
Thermodynamics Evaluation of Selective Hydride Reduction for α,β-Unsaturated Carbonyl Compounds

Abstract
:1. Introduction


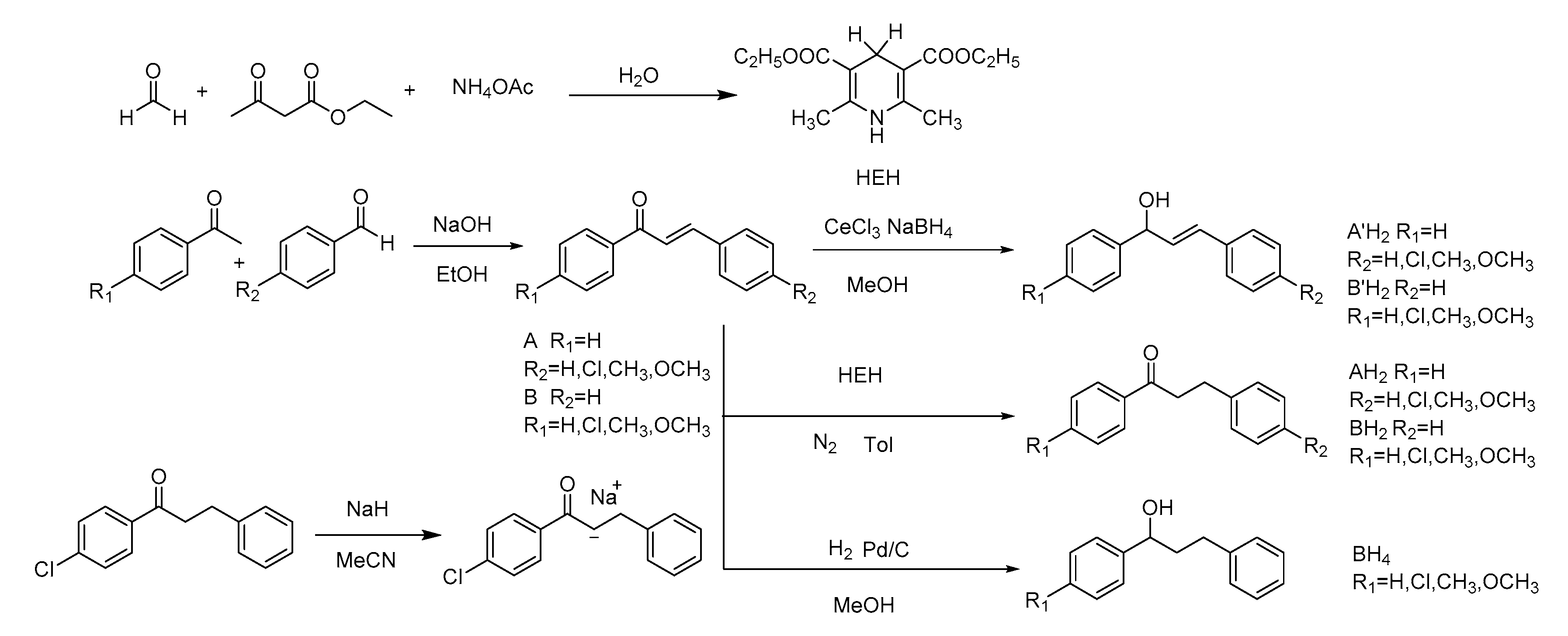
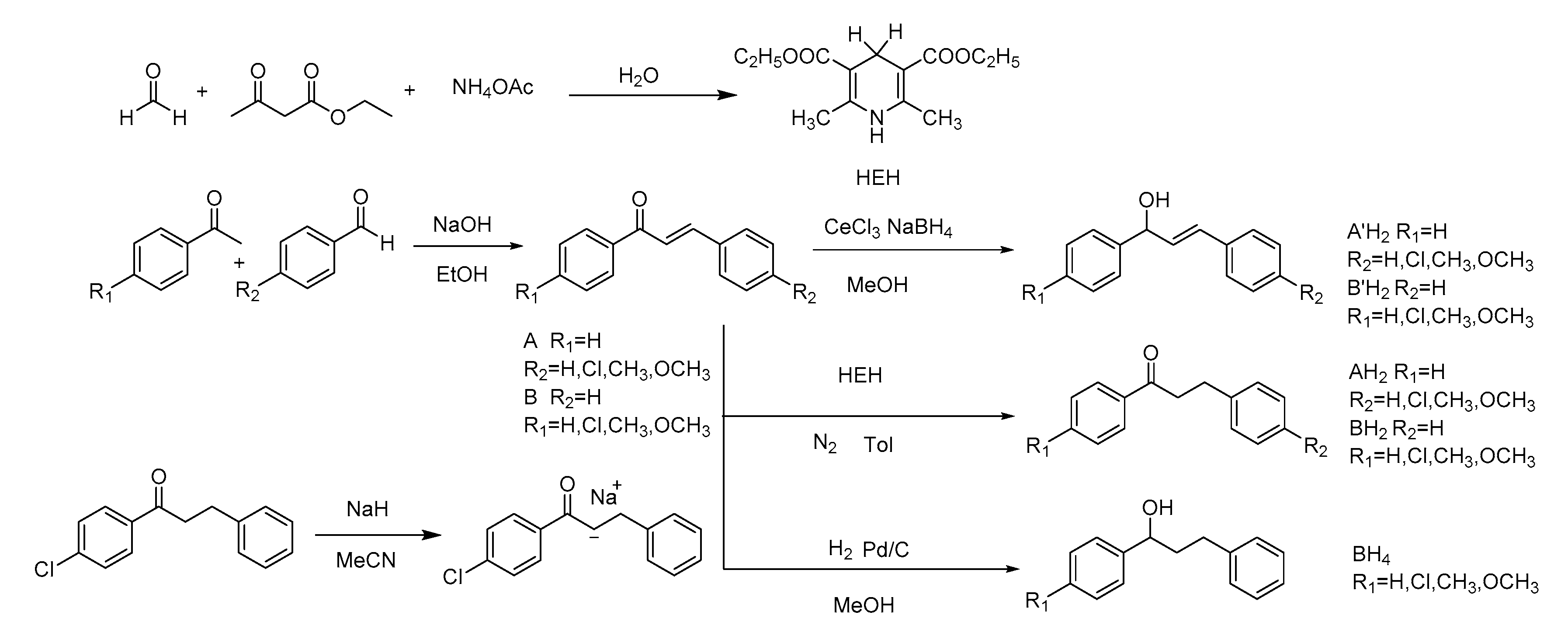
2. Results
3. Discussion
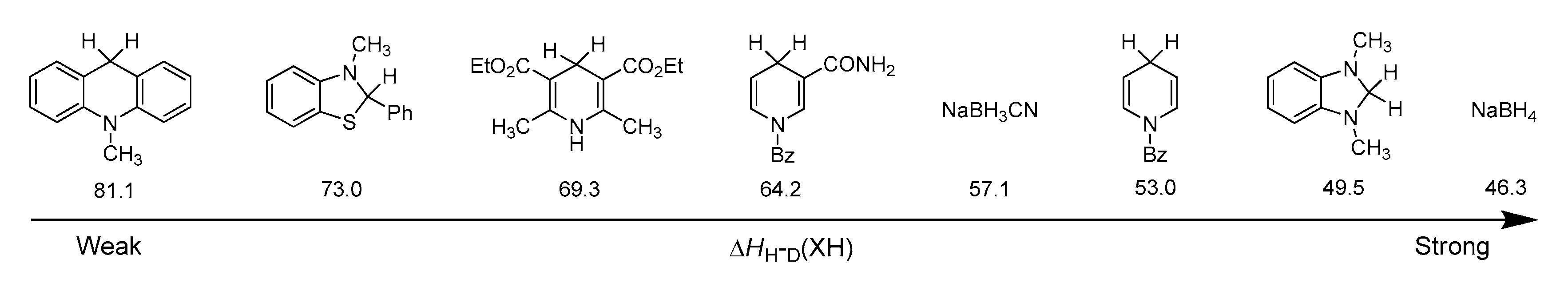
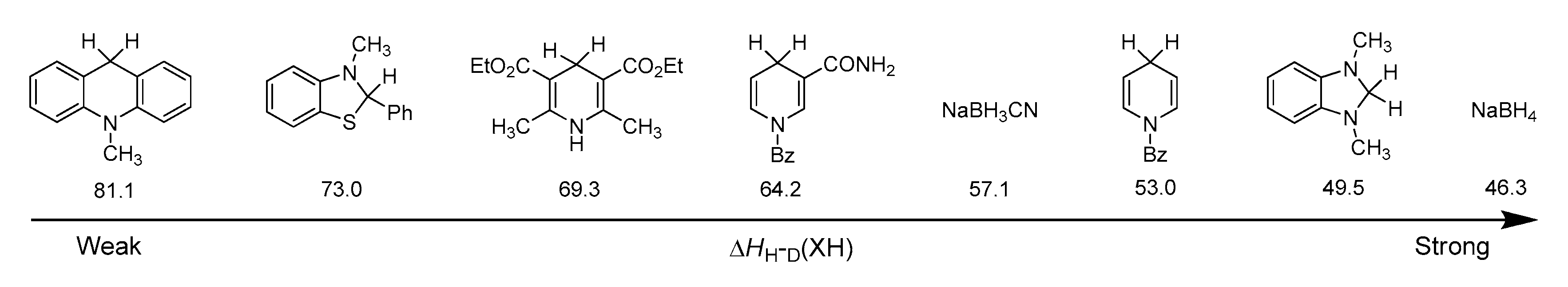
3.1. Hydride Affinity of α,β-Unsaturated Carbonyl Compounds
3.1.1. Hydride Affinity of Carbon–Oxygen Double Bond in α,β-Unsaturated Carbonyl Compounds
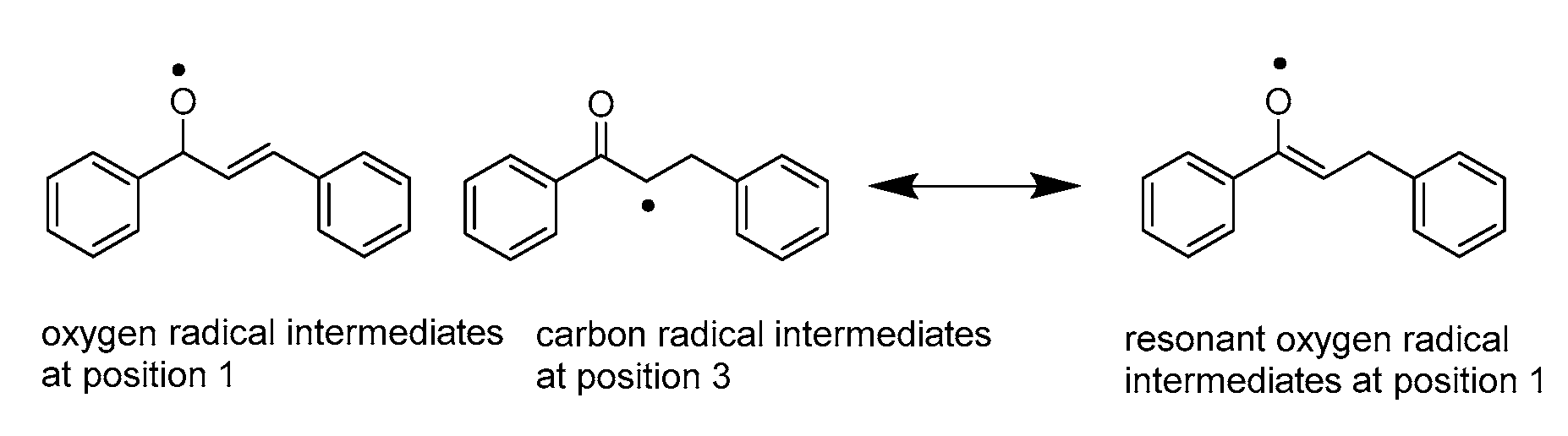
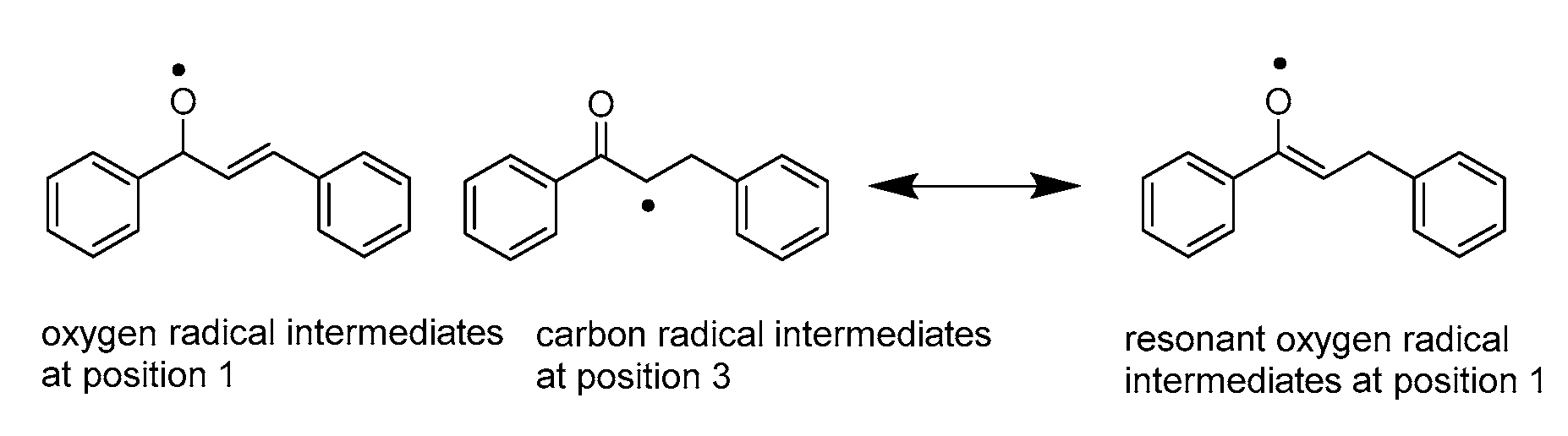
3.1.2. Hydride Affinity of Carbon–Carbon Double Bond in α,β-Unsaturated Carbonyl Compounds
3.1.3. Selective Reduction of α,β-Unsaturated Carbonyl Compound
