

Switching from Aromatase Inhibitors to Dual Targeting Flavonoid-Based Compounds for Breast Cancer Treatment

 ,
,  , ,
, ,  ,
,  ,
,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Results
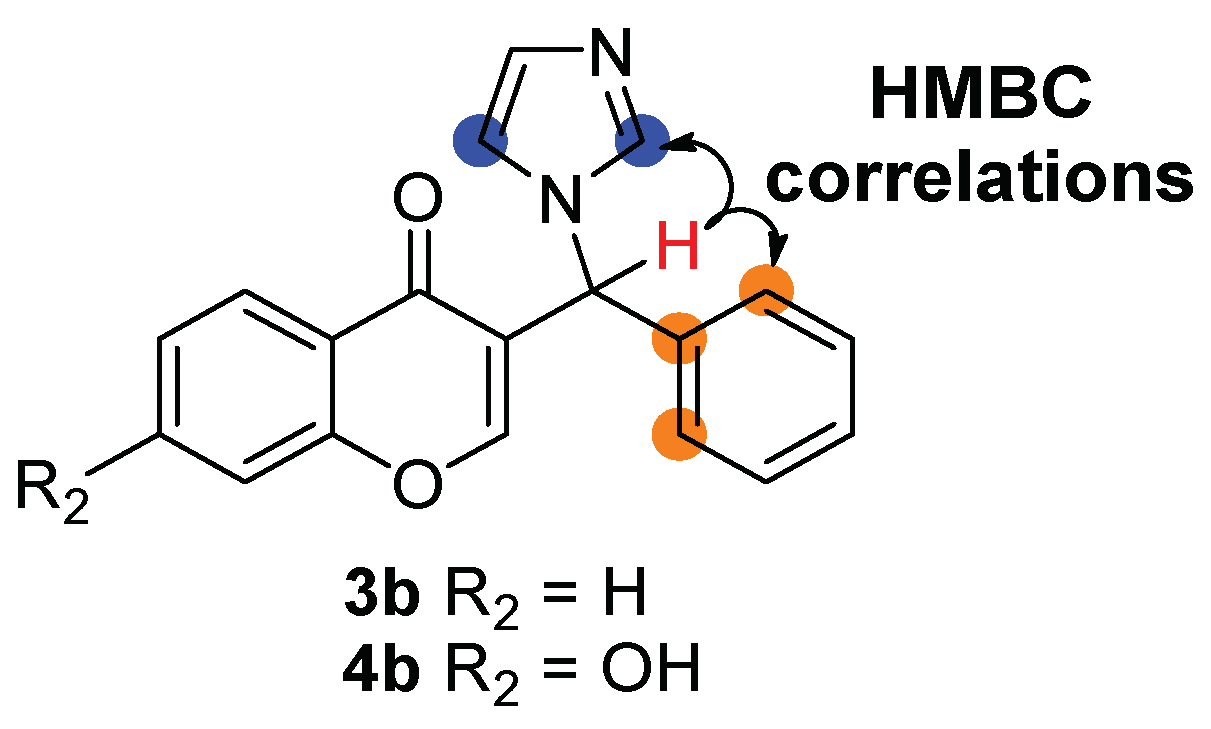
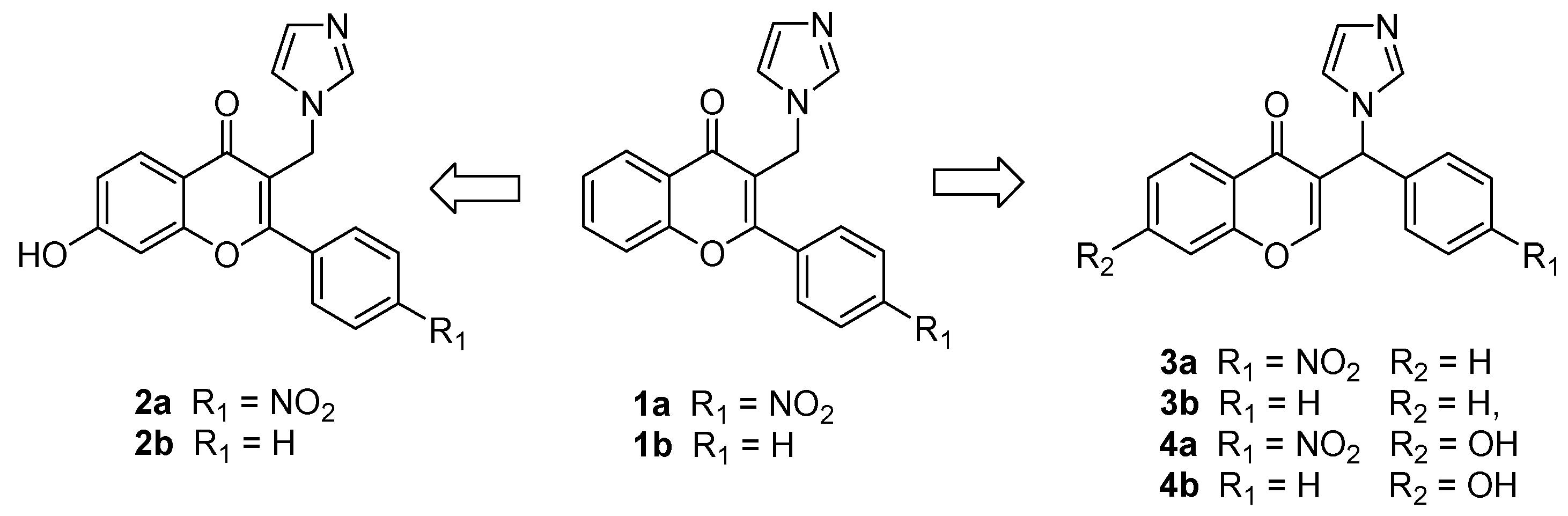
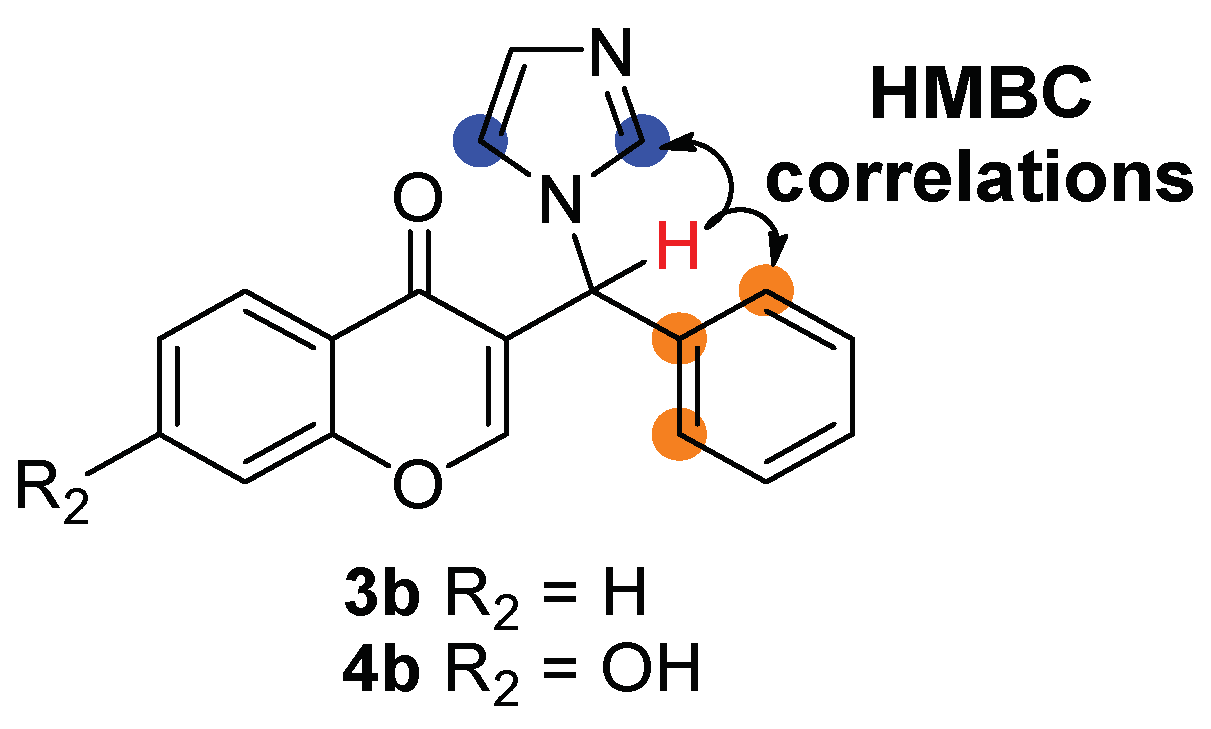
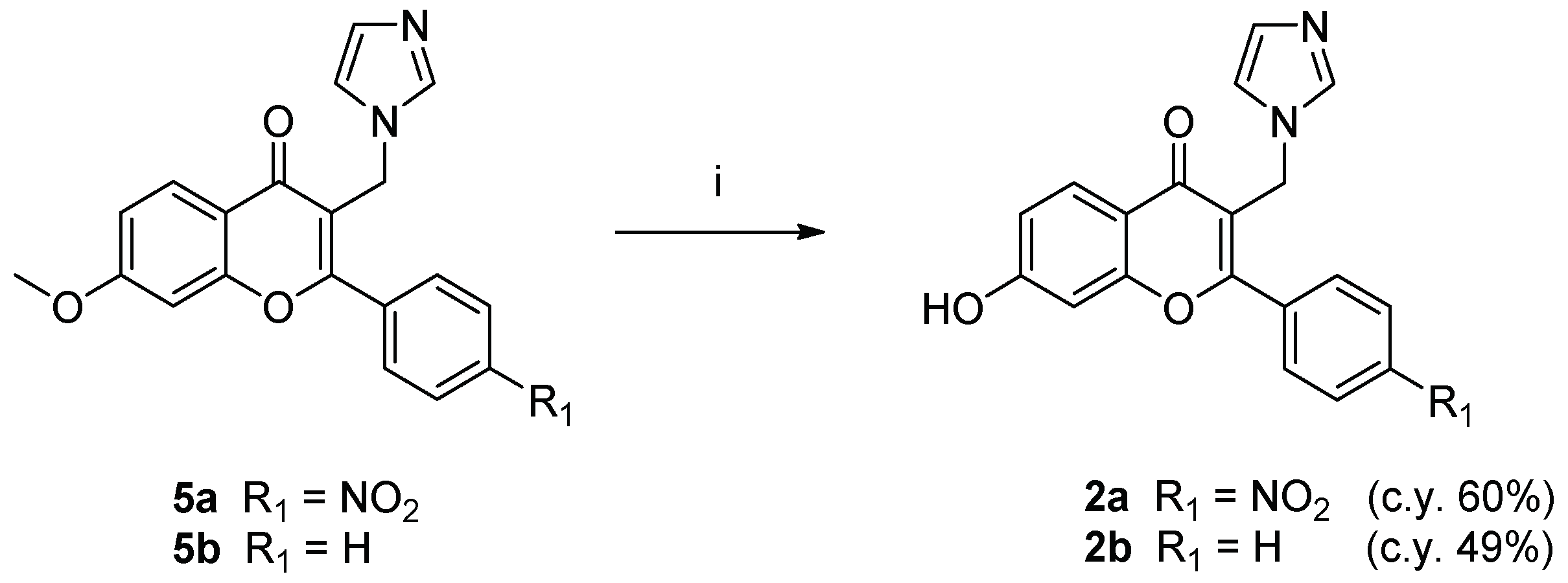
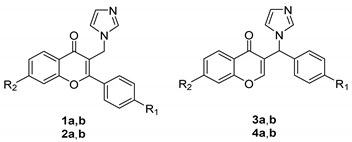
2.1. Chemistry
2.2. Biological Evaluation
2.3. Molecular Dynamics Simulations
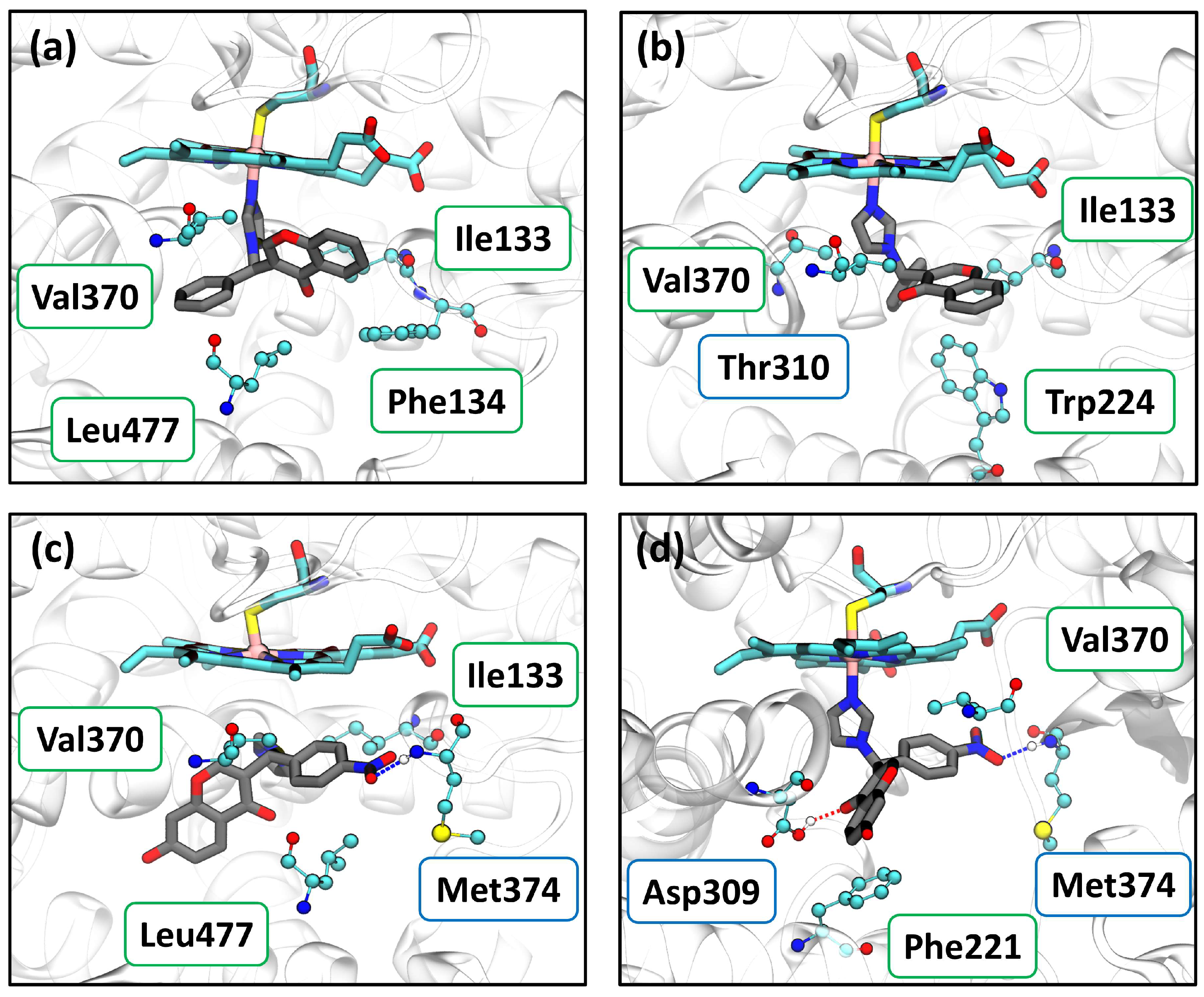
2.3.1. Aromatase
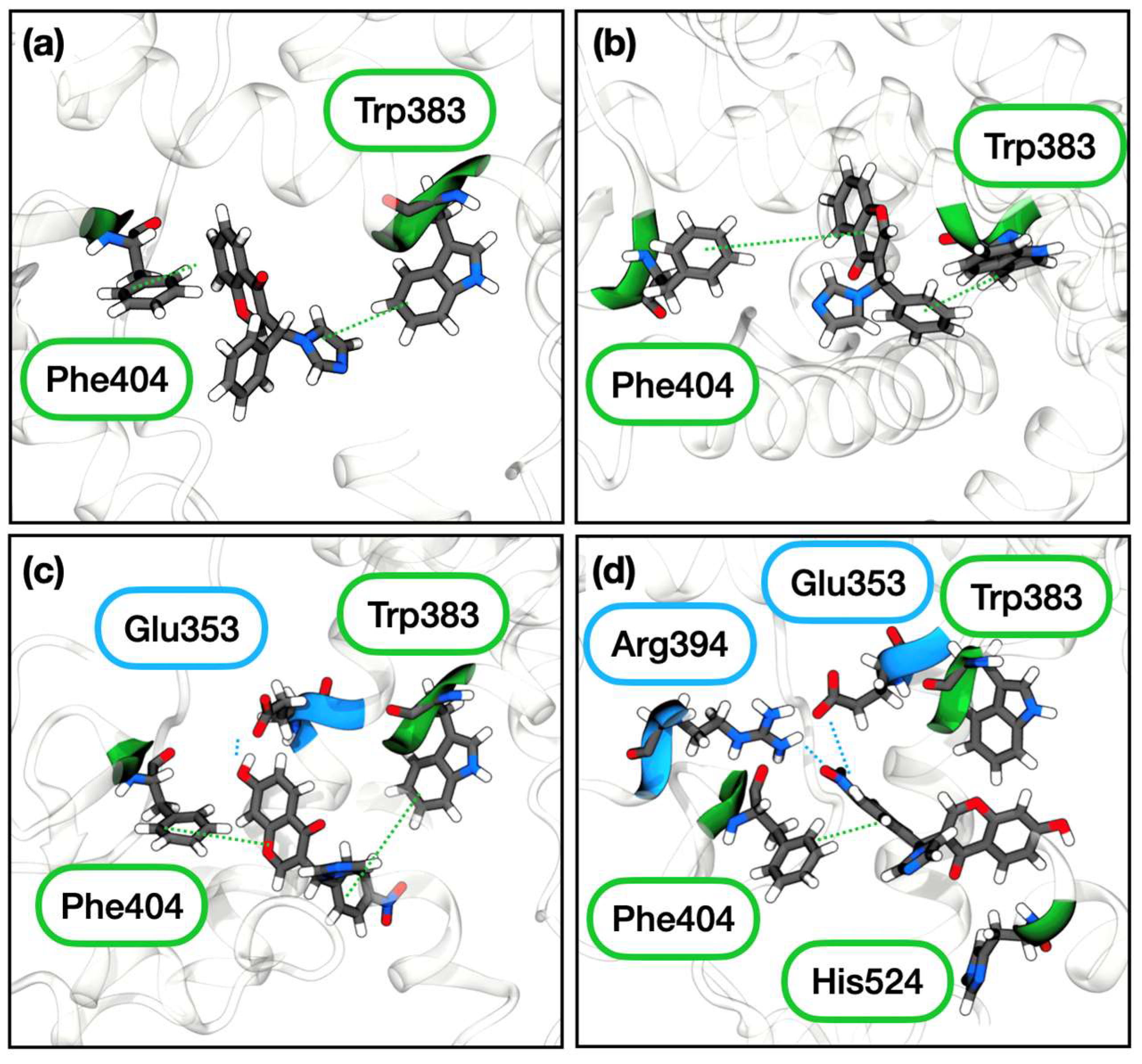
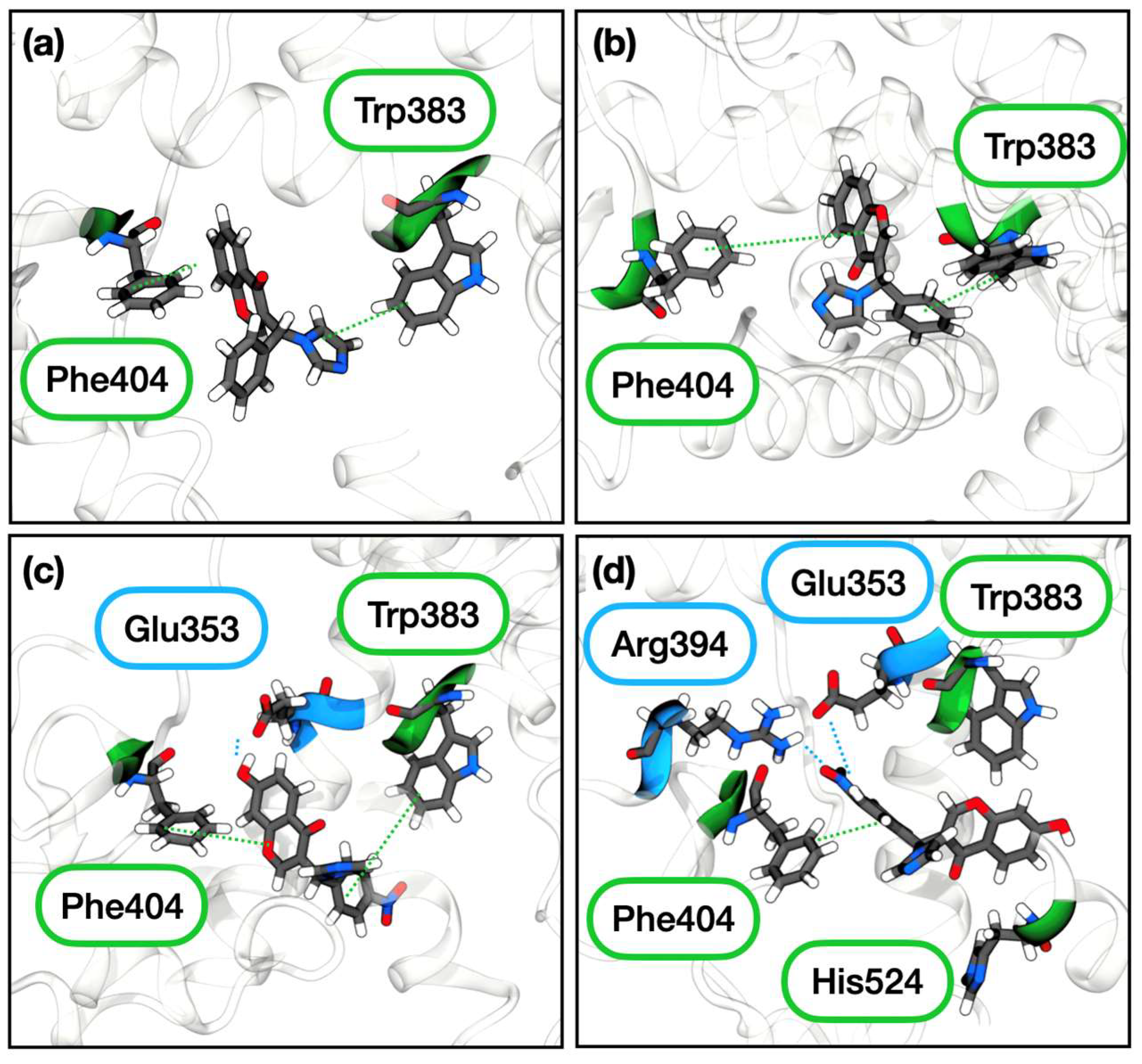
2.3.2. Estrogen Receptor α
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Materials and Methods
4.1.2. General Procedure I. (Synthesis of Compounds 2a,b)
4.1.3. Synthesis of 7-(methoxymethoxy)chroman-4-one (7)
4.1.4. General Procedure II. (Synthesis of Compounds 8a,b)
4.1.5. General Procedure III (Synthesis of Compounds 9a,b and 3b)
4.1.6. General Procedure IV. (Synthesis of Compounds 4a,b)
4.2. Biological Evaluation
4.2.1. Aromatase Inhibition Assay
4.2.2. Estrogen Receptor α Binding Assay
4.3. Computational Details
4.3.1. Docking Calculations
4.3.2. Model Building
4.3.3. Classical MD Simulations
4.3.4. QM/MM Molecular Dynamics Simulations
4.3.5. Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- American Cancer Society. Cancer Facts & Figures 2022; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current medical treatment of estrogen receptor-positive breast cancer. World J. Biol. Chem. 2015, 6, 231–239. [Google Scholar] [CrossRef]
- Gasco, M.; Argusti, A.; Bonanni, B.; Decensi, A. SERMs in chemoprevention of breast cancer. Eur. J. Cancer 2005, 41, 1980–1989. [Google Scholar] [CrossRef]
- Baumann, C.K.; Castiglione-Gertsch, M. Estrogen receptor modulators and down regulators: Optimal use in postmenopausal women with breast cancer. Drugs 2007, 67, 2335–2353. [Google Scholar] [CrossRef] [PubMed]
