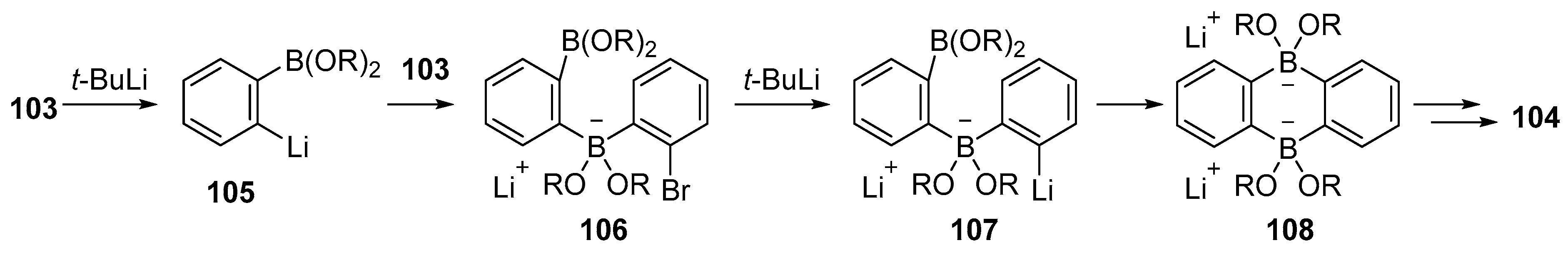{kind=link}
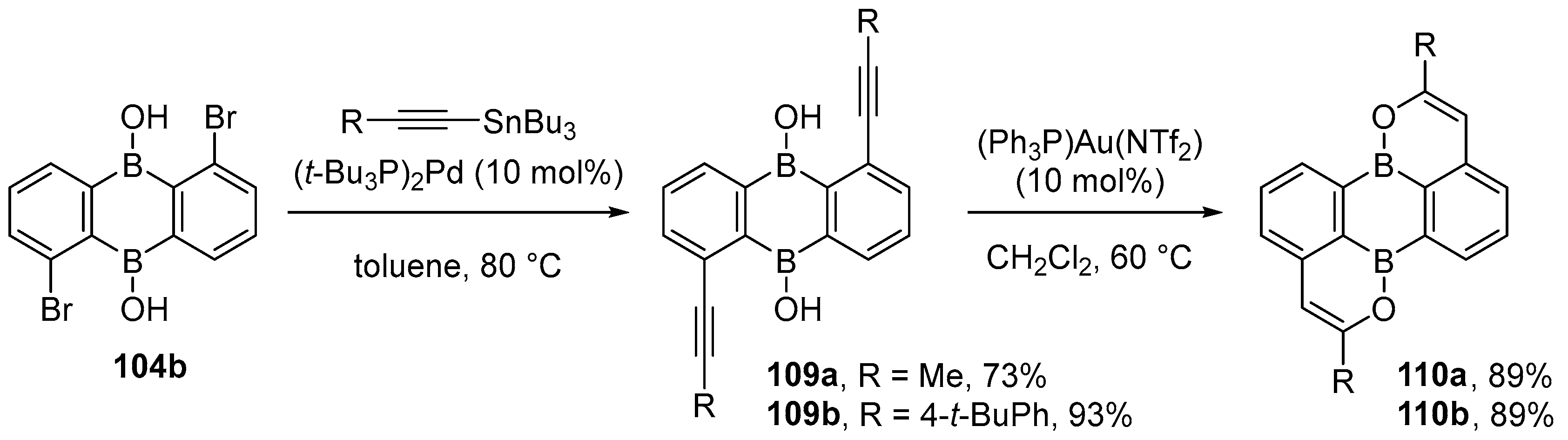{kind=link}
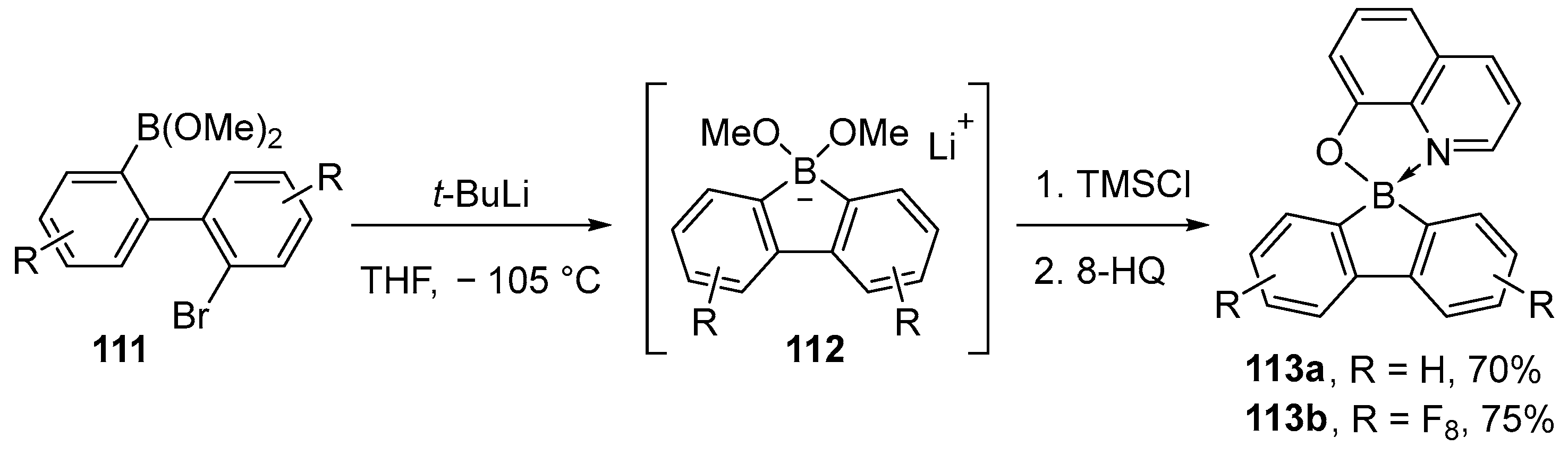{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
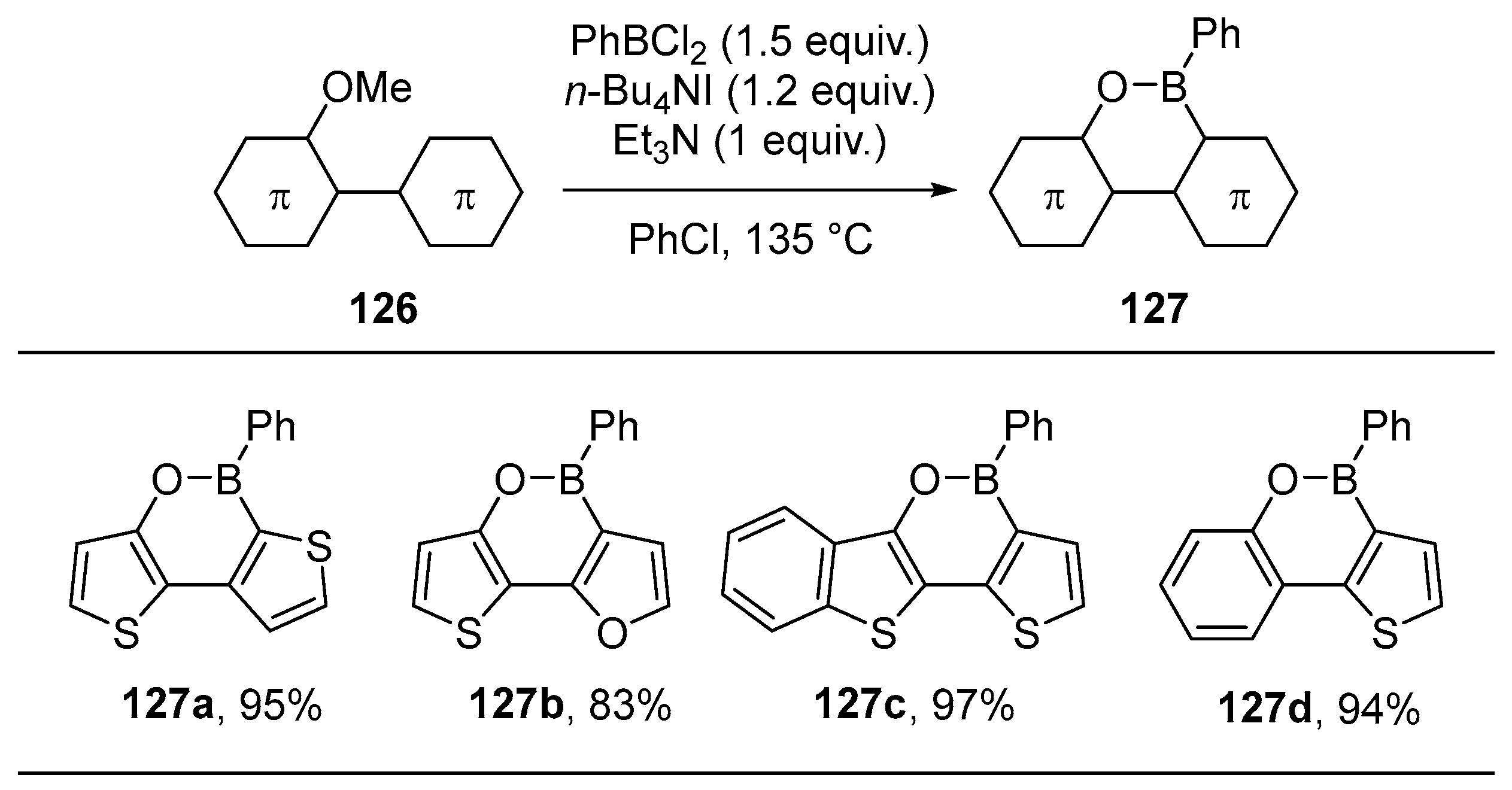{kind=link}
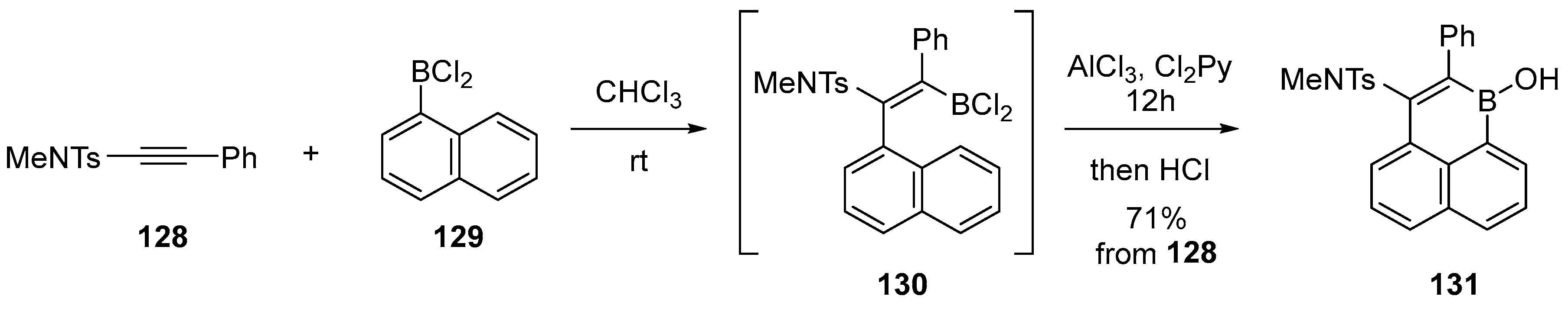{kind=link}
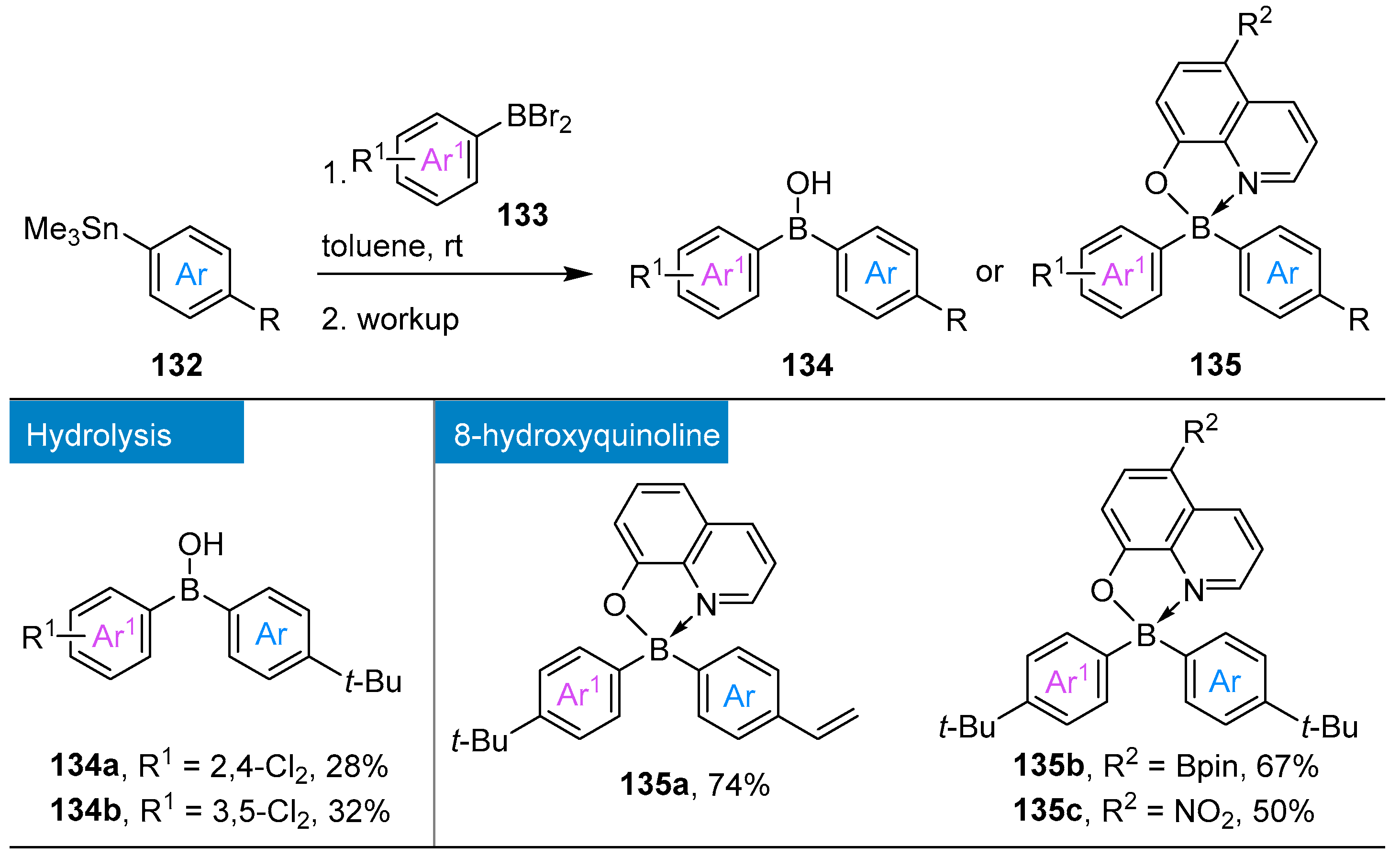{kind=link}