3.2. Hydrogen–Atom Affinity of α,β-Unsaturated Carbonyl Compounds
3.3. Hydrogen Atom Affinity and Proton Affinity of Radical Anion Intermediate in α,β-Unsaturated Carbonyl Compounds
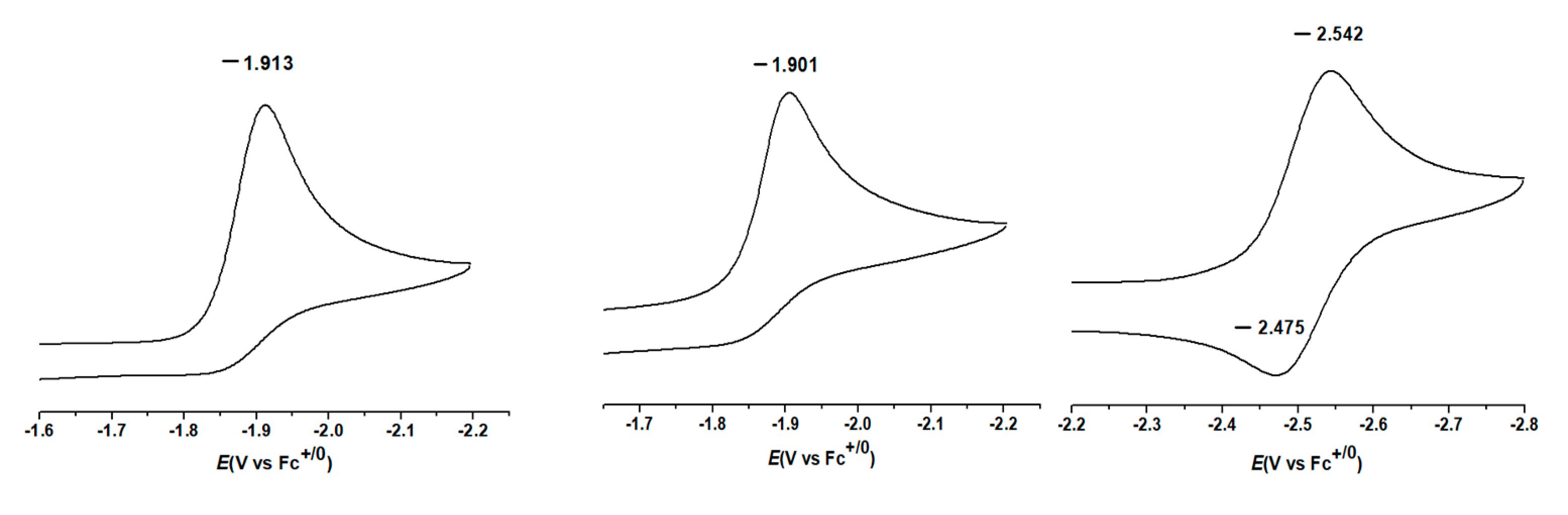
3.4. Electron Affinity of α,β-Unsaturated Carbonyl Compounds and Its Radical Intermediate
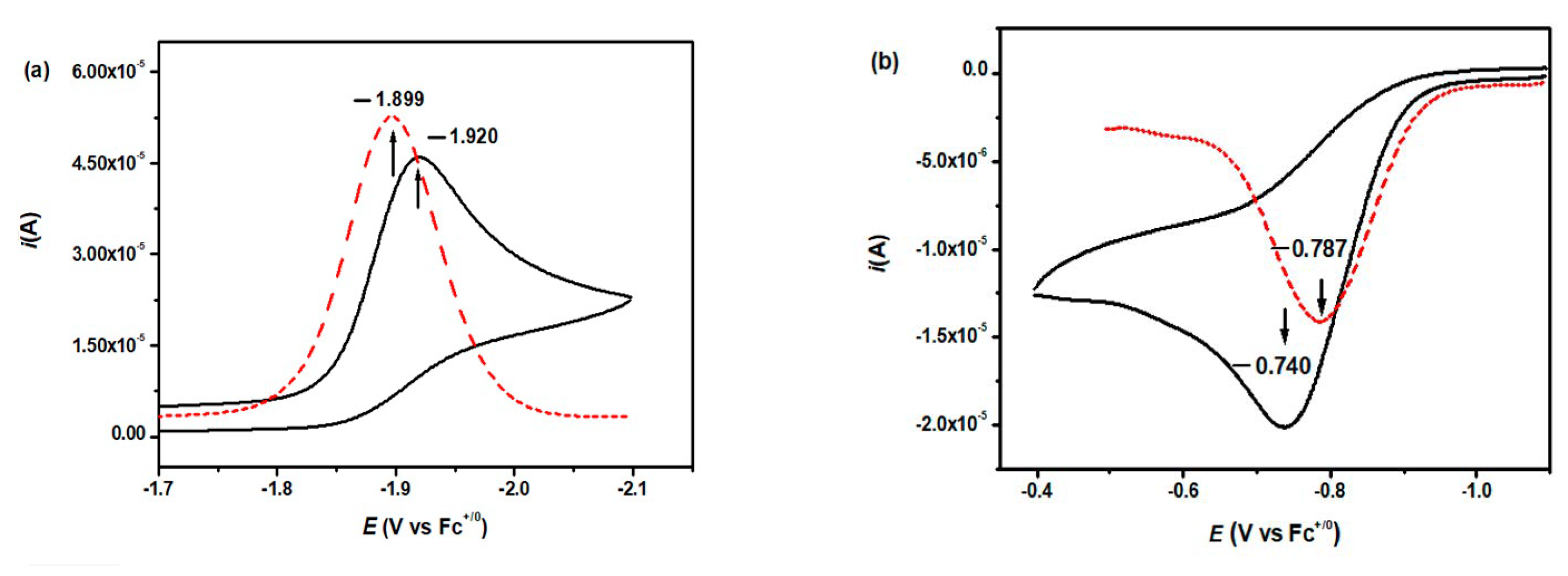
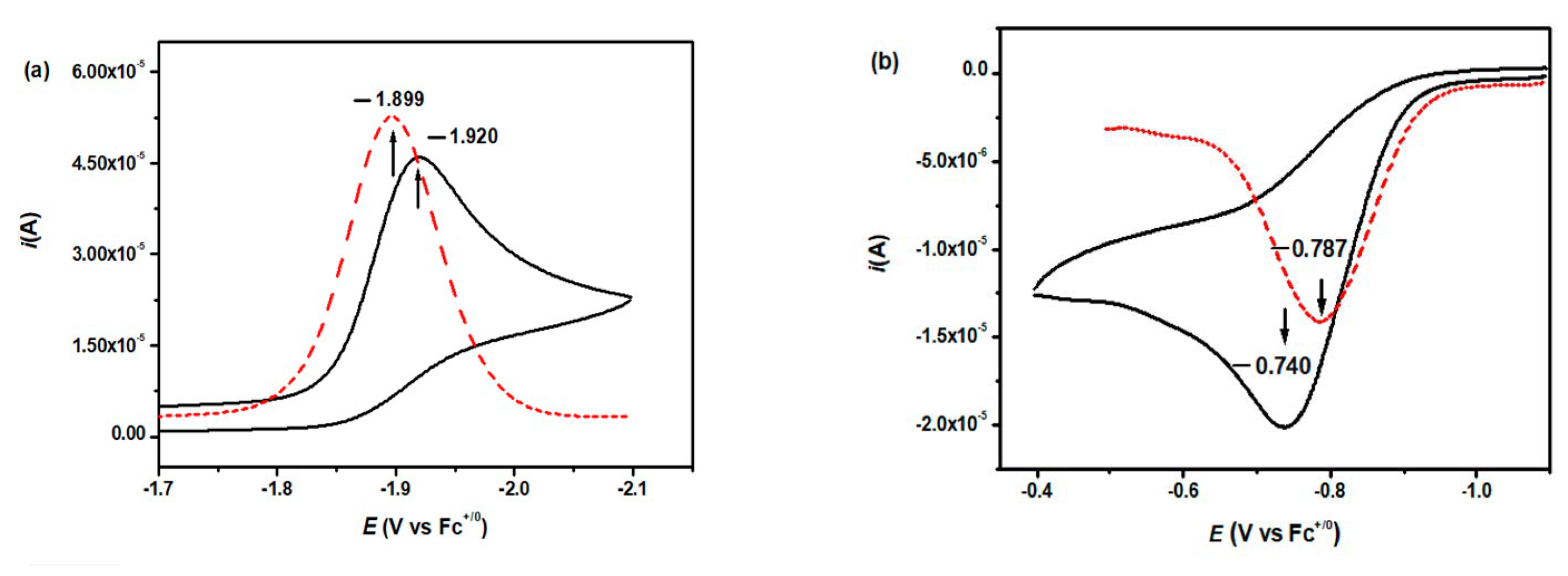
3.4.1. Single-Electron Reduction Potentials of α,β-Unsaturated Carbonyl Compounds
3.4.2. Single-Electron Reduction Potentials of Radical Intermediate in α,β-Unsaturated Carbonyl Compounds
3.5. Establishment of Molecule ID Card for Carbonyl Compounds
- (1)
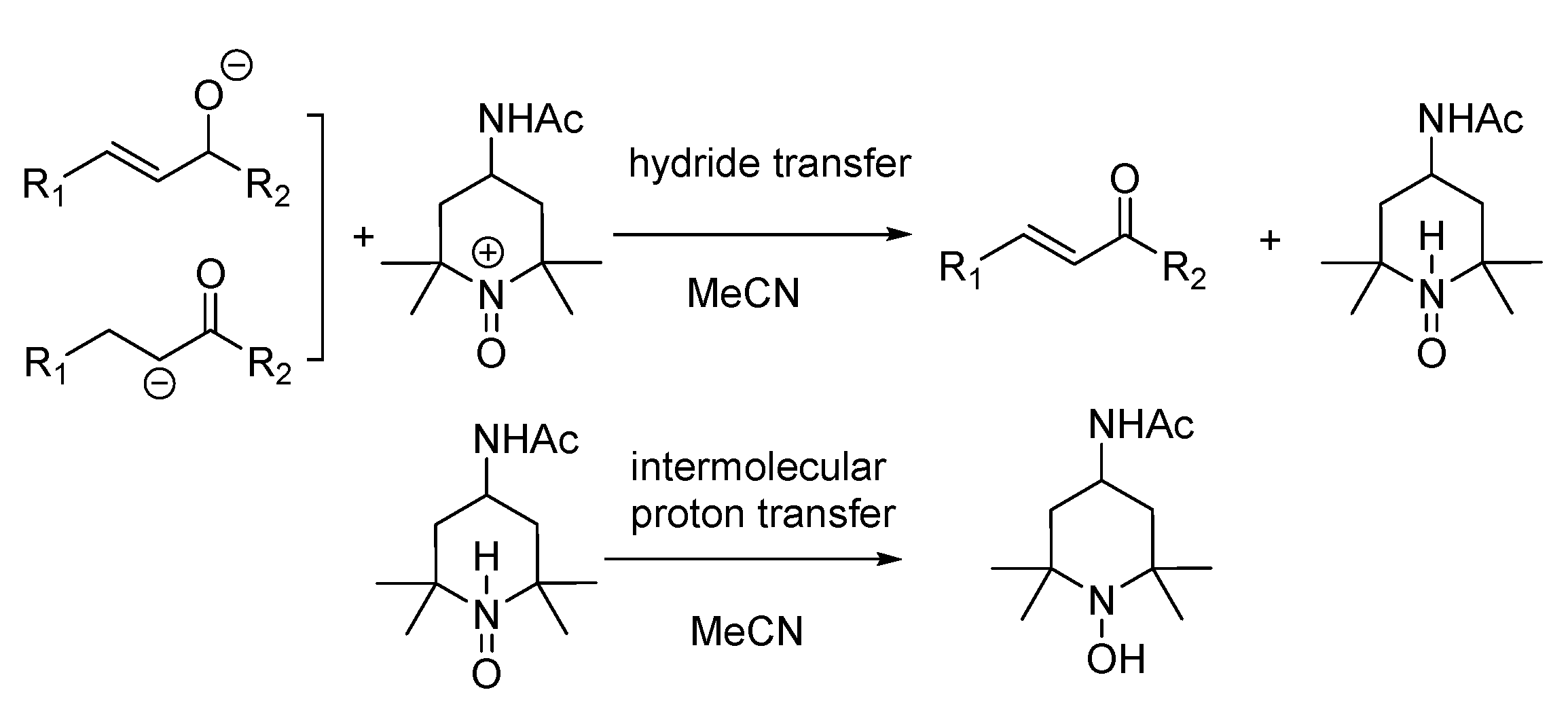