- Olson, E. Combination Therapies in Advanced, Hormone Receptor-Positive Breast Cancer. J. Adv. Pract. Oncol. 2018, 9, 43–54. [Google Scholar]
- Saatci, O.; Huynh-Dam, K.T.; Sahin, O. Endocrine resistance in breast cancer: From molecular mechanisms to therapeutic strategies. J. Mol. Med. 2021, 99, 1691–1710. [Google Scholar] [CrossRef]
- Ozyurt, R.; Ozpolat, B. Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies. Cancers 2022, 14, 5206. [Google Scholar] [CrossRef]
- Baum, M.; Buzdar, A.; Cuzick, J.; Forbes, J.; Houghton, J.; Howell, A.; Sahmoud, T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: Results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer 2003, 98, 1802–1810. [Google Scholar]
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J.F.; Hoctin-Boes, G.; Houghton, J.; Locker, G.Y.; Tobias, J.S.; et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005, 365, 60–62. [Google Scholar] [CrossRef]
- Jelovac, D.; Macedo, L.; Goloubeva, O.G.; Handratta, V.; Brodie, A.M. Additive antitumor effect of aromatase inhibitor letrozole and antiestrogen fulvestrant in a postmenopausal breast cancer model. Cancer Res. 2005, 65, 5439–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.J.; Desta, Z.; Flockhart, D.A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 131, 473–481. [Google Scholar] [CrossRef]
- Ferreira Almeida, C.; Oliveira, A.; João Ramos, M.; Fernandes, P.A.; Teixeira, N.; Amaral, C. Estrogen receptor-positive (ER+) breast cancer treatment: Are multi-target compounds the next promising approach? Biochem. Pharmacol. 2020, 177, 113989. [Google Scholar] [CrossRef]
- Lv, W.; Liu, J.; Skaar, T.C.; Flockhart, D.A.; Cushman, M. Design and synthesis of norendoxifen analogues with dual aromatase inhibitory and estrogen receptor modulatory activities. J. Med. Chem. 2015, 58, 2623–2648. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Liu, J.; Skaar, T.C.; O’Neill, E.; Yu, G.; Flockhart, D.A.; Cushman, M. Synthesis of Triphenylethylene Bisphenols as Aromatase Inhibitors That Also Modulate Estrogen Receptors. J. Med. Chem. 2016, 59, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Caciolla, J.; Martini, S.; Spinello, A.; Pavlin, M.; Turrini, E.; Simonelli, F.; Belluti, F.; Rampa, A.; Bisi, A.; Fimognari, C.; et al. Balanced dual acting compounds targeting aromatase and estrogen receptor α as an emerging therapeutic opportunity to counteract estrogen responsive breast cancer. Eur. J. Med. Chem. 2021, 224, 113733. [Google Scholar] [CrossRef]
- Almeida, C.F.; Teixeira, N.; Oliveira, A.; Augusto, T.V.; Correia-da-Silva, G.; Ramos, M.J.; Fernandes, P.A.; Amaral, C. Discovery of a multi-target compound for estrogen receptor-positive (ER+) breast cancer: Involvement of aromatase and ERs. Biochimie 2021, 181, 65–76. [Google Scholar] [CrossRef]
- Amaral, C.; Correia-da-Silva, G.; Almeida, C.F.; Valente, M.J.; Varela, C.; Tavares-da-Silva, E.; Vinggaard, A.M.; Teixeira, N.; Roleira, F.M.F. An Exemestane Derivative, Oxymestane-D1, as a New Multi-Target Steroidal Aromatase Inhibitor for Estrogen Receptor-Positive (ER+) Breast Cancer: Effects on Sensitive and Resistant Cell Lines. Molecules 2023, 28, 789. [Google Scholar] [CrossRef]
- Gobbi, S.; Cavalli, A.; Rampa, A.; Belluti, F.; Piazzi, L.; Paluszcak, A.; Hartmann, R.W.; Recanatini, M.; Bisi, A. Lead optimization providing a series of flavone derivatives as potent nonsteroidal inhibitors of the cytochrome P450 aromatase enzyme. J. Med. Chem. 2006, 49, 4777–4780. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Yearley, E.J.; Zhurova, E.A.; Zhurov, V.V.; Pinkerton, A.A. Binding of genistein to the estrogen receptor based on an experimental electron density study. J. Am. Chem. Soc. 2007, 129, 15013–15021. [Google Scholar] [CrossRef]
- Cavalli, A.; Bisi, A.; Bertucci, C.; Rosini, C.; Paluszcak, A.; Gobbi, S.; Giorgio, E.; Rampa, A.; Belluti, F.; Piazzi, L.; et al. Enantioselective Nonsteroidal Aromatase Inhibitors Identified through a Multidisciplinary Medicinal Chemistry Approach. J. Med. Chem. 2005, 48, 7282–7289. [Google Scholar] [CrossRef]
