
Impact of Ultrasonication on the Self-Assembly Behavior and Gel Properties of Bovine Bone Collagen I
 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Collagen Self-Assembly Behavior
2.1.1. Turbidity Assay
2.1.2. Fibrillogenesis Degree
2.2. Microstructure of Ultrasonic Treatment Collagen
2.2.1. SDS-PAGE
2.2.2. Fourier Transform Infrared Spectroscopy (FTIR)
2.2.3. X-ray Diffraction Spectra (XRD)
2.2.4. Fluorescence Emission Spectra
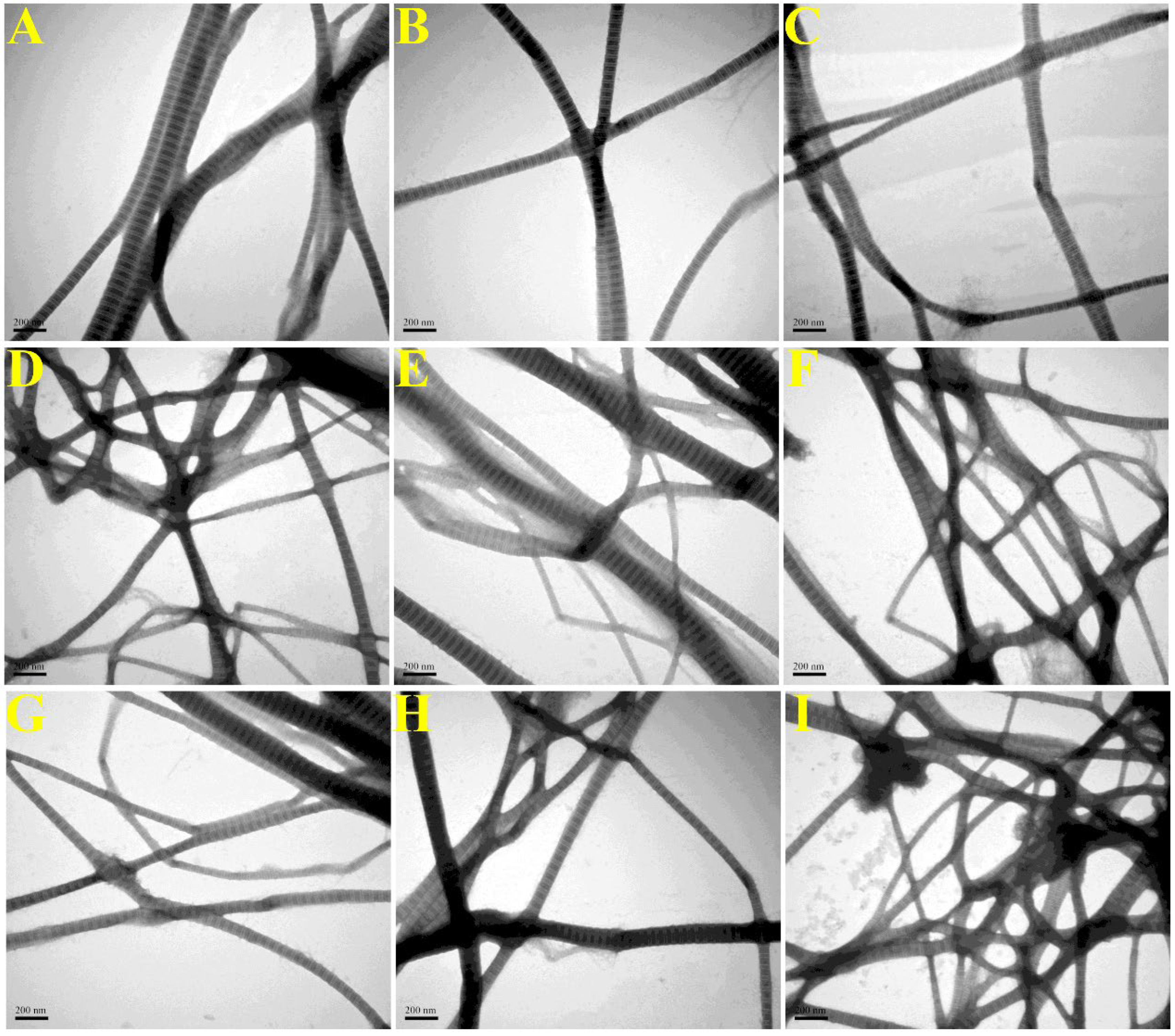
2.2.5. Collagen Fibril Morphology (SEM/TEM)
2.3. Thermal Stability of Collagen Fibril Gels
2.3.1. Differential Scanning Calorimetry (DSC)
2.3.2. Thermo-Gravimetric Analysis (TGA)
2.4. Viscoelasticity of Collagen Fibril Gels (Dynamic Frequency Sweep Test)
2.5. Gel Strength
2.6. Nitrogen Adsorption of Collagen Fibril Gels
3. Materials and Methods
3.1. Raw Materials and Chemical Reagents
3.2. Ultrasonic Treatment on Collagen
3.3. Turbidity Assay
3.4. Fibrillogenesis Degree
3.5. Microstructure of Ultrasonic Treatment Collagen
3.5.1. Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
3.5.2. Fourier Transform Infrared Spectroscopy (FT-IR)
3.5.3. X-ray Diffraction (XRD)
3.5.4. Fluorescence Emission Spectra
3.6. Microscopic Structure of Collagen Fibrils (SEM/TEM)
3.7. Preparation of Collagen Fibril Gels
3.8. Thermal Stability of Collagen Fibril Gels
3.8.1. Differential Scanning Calorimetry (DSC)
3.8.2. Thermogravimetric Analysis (TGA)
3.9. Collagen Fibril Gel Strength
3.10. Viscoelasticity Properties of Collagen Fibril Gels
3.11. Nitrogen Adsorption (BET) of Collagen Fibril Gels
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qin, L.; Bi, J.R.; Li, D.M.; Dong, M.; Zhao, Z.Y.; Dong, X.P.; Zhou, D.Y.; Zhu, B.W. Unfolding/refolding study on collagen from sea cucumber based on 2D fourier transform infrared spectroscopy. Molecules 2016, 21, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasekaran, D.; Thada, R.; Jeyakumar, G.F.S.; Manimegalai, N.P.; Shanmugam, G.; Sivagnanam, U.T. Physicochemical characterization and self-assembly of human amniotic membrane and umbilical cord collagen: A comparative study. Int. J. Biol. Macromol. 2020, 165, 2920–2933. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.E.; Xu, P.; Li, P.; Cai, P.; Zhang, M.; Sun, Z.L.; Sun, C.; Xu, W.M.; Wang, D.Y. Effect of ultrasound pre-treatment on the characterization and properties of collagen extracted from soft-shelled turtle (Pelodiscus sinensis). LWT Food Sci. Technol. 2017, 82, 72–81. [Google Scholar] [CrossRef]
- Tian, H.H.; Ren, Z.Y.; Shi, L.F.; Hao, G.X.; Chen, J.; Weng, W.Y. Self-assembly characterization of tilapia skin collagen in simulated body fluid with different salt concentrations. Process Biochem. 2021, 108, 153–160. [Google Scholar] [CrossRef]