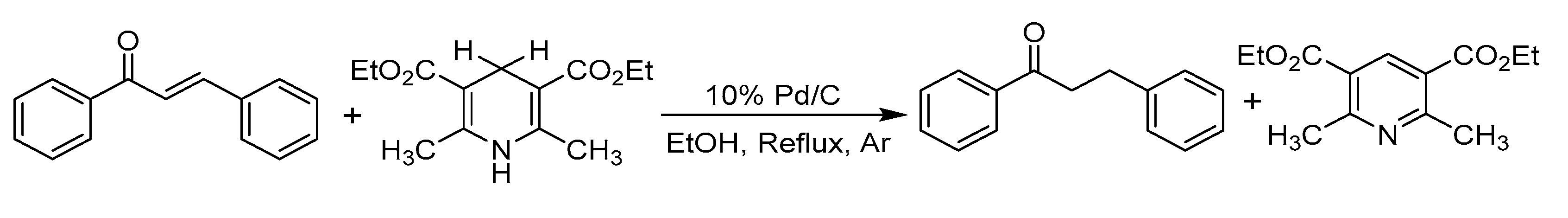
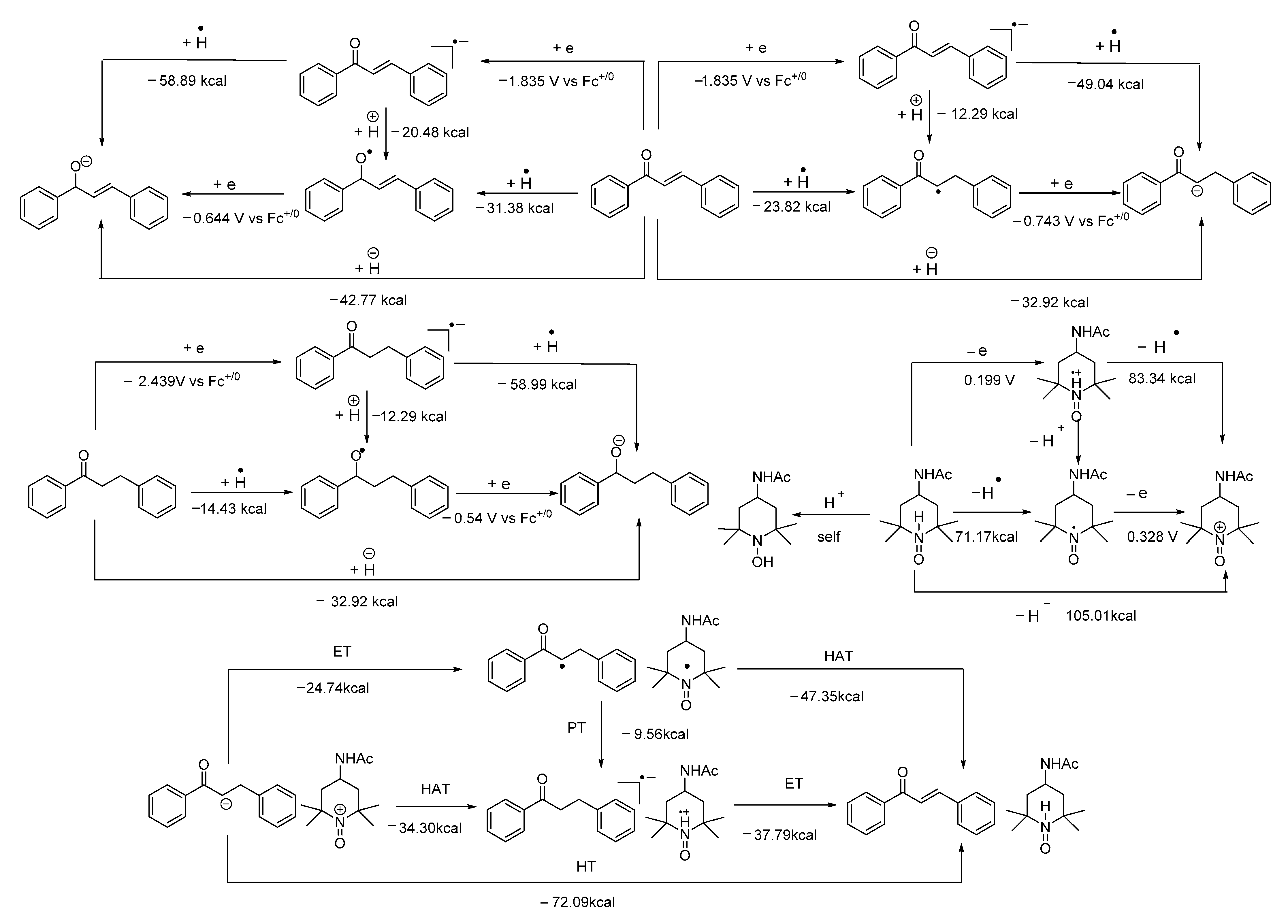
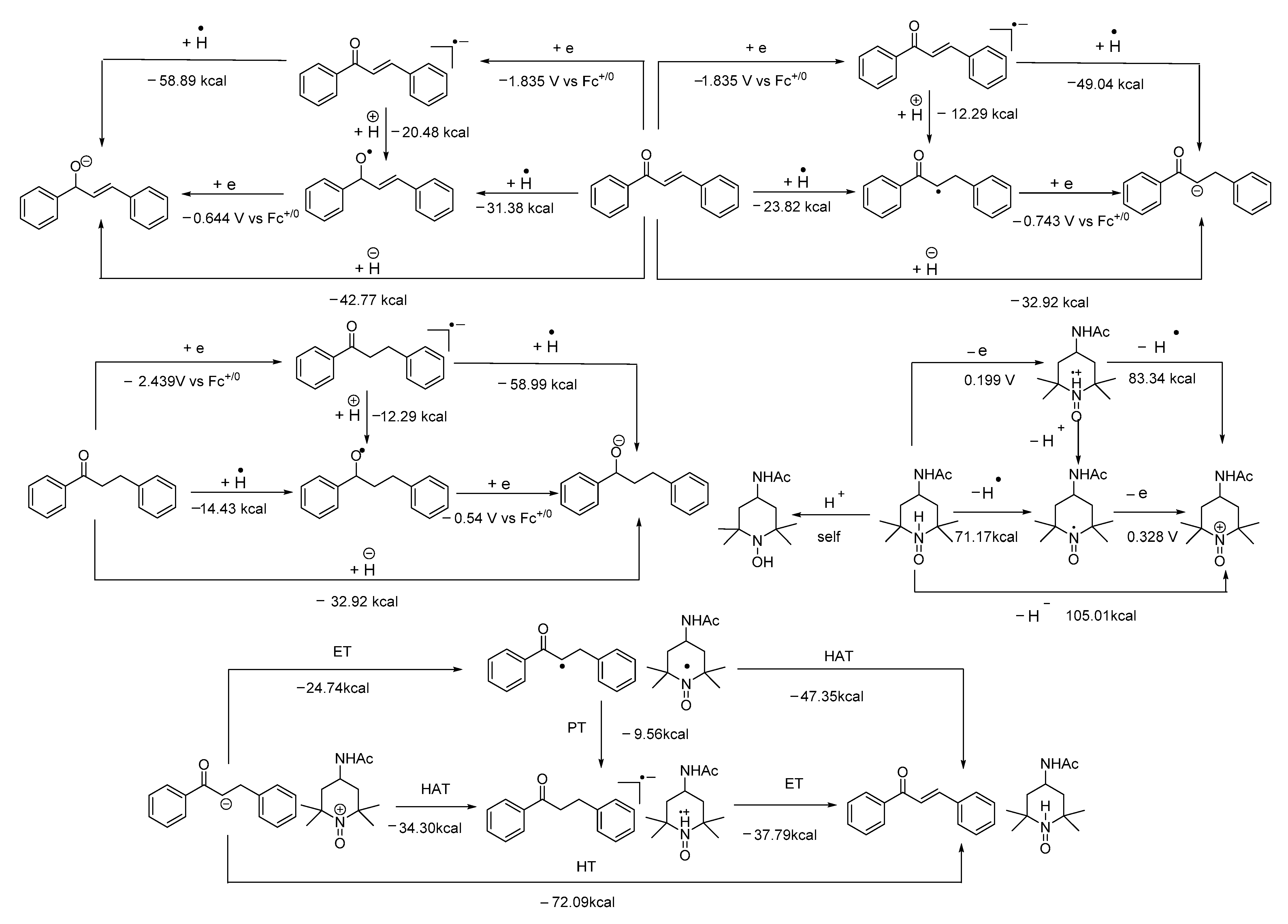
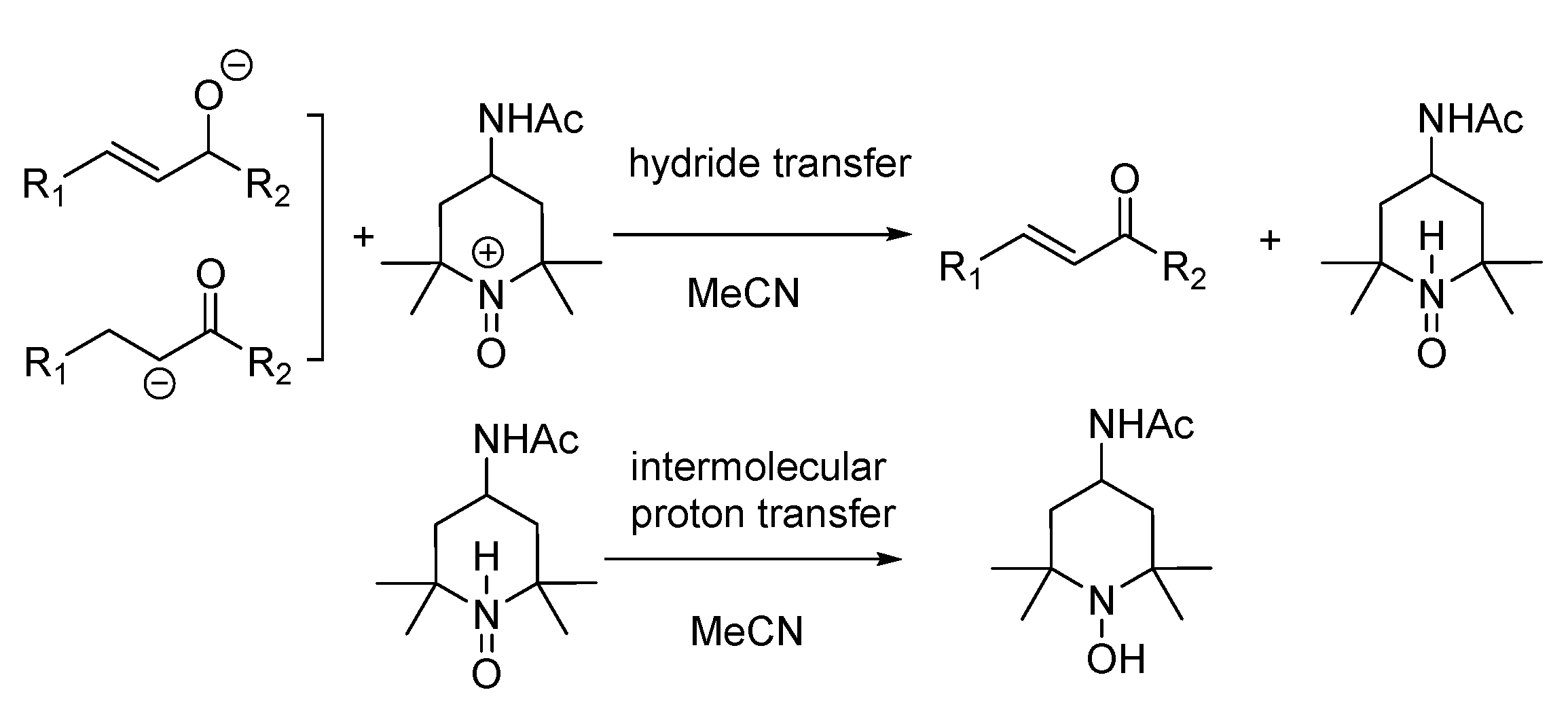
- The mixture (if any) of carbonyl hydride ions and Ac-tempo+ will be rapidly subject to hydride transfer to produce corresponding α,β-unsaturated carbonyl compounds and Ac-tempoH, as shown in the conducted experiments. A comparison of the energies between one-step hydride transfer and multi-step hydride transfer initiated by electrons or hydrogen atoms indicates that these three pathways are energetically favorable, and one-step hydride transfer has the greatest thermodynamic driving force;
- (2)
- When two types of radicals come in contact, either hydrogen–atom transfer or proton transfer may occur. Electron transfer is energetically analyzed as forbidden, and hydrogen atom transfer is significantly better than proton transfer;
- (3)
- When the radical anions and radical cations of two types of compounds meet, it is difficult for them to be inclined to hydrogen atom transfer or proton transfer but very easy for them to be stimulated to electron transfer, because the first two types of transfer are energetically forbidden, while electron transfer is a spontaneous process wherein these two types of intermediates can achieve a stable electronic structure;
- (4)
- When neutral α,β-unsaturated carbonyl compounds and Ac-tempoH are mixed, based on TCG analysis, the transfer in all steps is forbidden.
4. Conclusions
- (1)
- All three series compounds are weak hydride acceptors. The ability of unsaturated carbonyl compounds to accept hydride is better than that of saturated carbonyl compounds. All three series compounds are extremely weak hydrogen atom acceptors. Hydride transfer initiated by hydrogen atom transfer is not the best transfer path. Such compounds are extremely weak single-electron acceptors. Their corresponding anions X− are all extremely strong single-electron reducing agents. The radical anion X•− of carbonyl compounds is a strong hydrogen atom acceptor and weak proton acceptor, which is difficult to stabilize.