- Valkonen, A.; Laihia, K.; Kolehmainen, E.; Kauppinen, R.; Perjési, P. Structural studies of seven homoisoflavonoids, six thiohomoisoflavonoids, and four structurally related compounds. Struct. Chem. 2012, 23, 9. [Google Scholar] [CrossRef]
- Lucas, S.; Heim, R.; Negri, M.; Antes, I.; Ries, C.; Schewe, K.E.; Bisi, A.; Gobbi, S.; Hartmann, R.W. Novel aldosterone synthase inhibitors with extended carbocyclic skeleton by a combined ligand-based and structure-based drug design approach. J. Med. Chem. 2008, 51, 6138–6149. [Google Scholar] [CrossRef]
- Janoš, P.; Spinello, A.; Magistrato, A. All-atom simulations to studying metallodrugs/target interactions. Curr. Opin. Chem. Biol. 2020, 61, 1–8. [Google Scholar] [CrossRef]
- Spinello, A.; Borišek, J.; Pavlin, M.; Janoš, P.; Magistrato, A. Computing Metal-Binding Proteins for Therapeutic Benefit. ChemMedChem 2021, 16, 2034–2049. [Google Scholar] [CrossRef]
- Caciolla, J.; Spinello, A.; Martini, S.; Bisi, A.; Zaffaroni, N.; Gobbi, S.; Magistrato, A. Targeting Orthosteric and Allosteric Pockets of Aromatase via Dual-Mode Novel Azole Inhibitors. ACS Med. Chem. Lett. 2020, 11, 732–739. [Google Scholar] [CrossRef]
- Caciolla, J.; Martini, S.; Spinello, A.; Belluti, F.; Bisi, A.; Zaffaroni, N.; Magistrato, A.; Gobbi, S. Single-digit nanomolar inhibitors lock the aromatase active site via a dualsteric targeting strategy. Eur. J. Med. Chem. 2022, 244, 114802. [Google Scholar] [CrossRef]
- Egbuta, C.; Lo, J.; Ghosh, D. Mechanism of inhibition of estrogen biosynthesis by azole fungicides. Endocrinology 2014, 155, 4622–4628. [Google Scholar] [CrossRef]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme? Chemistry 2018, 24, 10840–10849. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Pavlin, M.; Spinello, A.; Pennati, M.; Zaffaroni, N.; Gobbi, S.; Bisi, A.; Colombo, G.; Magistrato, A. A Computational Assay of Estrogen Receptor α Antagonists Reveals the Key Common Structural Traits of Drugs Effectively Fighting Refractory Breast Cancers. Sci. Rep. 2018, 8, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauzá, A.; Frontera, A.; Mooibroek, T.J. π-Hole Interactions Involving Nitro Aromatic Ligands in Protein Structures. Chemistry 2019, 25, 13436–13443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Di Nardo, G.; Breitner, M.; Bandino, A.; Ghosh, D.; Jennings, G.K.; Hackett, J.C.; Gilardi, G. Evidence for an elevated aspartate pK(a) in the active site of human aromatase. J. Biol. Chem. 2015, 290, 1186–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalon, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Schmit, J.D.; Kariyawasam, N.L.; Needham, V.; Smith, P.E. SLTCAP: A Simple Method for Calculating the Number of Ions Needed for MD Simulation. J. Chem. Theory Comput. 2018, 14, 1823–1827. [Google Scholar] [CrossRef]
- Joung, I.; Cheatham, T. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, L.M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Shirts, M.R.; Klein, C.; Swails, J.M.; Yin, J.; Gilson, M.K.; Mobley, D.L.; Case, D.A.; Zhong, E.D. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput. Aided Mol. Des. 2017, 31, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1998, 18, 10. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Palermo, G.; Spinello, A.; Saha, A.; Magistrato, A. Frontiers of metal-coordinating drug design. Expert Opin. Drug Discov. 2021, 16, 497–511. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Sgrignani, J.; Iannuzzi, M.; Magistrato, A. Role of Water in the Puzzling Mechanism of the Final Aromatization Step Promoted by the Human Aromatase Enzyme. Insights from QM/MM MD Simulations. J. Chem. Inf. Model. 2015, 55, 2218–2226. [Google Scholar] [CrossRef]