- Ju, H.Y.; Liu, X.Y.; Zhang, G.; Liu, D.Z.; Yang, Y.S. Comparison of the structural characteristics of native collagen fibrils derived from bovine tendons using two different methods: Modified acid-solubilized and pepsin-aided extraction. Materials 2020, 13, 358. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.F.; Gao, Y.F.; Shao, J.H.; Tumarbekova, A.; Zhang, D.Q.; Zhu, J. Effects of ultrasound and thermal treatment on the ultrastructure of collagen fibers from bovine tendon using atomic force microscopy. Food Chem. 2021, 347, 128985. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, H.B.; Deng, M.X.; Wang, Z.W.; Zhang, J.T.; Wang, H.Y.; Zhang, H.J. Effect of ultrasonication on the fibril-formation and gel properties of collagen from grass carp skin. Mater. Sci. Eng. C 2016, 59, 1038–1046. [Google Scholar] [CrossRef]
- Akram, A.N.; Zhang, C.H. Extraction of collagen-II with pepsin and ultrasound treatment from chicken sternal cartilage; physicochemical and functional properties. Ultrason Sonochem. 2020, 64, 105053. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.N.; Zhang, C.H. Effect of ultrasonication on the yield, functional and physicochemical characteristics of collagen-II from chicken sternal cartilage. Food Chem. 2020, 307, 125544. [Google Scholar] [CrossRef]
- Ye, Y.K.; Dai, S.Q.; Zhang, H.Y.; He, S.D.; Hu, W.W.; Cao, X.D.; Wei, Z.J. Ultrasound-Assisted Preparation of Maillard Reaction Products Derived from Hydrolyzed Soybean Meal with Meaty Flavor in an Oil-In-Water System. Molecules 2022, 27, 7236. [Google Scholar] [CrossRef]
- Ata, O.; Kumcuoglu, S.; Tavman, S. Effects of sonication on the extraction of pepsin-soluble collagens from lamb feet and product characterization. LWT Food Sci. Technol. 2022, 159, 113253. [Google Scholar] [CrossRef]
- Li, G.X.; Wan, Y.F.; Song, X.; Wang, Y.; Zan, L.S.; Zhu, J. Effects of various processing methods on the ultrastructure of tendon collagen fibrils from Qinchuan beef cattle observed with atomic force microscopy. J. Food Qual. 2018, 2018, 9090831. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.J.; Tu, S.S.; Li, Y.H.; Yagoub, A.E.A.; Ma, H.L.; Zhou, C.S. Effects of single-and tri-frequency ultrasound on self-assembly and characterizations of bionic dynamic rat stomach digestion of pepsin-soluble collagen from chicken leg skin. Food Res. Int. 2020, 137, 109710. [Google Scholar] [CrossRef]
- Liu, H.; Xu, X.; Liu, J.Q.; Zhang, H.R.; Qi, L.W.; Zhang, C.H. Comparative assessment of bone collagen recovered from different livestock and poultry species: Microstructure, physicochemical characteristics and functional properties. Int. J. Food Sci. Tech. 2023, 58, 1597–1610. [Google Scholar] [CrossRef]