- (2)
- A comparison of hydride affinity and hydrogen atom affinity between two types of unsaturated bonds in α,β-unsaturated carbonyl compounds indicates that the absolute value of them for carbonyl groups is slightly larger than that of carbon–carbon double bonds. The selective reduction of them is more difficult to control. However, the presence of a conjugated system activates carbonyl groups and enhances the capability of carbonyl groups to accept hydride ions and hydrogen atoms.
- (3)
- We constructed “Molecule ID Cards” for carbonyl compounds by means of thermodynamic and electrochemical data to not only quantitatively determine the chemical properties of carbonyl compounds and their reaction intermediates but also easily predict the reaction mechanism.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board State
Data Availability Statement
Conflicts of Interest
References
- Hamlin, T.A.; Fernandez, I.; Bickelhaupt, F.M. How Dihalogens Catalyze Michael Addition Reactions. Angew. Chem. Int. Ed. 2019, 58, 8922–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wonner, P.; Steinke, T.; Vogel, L.; Huber, S.M. Carbonyl Activation by Selenium and Tellurium-based Chalcogen Bonding in a Michael Addition Reaction. Chem. Eur. J. 2020, 26, 1258–1262. [Google Scholar] [CrossRef]
- Reznikov, A.N.; Klimochkin, Y.N. Recent Developments in Highly Stereoselective Michael Addition Reactions Catalyzed by Metal Complexes. Synthesis 2020, 52, 781–795. [Google Scholar] [CrossRef]
- Malkar, R.S.; Jadhav, A.L.; Yadav, G.D. Innovative catalysis in Michael addition reactions for C-X bond formation. Mol. Catal. 2020, 485, 110814. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Krishnakumar, V.; Gunanathan, C. KOtBu-Catalyzed Michael Addition Reactions under Mild and Solvent-Free Conditions. Chem. Asian. J. 2020, 15, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, F.-M.; Tu, Y.-Q. Enantioselective synthesis of cis-hydrobenzofurans bearing all-carbon quaternary stereocenters and application to total synthesis of (–)-morphine. Nat. Commun. 2019, 10, 2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhang, F.-M.; Tu, Y.-Q. Catalytic Asymmetric Total Syntheses of (−) -Galanthamine and (−)-Lycoramine. J. Org. Chem. 2019, 84, 12664–12671. [Google Scholar] [CrossRef]
- Yamabe, S.; Tsuchida, N.; Yamazaki, S. Keto-Enol Tautomerization Controls the Acid- Catalyzed Robinson Annulation—A DFT Study. Chemistryselect 2019, 4, 4962–4966. [Google Scholar] [CrossRef]
- Xu, B.; Wang, B.-Y.; Xun, W.; Qiu, F.-Y. Total Synthesis of (−)-Daphenylline. Angew. Chem. Int. Ed. 2019, 58, 5754–5757. [Google Scholar] [CrossRef]
- Mojtabaei, F.; Mokhtary, M. One-pot Synthesis and Antibacteial Study of New Cyclohexenone Derivatives Catalyzed by Tetramethylammonium Hydroxide. Polycycl. Aromat. Comp. 2020, 40, 585–593. [Google Scholar] [CrossRef]
- Vermeeren, P.; Hamlin, T.A.; Fernandez, I.; Bickelhaupt, F.M. How Acid Lewis Catalyze Diels–Alder Reactions. Angew. Chem. Int. Ed. 2020, 59, 6201–6206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-B.; Gao, J.; Hu, Y.-C. Enzymatic Intermolecular Hetero-Diels–Alder Reaction in the Biosynthesis of Tropolonic Sesquiterpenes. J. Am. Chem. Soc. 2019, 141, 14052–14056. [Google Scholar] [CrossRef] [PubMed]
- Fluegel, L.L.; Hoye, T.R. Hexadehydro-Diels–Alder Reaction: Benzyne Generation via Cycloisomerization of Tethered Triynes. Chem. Rev. 2021, 121, 2413–2444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Shukla, V.; Boger, D.L. Inverse Electron Demand Diels–Alder Reactions of Heterocyclic Azadienes, 1-Aza-1,3-Butadienes, Cyclopropenone Ketals and Related Systems. A Retrospective. J. Org. Chem. 2019, 84, 9397–9445. [Google Scholar] [CrossRef] [PubMed]
- Beker, W.; Gajewska, E.P.; Badowski, T.; Grzybowski, B.A. Prediction of Major Regio-,Site-, and Diastereoisomers in Diels-Alder Reactions by Using Machine-Learning: The Importance of Physically Meaningful Descriptors. Angew. Chem. Int. Ed. 2019, 58, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
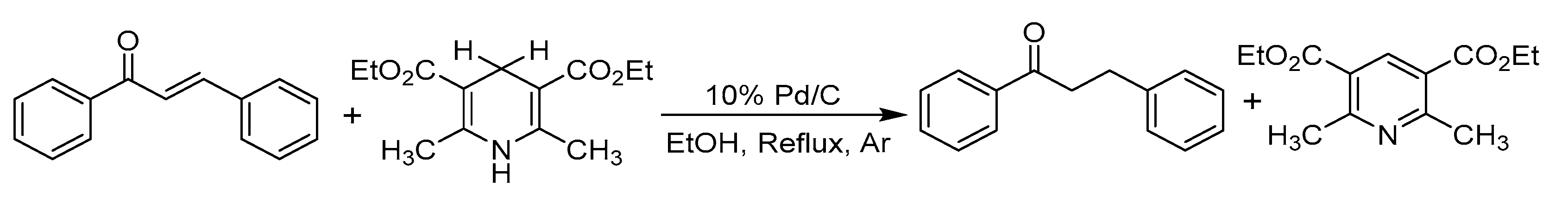
- Lee, H.Y.; An, M. Selective 1,4-reduction of unsaturated carbonyl compounds using Co2(CO)8–H2O. Tetrahedron Lett. 2003, 44, 2775–2778. [Google Scholar] [CrossRef]