- Ritacco, I.; Spinello, A.; Ippoliti, E.; Magistrato, A. Post-Translational Regulation of CYP450s Metabolism As Revealed by All-Atoms Simulations of the Aromatase Enzyme. J. Chem. Inf. Model. 2019, 59, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Nose, S.A. Unified Formulation of the Constant Temperature Molecular-Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Spinello, A.; Ritacco, I.; Magistrato, A. The Catalytic Mechanism of Steroidogenic Cytochromes P450 from All-Atom Simulations: Entwinement with Membrane Environment, Redox Partners, and Post-Transcriptional Regulation. Catalysts 2019, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Bouysset, C.; Fiorucci, S. ProLIF: A library to encode molecular interactions as fingerprints. J. Cheminform. 2021, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Van Gunsteren, W.F.; Mark, A.E. Folding–unfolding thermodynamics of a β-heptapeptide from equilibrium simulations. Proteins 1999, 34, 147. [Google Scholar] [CrossRef]
- Miller, B.; McGee, T.; Swails, J.; Homeyer, N.; Gohlke, H.; Roitberg, A. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Borisek, J.; Saltalamacchia, A.; Spinello, A.; Magistrato, A. Exploiting Cryo-EM Structural Information and All-Atom Simulations to Decrypt the Molecular Mechanism of Splicing Modulators. J. Chem. Inf. Model. 2020, 60, 2510–2521. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | R2 | R1 | AR Inhibition IC50 μM 1 | ERα Binding IC50 μM 1 |
| 1a | H | NO2 | 0.045 2 | >10 |
| 1b | H | H | 0.072 2 | >10 |
| 2a | OH | NO2 | 2.1 | >10 |
| 2b | OH | H | 4.0 | >10 |
| 3a 3 | H | NO2 | 0.063 | >10 |
| 3b | H | H | 0.50 | 0.310 |
| 4a | OH | NO2 | 0.122 | 0.595 |
| 4b | OH | H | 2.2 | 0.203 |
| Letrozole | - | - | 0.005 | >10 |
| Endoxifen | - | - | >10 | 0.043 |
| IC50 (µm) | Distance (Fe-N) Å | Angle (Planes) Deg | |
|---|---|---|---|
| LTZ | 0.010 | 2.33 ± 0.15 | 91.8 ± 2.7 |
| R-3b | 0.50 1 | 2.23 ± 0.10 | 94.7 ± 2.2 |
| S-3b | 0.50 1 | 2.16 ± 0.08 | 87.0 ± 2.2 |
| R-4a | 0.122 1 | - | - |