- Thankachan, S.N.; Ilamaran, M.; Ayyadurai, N.; Shanmugam, G. Insights into the effect of artificial sweeteners on the structure, stability, and fibrillation of type I collagen. Int. J. Biol. Macromol. 2020, 164, 748–758. [Google Scholar] [CrossRef]
- Ran, Y.Q.; Su, W.; Ma, L.; Wang, X.L.; Li, X.D. Insight into the effect of sulfonated chitosan on the structure, rheology and fibrillogenesis of collagen. Int. J. Biol. Macromol. 2021, 166, 1480–1490. [Google Scholar] [CrossRef]
- Chandrapala, J.; Oliver, C.; Kentish, S.; Ashokkumar, M. Ultrasonics in food processing. Ultrason. Sonochem. 2012, 19, 975–983. [Google Scholar] [CrossRef]
- Liao, W.; Xia, G.H.; Li, Y.C.; Shen, X.R.; Li, C. Comparison of characteristics and fibril-forming ability of skin collagen from barramundi (Lates calcarifer) and tilapia (Oreochromis niloticus). Int. J. Biol. Macromol. 2018, 107, 549–559. [Google Scholar] [CrossRef]
- Li, Y.P.; Asadi, A.; Monroe, M.R.; Douglas, E.P. pH effects on collagen fibrillogenesis in vitro: Electrostatic interactions and phosphate binding. Mater. Sci. Eng. C 2009, 29, 1643–1649. [Google Scholar] [CrossRef]
- Tang, L.L.; Chen, S.L.; Su, W.J.; Weng, W.Y.; Osako, K.; Tanaka, M. Physicochemical properties and film-forming ability of fish skin collagen extracted from different freshwater species. Process Biochem. 2015, 50, 148–155. [Google Scholar] [CrossRef]
- Zhang, X.; Ookawa, M.; Tan, Y.K.; Ura, K.; Adachi, S.; Takagi, Y. Biochemical characterisation and assessment of fibril-forming ability of collagens extracted from Bester sturgeon Huso huso× Acipenser ruthenus. Food Chem. 2014, 160, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.W.; Tanaka, H.; Kobayashi, T.; Hatayama, H.; Zhang, X.; Ura, K.; Yunoki, S.; Takagi, Y. The effect of alkaline pretreatment on the biochemical characteristics and fibril-forming abilities of types I and II collagen extracted from bester sturgeon by-products. Int. J. Biol. Macromol. 2019, 131, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, V.; Gaillard-Martinie, B.; Sayd, T.; Chambon, C.; Anton, M.; Santé-Lhoutellier, V. Collagen type I from bovine bone. Effect of animal age, bone anatomy and drying methodology on extraction yield, self-assembly, thermal behaviour and electrokinetic potential. Int. J. Biol. Macromol. 2017, 97, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, M.; Ariffin, F.; Huda, N. An alternative source of type I collagen based on by-product with higher thermal stability. Food Hydrocolloid 2017, 63, 372–382. [Google Scholar] [CrossRef]
- Andonegi, M.; Las Heras, K.; Santos-Vizcaíno, E.; Igartua, M.; Hernandez, R.M.; de la Caba, K.; Guerrero, P. Structure-properties relationship of chitosan/collagen films with potential for biomedical applications. Carbohyd. Polym. 2020, 237, 116159. [Google Scholar] [CrossRef]