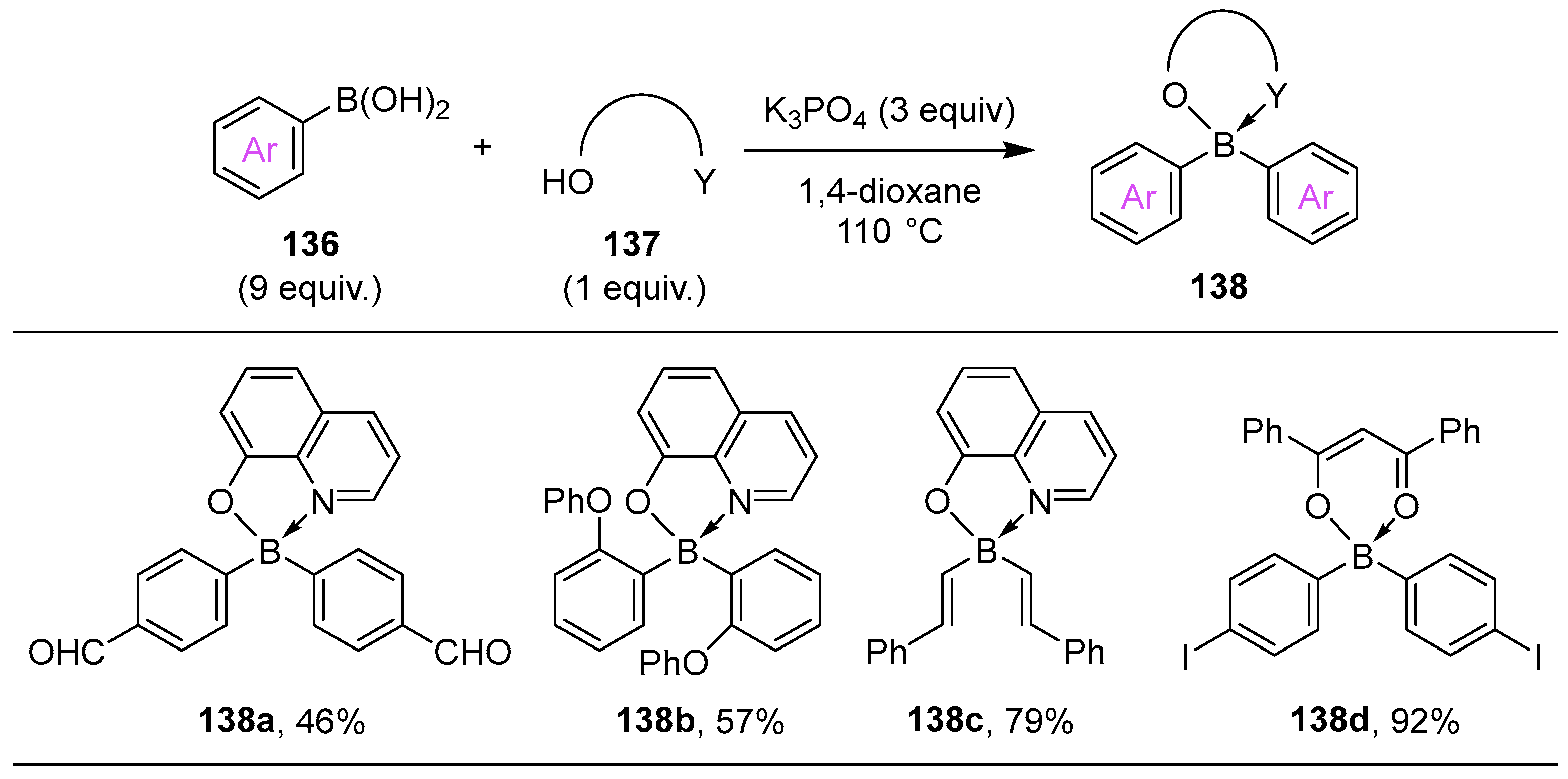{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
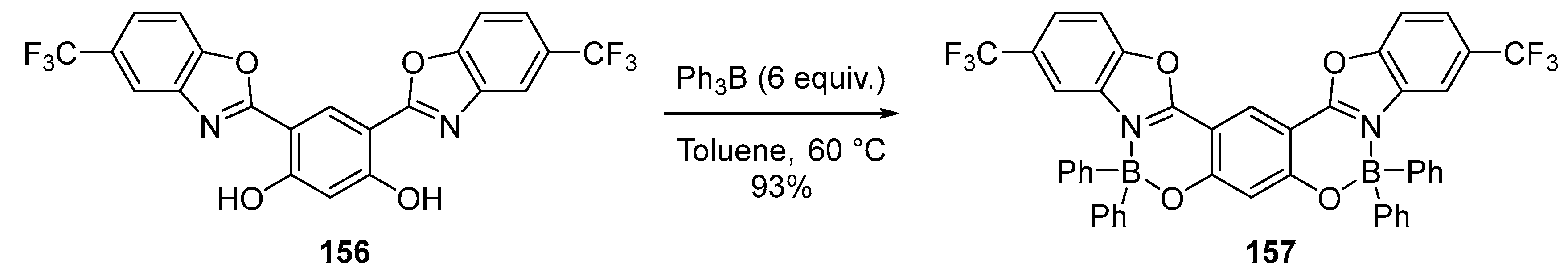{kind=link}
{kind=link}
{kind=link}
{kind=link}
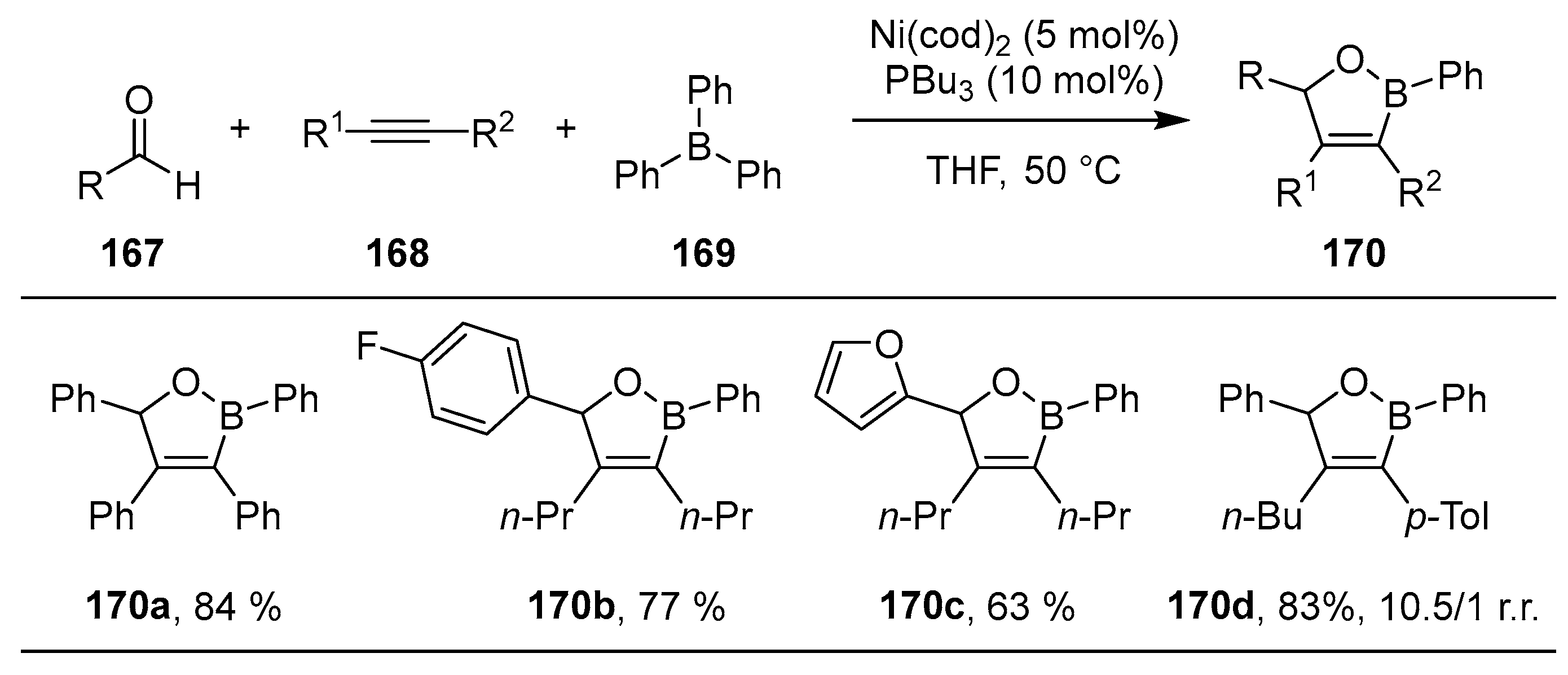{kind=link}
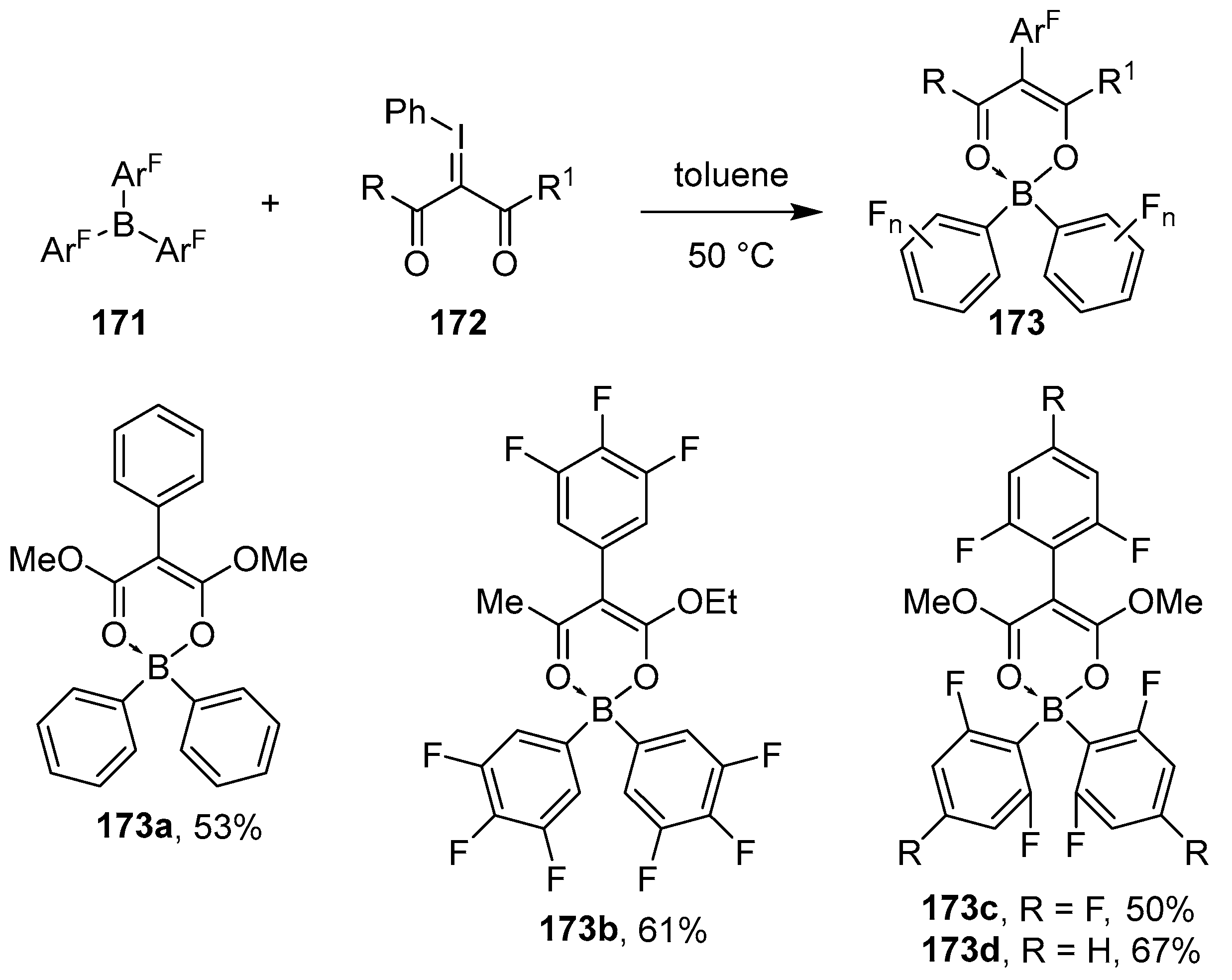{kind=link}
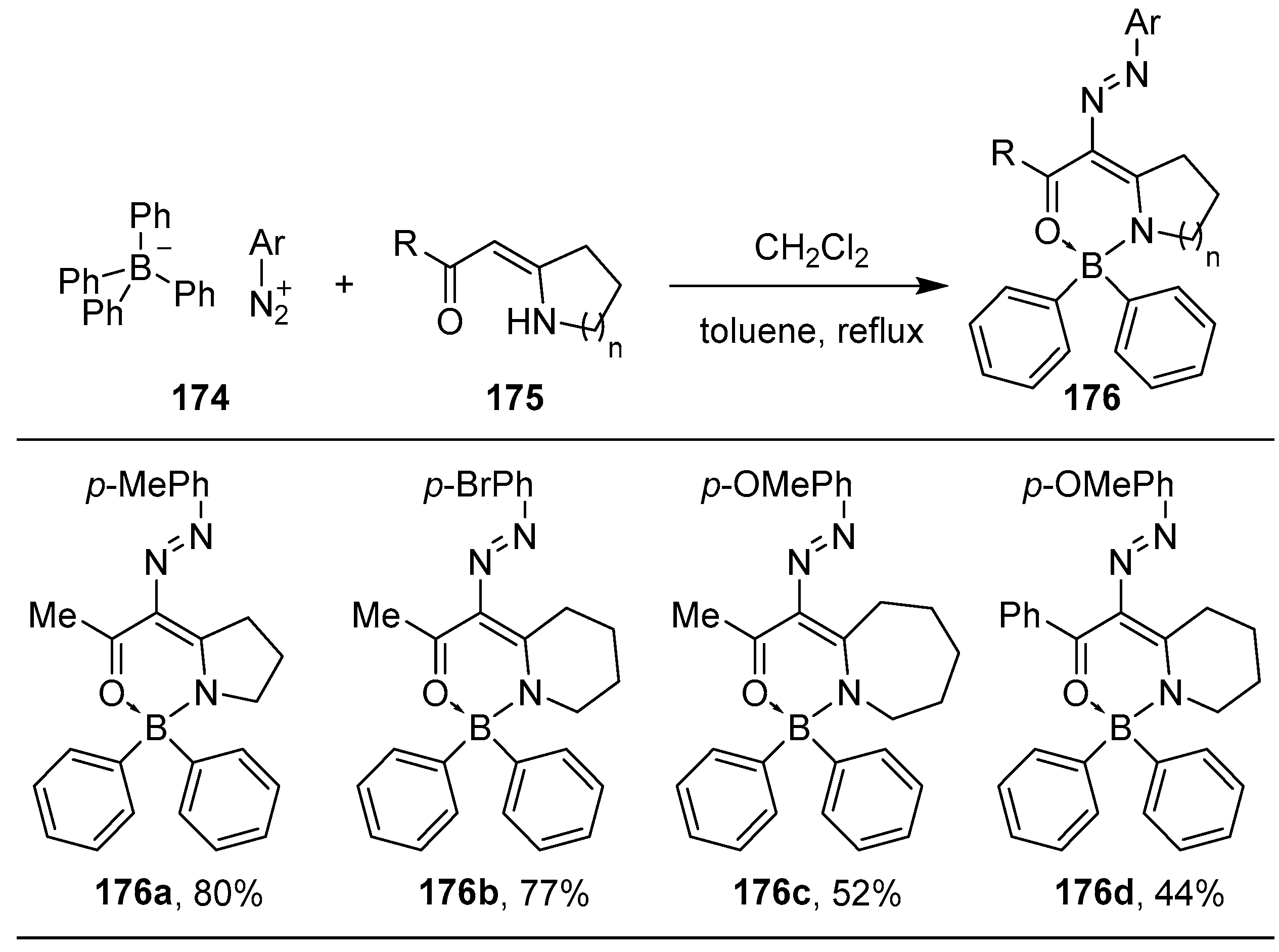{kind=link}