| S-4a | 0.122 1 | 2.30 ± 0.12 | 93.2 ± 2.7 |
| R-3b | S-3b | R-4a | S-4a | |
|---|---|---|---|---|
| IC50s | 0.50 | 0.12 | ||
| MM-GBSA ΔGb | −10.95 ± 0.93 | −10.74 ± 1.05 | −18.24 ± 2.06 | −14.73 ± 0.96 |
| Average | −10.84 | −16.48 | ||
| ΔGb per-residue | ||||
| Arg115 | −0.70 ± 0.08 | −0.54 ± 0.03 | −0.95 ± 0.04 | −1.15 ± 0.03 |
| Ile133 | −1.77 ± 0.03 | −1.53 ± 0.04 | −1.13 ± 0.03 | −1.27 ± 0.02 |
| Phe134 | −1.10 ± 0.04 | −0.99 ± 0.03 | −0.65 ± 0.02 | −0.76 ± 0.02 |
| Phe221 | −0.57 ± 0.03 | −0.32 ± 0.02 | −1.07 ± 0.03 | −1.52 ± 0.04 |
| Trp224 | −0.89 ± 0.03 | −1.52 ± 0.03 | −0.68 ± 0.02 | −1.01 ± 0.03 |
| Asp309 | −0.16 ± 0.03 | −0.26 ± 0.03 | −0.60 ± 0.04 | −1.33 ± 0.07 |
| Thr310 | −0.46 ± 0.05 | −1.42 ± 0.03 | −1.14 ± 0.05 | −1.03 ± 0.06 |
| Val370 | −1.75 ± 0.04 | −1.49 ± 0.05 | −1.90 ± 0.04 | −1.28 ± 0.03 |
| Val373 | −0.07. ± 0.03 | −0.14 ± 0.01 | −1.13 ± 0.02 | −1.30 ± 0.02 |
| Met374 | −0.90 ± 0.05 | −0.59 ± 0.02 | −1.18 ± 0.02 | −1.15 ± 0.02 |
| Leu477 | −1.64 ± 0.03 | −0.81 ± 0.04 | −1.35 ± 0.03 | −1.23 ± 0.03 |
| R-3b | S-3b | R-4a | S-4a | |
|---|---|---|---|---|
| Glu353 | 92 | 90 | ||
| Trp383 | 29 | 51 | 31 | 15 |
| Arg394 | 98 | |||
| Phe404 | 41 | 18 | 39 | 62 |
| His524 | 17 |
| R-3b | S-3b | R-4a | S-4a | |
|---|---|---|---|---|
| IC50 | 0.310 | 0.595 | ||
| Docking score | −9.84 | −8.68 | −10.49 | −8.96 |
| MM-GBSA ΔGb | −29.36 ± 0.26 | −27.64 ± 0.38 | −30.17 ± 0.37 | −48.03 ± 0.30 |
| Average | −28.50 ± 0.46 | −39.10 ± 0.48 | ||
| ΔGb, Per-Residue Decomposition | ||||
| Met343 | −0.66 ± 0.03 | |||
| Leu346 | −0.72 ± 0.05 | −1.50 ± 0.12 | −0.88 ± 0.05 | −2.00 ± 0.05 |
| Thr347 | −1.74 ± 0.06 | −0.63 ± 0.05 | −0.71 ± 0.04 | −1.27 ± 0.03 |
| Leu349 | −0.51 ± 0.03 | −0.56 ± 0.03 | −1.07 ± 0.03 | −0.97 ± 0.02 |
| Ala350 | −0.88 ± 0.03 | −1.31 ± 0.05 | −1.81 ± 0.05 | −1.43 ± 0.03 |
| Glu353 | −2.04 ± 0.05 | −0.74 ± 0.12 | −1.58 ± 0.09 | |
| Trp383 | −0.90 ± 0.05 | −0.90 ± 0.04 | ||
| Leu384 | −1.06 ± 0.05 | −1.02 ± 0.05 | −1.69 ± 0.04 | −0.63 ± 0.03 |