- Valencia-Llano, C.H.; López-Tenorio, D.; Saavedra, M.; Zapata, P.A.; Grande-Tovar, C.D. Comparison of Two Bovine Commercial Xenografts in the Regeneration of Critical Cranial Defects. Molecules 2022, 27, 5745. [Google Scholar] [CrossRef]
- Ion, A.; Andronescu, E.; Rădulescu, D.; Rădulescu, M.; Iordache, F.; Vasile, B.Ș.; Surdu, A.V.; Albu, M.G.; Maniu, H.; Chifiriuc, M.C.; et al. Biocompatible 3d matrix with antimicrobial properties. Molecules 2016, 21, 115. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Li, H.J.; Nuerjiang, M.; Shi, S.; Kong, B.H.; Liu, Q.; Xia, X.F. Application of ultrasound treatment in chicken gizzards tenderization: Effects on muscle fiber and connective tissue. Ultrason. Sonochem. 2021, 79, 105786. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Zhou, P.; Li, T.C.; Regenstein, J.M. Comparison of acid-soluble collagens from the skins and scales of four carp species. Food Hydrocolloid 2014, 41, 290–297. [Google Scholar] [CrossRef]
- Pal, G.K.; Suresh, P.V. Comparative assessment of physico-chemical characteristics and fibril formation capacity of thermostable carp scales collagen. Mater. Sci. Eng. C 2017, 70, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.F.; Shen, P.; Yu, M.Q.; Yu, C.X.; Zhu, B.W.; Qi, H. (−)-Epigallocatechin gallate protected molecular structure of collagen fibers in sea cucumber Apostichopus japonicus body wall during thermal treatment. LWT Food Sci. Technol. 2020, 123, 109076. [Google Scholar] [CrossRef]
- Adamiak, K.; Lewandowska, K.; Sionkowska, A. The infuence of salicin on rheological and film-forming properties of collagen. Molecules 2021, 26, 1661. [Google Scholar] [CrossRef]
- Li, Y.S.; Yang, L.H.; Wu, S.J.; Chen, J.D.; Lin, H.W. Structural, functional, rheological, and biological properties of the swim bladder collagen extracted from grass carp (Ctenopharyngodon idella). LWT Food Sci. Technol. 2022, 153, 112518. [Google Scholar] [CrossRef]
- Song, X.; Si, L.L.; Sun, X.; Zhu, X.; Li, Z.X.; Li, Y.Y.; Wang, Y.F.; Hou, H. Rheological properties, thermal stability and conformational changes of collagen from sea cucumber (Apostichopus japonicas). Food Chem. 2022, 389, 133033. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Li, J.H.; Li, C.H.; Li, G.Y. Effects of NaCl on the rheological behavior of collagen solution. Korea-Aust. Rheol. J. 2013, 25, 137–144. [Google Scholar] [CrossRef]
- Lai, G.L.; Li, Y.; Li, G.Y. Effect of concentration and temperature on the rheological behavior of collagen solution. Int. J. Biol. Macromol. 2008, 42, 285–291. [Google Scholar] [CrossRef]
- Pudło, A.; Juchniewicz, S.; Kopeć, W. Characteristics of Reconstituted Collagen Fibers from Chicken Keel Cartilage Depends on Salt Type for Removal of Proteoglycans. Molecules 2021, 26, 3538. [Google Scholar] [CrossRef]