- Deursen, R.; Stampfer, W.; Edegger, K.; Faber, K.; Wolfgang, K. Chemo- and stereo-selective biocatalytic reduction of α,β-unsaturated ketones employing a chemo-tolerant ADH from Rhodococcus ruber DSM 44541. J. Mol. Catal. B Enzym. 2004, 31, 159–163. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Selective, C,C-double bond reduction of α,β- unsaturated carbonyl compounds with cyclohexane using zeolites. J. Mol. Catal. A Chem. 2006, 245, 231–234. [Google Scholar] [CrossRef]
- Daimon, A.; Kamitanaka, T.; Kishida, N.; Matsuda, T.; Harada, T. Selective reduction of unsaturated aldehydes to unsaturated alcohols using supercritical 2-propanol. J. Supercrit. Fluids 2006, 37, 215–219. [Google Scholar] [CrossRef]
- Zhu, Y.-Z.; Chuah, G.K.; Jaenicke, S. Selective Meerwein-Ponndorf-Verley reduction of α,β-unsaturated aldehydes over Zr-zeolite beta. J. Catal. 2006, 241, 25–33. [Google Scholar] [CrossRef]
- Heydari, A.; Khaksar, S.; Akbari, J.; Esfandyari, M.; Tajbakhsh, M. Direct reductive amination and selective1,2- reduction of α,β-unsaturated aldehydes and ketones by NaBH4 using H3PW12O40 as catalyst. Tetrahedron Lett. 2007, 48, 1135–1138. [Google Scholar] [CrossRef]
- Bobbitt, J.M. Explosion of 4-Acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium Perchlorate and Replacement by Tetrafluoroborate. Molecules 1999, 4, M102. [Google Scholar]
- Bobbitt, J.M. Oxoammonium Salts. 6. 4-Acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium Perchlorate: A Stable and Convenient Reagent for the Oxidation of Alcohols. Silica Gel Catalysis. J. Org. Chem. 1998, 63, 9367–9374. [Google Scholar] [CrossRef]
- Wayner, D.D.M.; Parker, V.D. Bond energies in solution from electrode potentials and thermochemical cycles. A simplified and general approach. Acc. Chem. Res. 1993, 26, 287–294. [Google Scholar]
- Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. Hydride, Hydrogen Atom, Proton and Electron Transfer Driving Forces of Various Five-membered Heterocyclic Organic Hydrides and Their Reaction Intermediates in Acetonitrile. J. Am. Chem. Soc. 2008, 130, 2501–2516. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-P.; Handoo, K.L.; Parker, V.D. Hydride Affinities of Carbenium Ions in Acetonitrile and Dimethyl Sulfoxide Solution. J. Am. Chem. Soc. 1993, 115, 2655–2660. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Zhang, M.; Liu, Q.-Y. A facile experimental method to determine the hydride affinity of polarized olefins in acetonitrile. Angew. Chem. Int. Ed. 2006, 45, 3954–3957. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Shen, X.-X.; Zhou, B. Hydrogenation of olefins using Hantzsch ester catalyzed by palladium on carbon. Tetrahedron Lett. 2009, 50, 1026–1028. [Google Scholar] [CrossRef]
- Stroba, A.; Schaeffer, F.; Hindie, V.; Lopez-Garcia, L.; Adrian, I.; Frohner, W.; Hartmann, R.W.; Biondi, R.M.; Engel, M. 3,5-Diphenylpent-2-enoic Acids as Allosteric Activators of the Protein Kinase PDK1: Structure−Activity Relationships and Thermodynamic Characterization of Binding as Paradigms for PIF-Binding Pocket-Targeting Compounds. J. Med. Chem. 2009, 52, 4683–4693. [Google Scholar] [CrossRef]
- Smith, C.D.; Rosocha, G.; Mui, L.; Batey, R.A. Investigation of Substituent Effects on the Selectivity of 4π-Electrocyclization of 1,3-Diarylallylic Cations for the Formation of Highly Substituted Indenes. J. Org. Chem. 2010, 75, 4716–4727. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Liu, Q.-Y.; Chen, Q.; Mei, L.-R. Hydride, Hydrogen, Proton, and Electron Affinities of Imines and Their Reaction Intermediates in Acetonitrile and Construction of Thermodynamic Characteristic Graphs (TCGs) of Imines as a “Molecule ID Card”. J. Org. Chem. 2010, 75, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Sawle, P.; Moulton, B.E.; Jarzykowska, M.; Green, C.J.; Foresti, R.; Fairlamb, J.S.; Motterlini, R. Structure−Activity Relationships of Methoxychalcones as Inducers of Heme Oxygenase-1. Chem. Res. Toxicol. 2008, 21, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.T.; Kang, S.K.; Kim, M.S.; Ryu, S.R.; An, D.K. Solvent-free reduction of aldehydes and ketones using solid acid-activated sodium borohydride. Tetrahedron 2006, 62, 8164–8168. [Google Scholar] [CrossRef]