| Leu387 | −2.50 ± 0.06 | −1.60 ± 0.06 | −0.96 ± 0.05 | −1.71 ± 0.04 |
| Met388 | −1.33 ± 0.06 | −0.72 ± 0.05 | −0.65 ± 0.04 | −0.59 ± 0.03 |
| Leu391 | −1.00 ± 0.04 | −0.57 ± 0.07 | −0.89 ± 0.04 | −0.93 ± 0.02 |
| Phe404 | −1.96 ± 0.07 | −0.69 ± 0.07 | −1.15 ± 0.03 | −1.54 ± 0.03 |
| Val418 | −0.51 ± 0.03 | −1.11 ± 0.09 | ||
| Glu419 | −0.96 ± 0.20 | |||
| Gly420 | −0.61 ± 0.06 | |||
| Met421 | −1.61 ± 0.06 | −1.69 ± 0.07 | −1.76 ± 0.06 | −0.90 ± 0.03 |
| Met522 | −0.51 ± 0.04 | |||
| Leu525 | −1.11 ± 0.05 | −0.83 ± 0.04 | −2.12 ± 0.05 | −1.94 ± 0.05 |
| Met528 | −1.05 ± 0.08 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gobbi, S.; Martini, S.; Rozza, R.; Spinello, A.; Caciolla, J.; Rampa, A.; Belluti, F.; Zaffaroni, N.; Magistrato, A.; Bisi, A. Switching from Aromatase Inhibitors to Dual Targeting Flavonoid-Based Compounds for Breast Cancer Treatment. Molecules 2023, 28, 3047. https://doi.org/10.3390/molecules28073047
Gobbi S, Martini S, Rozza R, Spinello A, Caciolla J, Rampa A, Belluti F, Zaffaroni N, Magistrato A, Bisi A. Switching from Aromatase Inhibitors to Dual Targeting Flavonoid-Based Compounds for Breast Cancer Treatment. Molecules. 2023; 28(7):3047. https://doi.org/10.3390/molecules28073047
Chicago/Turabian StyleGobbi, Silvia, Silvia Martini, Riccardo Rozza, Angelo Spinello, Jessica Caciolla, Angela Rampa, Federica Belluti, Nadia Zaffaroni, Alessandra Magistrato, and Alessandra Bisi. 2023. "Switching from Aromatase Inhibitors to Dual Targeting Flavonoid-Based Compounds for Breast Cancer Treatment" Molecules 28, no. 7: 3047. https://doi.org/10.3390/molecules28073047
APA StyleGobbi, S., Martini, S., Rozza, R., Spinello, A., Caciolla, J., Rampa, A., Belluti, F., Zaffaroni, N., Magistrato, A., & Bisi, A. (2023). Switching from Aromatase Inhibitors to Dual Targeting Flavonoid-Based Compounds for Breast Cancer Treatment. Molecules, 28(7), 3047. https://doi.org/10.3390/molecules28073047