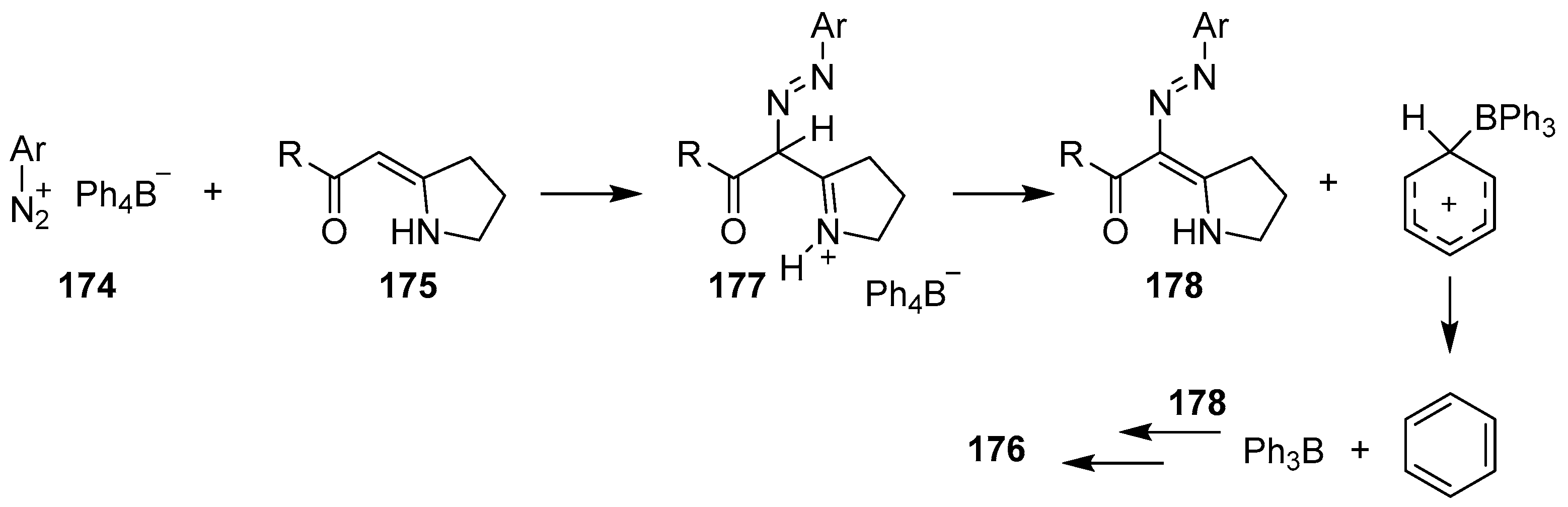{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Boron Reagent |  |  |  |  |  | |
|---|---|---|---|---|---|---|
| Methods | ||||||
| Organometallic pathway: poor functional compatibility | + symmetric/non-symmetric + cyclic/acyclic − moisture sensitive reagents − possible polyaddition of organometallic reagent | + symmetric + cyclic/acyclic − moisture sensitive reagents − possible polyaddition of organometallic reagent | + symmetric/non-symmetric + cyclic/acyclic + no polyaddition + catalytic methods exist, tolerant with functional groups − moisture sensitive boron reagents | + symmetric/non-symmetric + cyclic/acyclic + stable reagent + no polyaddition | ||
| Electrophilic borylation/ Transmetalation: good functional compatibility, no polyaddition to boron reagent | + symmetric/non-symmetric + cyclic/acyclic − some examples with tin reagents − sometimes requires a chelating group or limited to specific substrates | + symmetric/non-symmetric + cyclic/acyclic + stable reagent − some examples with tin reagents − sometimes requires a chelating group or limited to specific substrates | ||||
| Ligands exchange: good functional compatibility | ArB(OH)2 or ArBF3K + symmetric + acyclic + wide range of ligands + stable reagent − excess reagent | + symmetric + acyclic + wide range of ligands − usually if Ar ≠ Ph, boron reagents need to be prepared | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyet, M.; Chabaud, L.; Pucheault, M. Recent Advances in the Synthesis of Borinic Acid Derivatives. Molecules 2023, 28, 2660. https://doi.org/10.3390/molecules28062660
Boyet M, Chabaud L, Pucheault M. Recent Advances in the Synthesis of Borinic Acid Derivatives. Molecules. 2023; 28(6):2660. https://doi.org/10.3390/molecules28062660
Chicago/Turabian StyleBoyet, Marion, Laurent Chabaud, and Mathieu Pucheault. 2023. "Recent Advances in the Synthesis of Borinic Acid Derivatives" Molecules 28, no. 6: 2660. https://doi.org/10.3390/molecules28062660
APA StyleBoyet, M., Chabaud, L., & Pucheault, M. (2023). Recent Advances in the Synthesis of Borinic Acid Derivatives. Molecules, 28(6), 2660. https://doi.org/10.3390/molecules28062660