- Mori, A.; Mizusaki, T.; Miyakawa, Y.; Ohashi, E.; Haga, T.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Chemoselective hydrogenation method catalyzed by Pd/C using diphenylsulfide as a reasonable catalyst poison. Tetrahedron 2006, 62, 11925–11932. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Li, Q.; Hao, W.-F.; Cheng, J.-P. Dissociation Energies and Charge Distribution of the Co? NO Bond for Nitrosyl-α,β,γ,δ-tetraphenylporphinatocobalt(II) and Nitrosyl-α,β,γ,δ-tetraphenylorphinatocobalt(III) in Benzonitrile Solution. J. Am. Chem. Soc. 2002, 124, 9887–9893. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-Q.; Hao, W.-F.; Tang, H.; Wang, C.-H.; Cheng, J.-P. Determination of N-NO Bond Dissociation Energies of N-Methyl-N-Nitrosobenzenesulfonamindes in Acetonitrile and Application in the Mechanism Analyses on NO Transfer. J. Am. Chem. Soc. 2005, 127, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
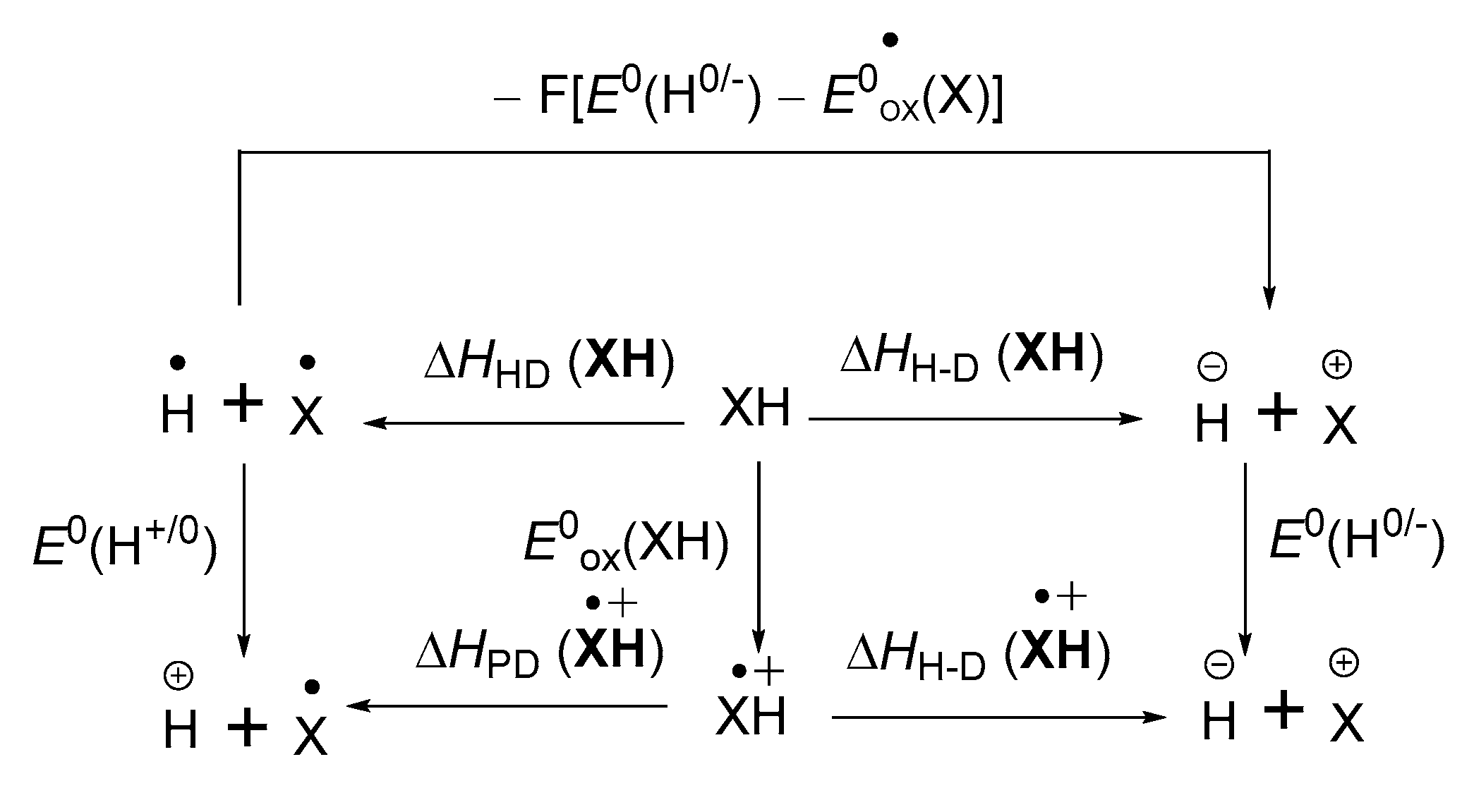


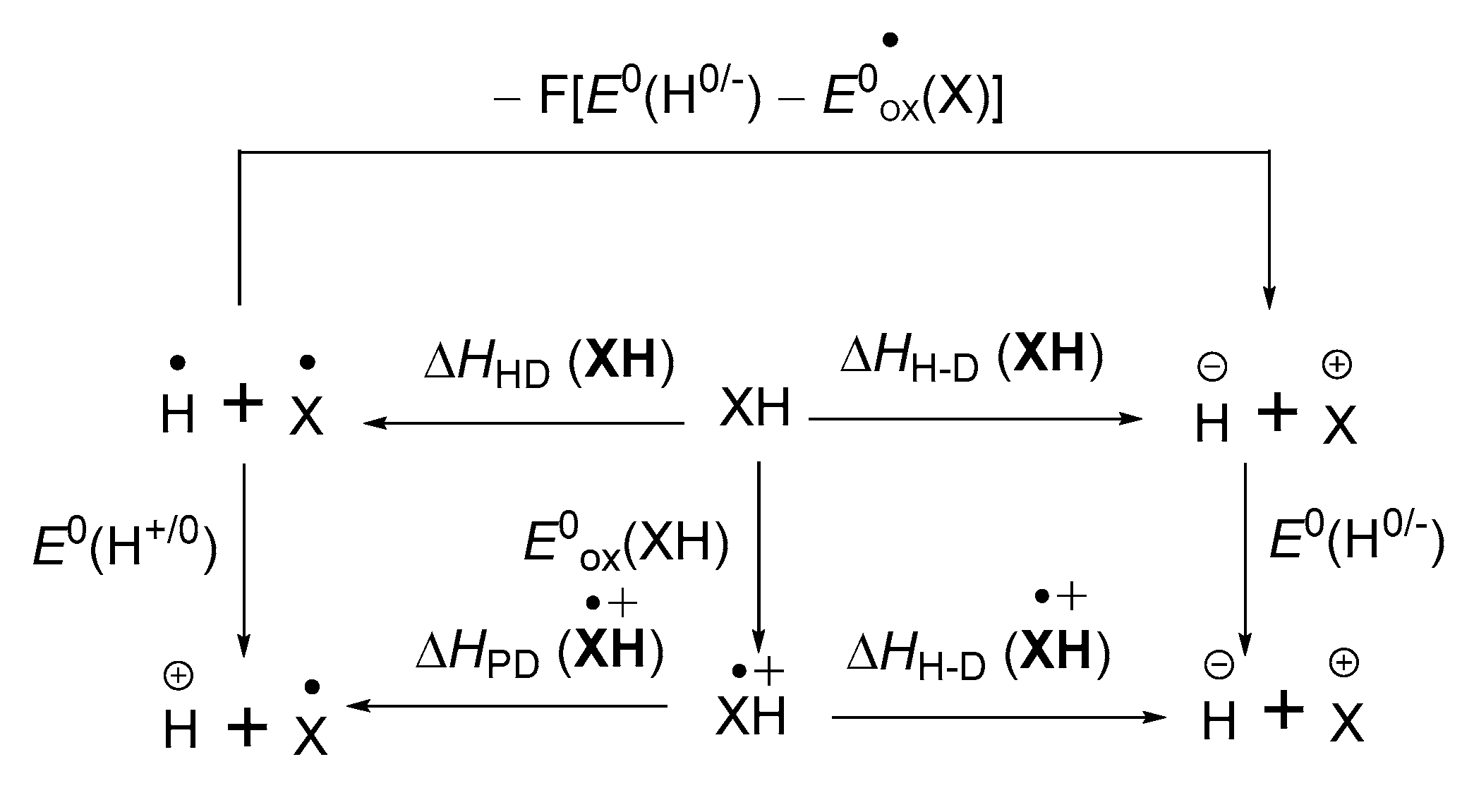{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituent | ∆Hrxn a | Eredb | ||||||
|---|---|---|---|---|---|---|---|---|
| ∆HH-A2 | ∆HH-A4 | CV | OSWV | CV | OSWV | CV | OSWV | |
| A | Ered(A) | Ered(A′H2) | Ered(AH2) | |||||
| CH3O | −70.68 | −75.35 | −1.920 | −1.899 | −0.672 | −0.700 | −0.740 | −0.787 |
| CH3 | −65.26 | −73.36 | −1.913 | −1.889 | −0.660 | −0.680 | −0.726 | −0.763 |
| H | −62.24 | −72.09 | −1.876 | −1.835 | −0.609 | −0.644 | −0.690 | −0.743 |
| Cl | −52.27 | −64.93 | −1.798 | −1.771 | −0.587 | −0.612 | −0.671 | −0.719 |
| B | Ered(B) | Ered(B′H2) | Ered(BH2) c | |||||
| CH3O | −72.65 | −77.35 | −1.929 | −1.904 | −0.651 | −0.692 | −0.723 | −0.781 |
| CH3 | −66.01 | −74.64 | −1.901 | −1.876 | −0.643 | −0.672 | −0.714 | −0.761 |
| H | −62.24 | −72.09 | −1.876 | −1.835 | −0.609 | −0.644 | −0.690 | −0.743 |
| Cl | −53.27 | −69.33 | −1.811 | −1.784 | −0.577 | −0.608 | −0.682 | −0.717 |
| BH2 | E′red(BH2) c | Ered(BH4) | ||||||
| CH3O | −86.24 | −2.586 | −2.595 | −0.500 | −0.538 | |||
| CH3 | −81.55 | −2.509 | −2.515 | −0.489 | −0.526 | |||
| H | −76.10 | −2.437 | −2.439 | −0.478 | −0.510 | |||
| Cl | −64.90 | −2.244 | −2.215 | −0.457 | −0.486 | |||
| Position | 2 | 4 | 2 | 4 | 2 | 4 | 2 | 4 |
|---|---|---|---|---|---|---|---|---|
| Substituent | ∆HH-A(X) a | ∆HHA(X) b | ∆HHA(X•−) b | ∆HPA(X•−) b | ||||
| A | ||||||||
| CH3O | −34.33 | −29.66 | −24.24 | −21.58 | −51.93 | −47.26 | −14.81 | −12.15 |
| CH3 | −39.75 | −31.65 | −29.19 | −23.01 | −57.12 | −49.02 | −19.54 | −13.35 |
| H | −42.77 | −32.92 | −31.38 | −23.82 | −58.89 | −49.04 | −20.48 | −12.92 |
| Cl | −52.74 | −40.08 | −40.61 | −30.42 | −67.39 | −54.73 | −28.23 | −18.04 |
| B | ||||||||
| CH3O | −32.36 | −27.66 | −22.08 | −19.44 | −50.08 | −45.38 | −12.77 | −10.13 |
| CH3 | −39.00 | −30.37 | −28.26 | −21.68 | −56.07 | −47.44 | −18.30 | −11.73 |
| H | −42.77 | −32.92 | −31.38 | −23.82 | −58.89 | −49.04 | −20.48 | −12.92 |
| Cl | −51.74 | −35.68 | −39.52 | −25.98 | −66.69 | −50.63 | −27.44 | −13.90 |
| BH2 | ||||||||
| CH3O | −18.77 | −4.93 | −52.45 | 3.22 | −11.59 | −14.81 | ||
| CH3 | −23.46 | −9.35 | −55.29 | 5.39 | −14.15 | −19.54 | ||
| H | −28.91 | −14.43 | −58.99 | 3.00 | −17.48 | −20.48 | ||
| Cl | −40.11 | −25.07 | −65.01 | 5.28 | −22.95 | −28.23 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.-L.; Jing, S.; Zhu, X.-Q. Thermodynamics Evaluation of Selective Hydride Reduction for α,β-Unsaturated Carbonyl Compounds. Molecules 2023, 28, 2862. https://doi.org/10.3390/molecules28062862
Chen B-L, Jing S, Zhu X-Q. Thermodynamics Evaluation of Selective Hydride Reduction for α,β-Unsaturated Carbonyl Compounds. Molecules. 2023; 28(6):2862. https://doi.org/10.3390/molecules28062862
Chicago/Turabian StyleChen, Bao-Long, Sha Jing, and Xiao-Qing Zhu. 2023. "Thermodynamics Evaluation of Selective Hydride Reduction for α,β-Unsaturated Carbonyl Compounds" Molecules 28, no. 6: 2862. https://doi.org/10.3390/molecules28062862
APA StyleChen, B.-L., Jing, S., & Zhu, X.-Q. (2023). Thermodynamics Evaluation of Selective Hydride Reduction for α,β-Unsaturated Carbonyl Compounds. Molecules, 28(6), 2862. https://doi.org/10.3390/molecules28062862