- Kang, D.C.; Wang, A.R.; Zhou, G.H.; Zhang, W.G.; Xu, S.M.; Guo, G.P. Power ultrasonic on mass transport of beef: Effects of ultrasound intensity and NaCl concentration. Innov. Food Sci. Emerg. 2016, 35, 36–44. [Google Scholar] [CrossRef]
- Thuy, L.T.M.; Okazaki, E.; Osako, K. Isolation and characterization of acid-soluble collagen from the scales of marine fishes from Japan and Vietnam. Food Chem. 2014, 149, 264–270. [Google Scholar] [CrossRef]
- Kang, D.C.; Zou, Y.H.; Cheng, Y.P.; Xing, L.J.; Zhou, G.H.; Zhang, W.G. Effects of power ultrasound on oxidation and structure of beef proteins during curing processing. Ultrason. Sonochem. 2016, 33, 47–53. [Google Scholar] [CrossRef]
- Pezeshk, S.; Rezaei, M.; Abdollahi, M. Impact of ultrasound on extractability of native collagen from tuna by-product and its ultrastructure and physicochemical attributes. Ultrason. Sonochem. 2022, 89, 106129. [Google Scholar] [CrossRef] [PubMed]
- Monago-Maraña, O.; Wold, J.P.; Rødbotten, R.; Dankel, K.R.; Afseth, N.K. Raman, near-infrared and fluorescence spectroscopy for determination of collagen content in ground meat and poultry by-products. LWT-Food Sci. Technol. 2021, 140, 110592. [Google Scholar] [CrossRef]
- Qu, W.J.; Guo, T.T.; Zhang, X.X.; Jin, Y.T.; Wang, B.; Wahia, H.; Ma, H.L. Preparation of tuna skin collagen-chitosan composite film improved by sweep frequency pulsed ultrasound technology. Ultrason. Sonochem. 2022, 82, 105880. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Power Setting (W) | Actual Power Output (W) | Ultrasonic Time at 30 °C (min) | Ultrasonication Intensity (W/cm2) a | Incubation Time at 30 °C in the Absence of Ultrasonic (min) |
|---|---|---|---|---|---|
| C0W0m | 0 | 0 | 0 | 0 | 60 |
| C50W10m | 50 | 0.82 | 10 | 0.62 | 50 |
| C100W10m | 100 | 1.56 | 10 | 1.15 | 50 |
| C200W10m | 200 | 8.32 | 10 | 6.27 | 50 |
| C400W10m | 400 | 51.05 | 10 | 38.47 | 50 |
| C200W5m | 200 | 8.32 | 5 | 6.27 | 55 |
| C200W15m | 200 | 8.32 | 15 | 6.27 | 45 |
| C200W30m | 200 | 8.32 | 30 | 6.27 | 30 |
| C200W60m | 200 | 8.32 | 60 | 6.27 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Zhang, H.; Wang, K.; Qi, L.; Guo, Y.; Zhang, C.; Xu, Y. Impact of Ultrasonication on the Self-Assembly Behavior and Gel Properties of Bovine Bone Collagen I. Molecules 2023, 28, 3096. https://doi.org/10.3390/molecules28073096
Liu H, Zhang H, Wang K, Qi L, Guo Y, Zhang C, Xu Y. Impact of Ultrasonication on the Self-Assembly Behavior and Gel Properties of Bovine Bone Collagen I. Molecules. 2023; 28(7):3096. https://doi.org/10.3390/molecules28073096
Chicago/Turabian StyleLiu, Hong, Hongru Zhang, Kangyu Wang, Liwei Qi, Yujie Guo, Chunhui Zhang, and Yang Xu. 2023. "Impact of Ultrasonication on the Self-Assembly Behavior and Gel Properties of Bovine Bone Collagen I" Molecules 28, no. 7: 3096. https://doi.org/10.3390/molecules28073096