
Synthesis of Tetrasubstituted Nitroalkenes and Preliminary Studies of Their Enantioselective Organocatalytic Reduction

 , , and
, , and
Abstract
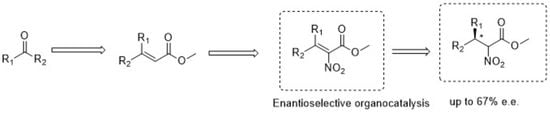
:
1. Introduction
2. Results and Discussion
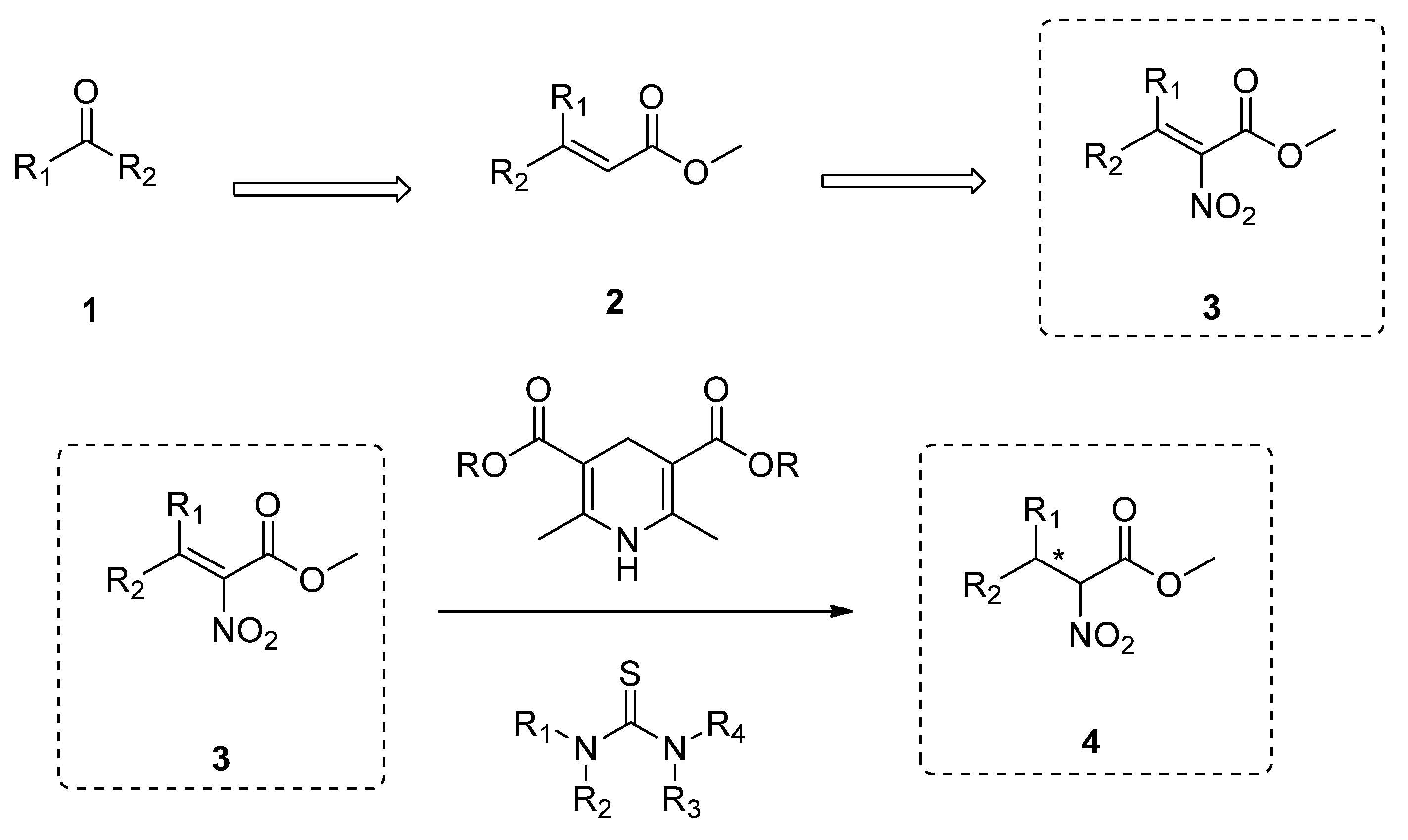
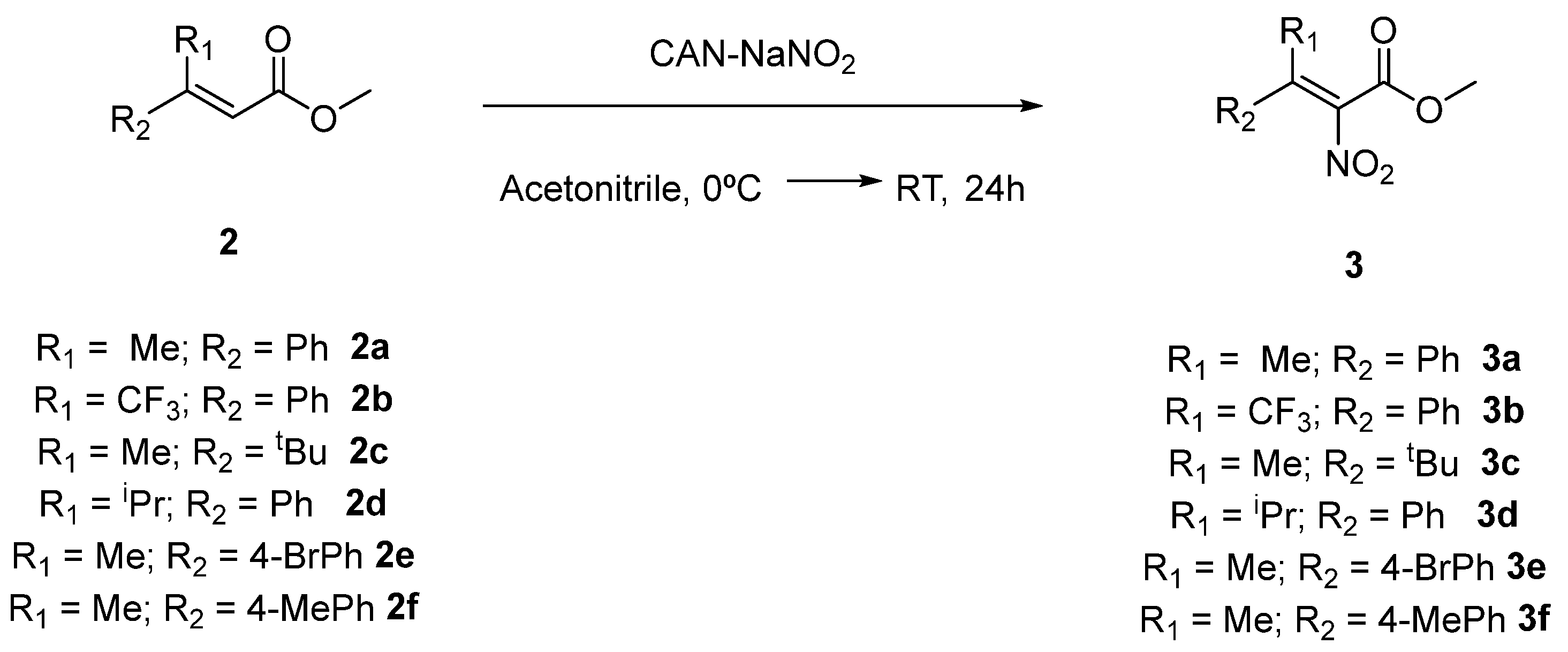
2.1. Synthesis of Tetrasubstituted Nitroalkenes
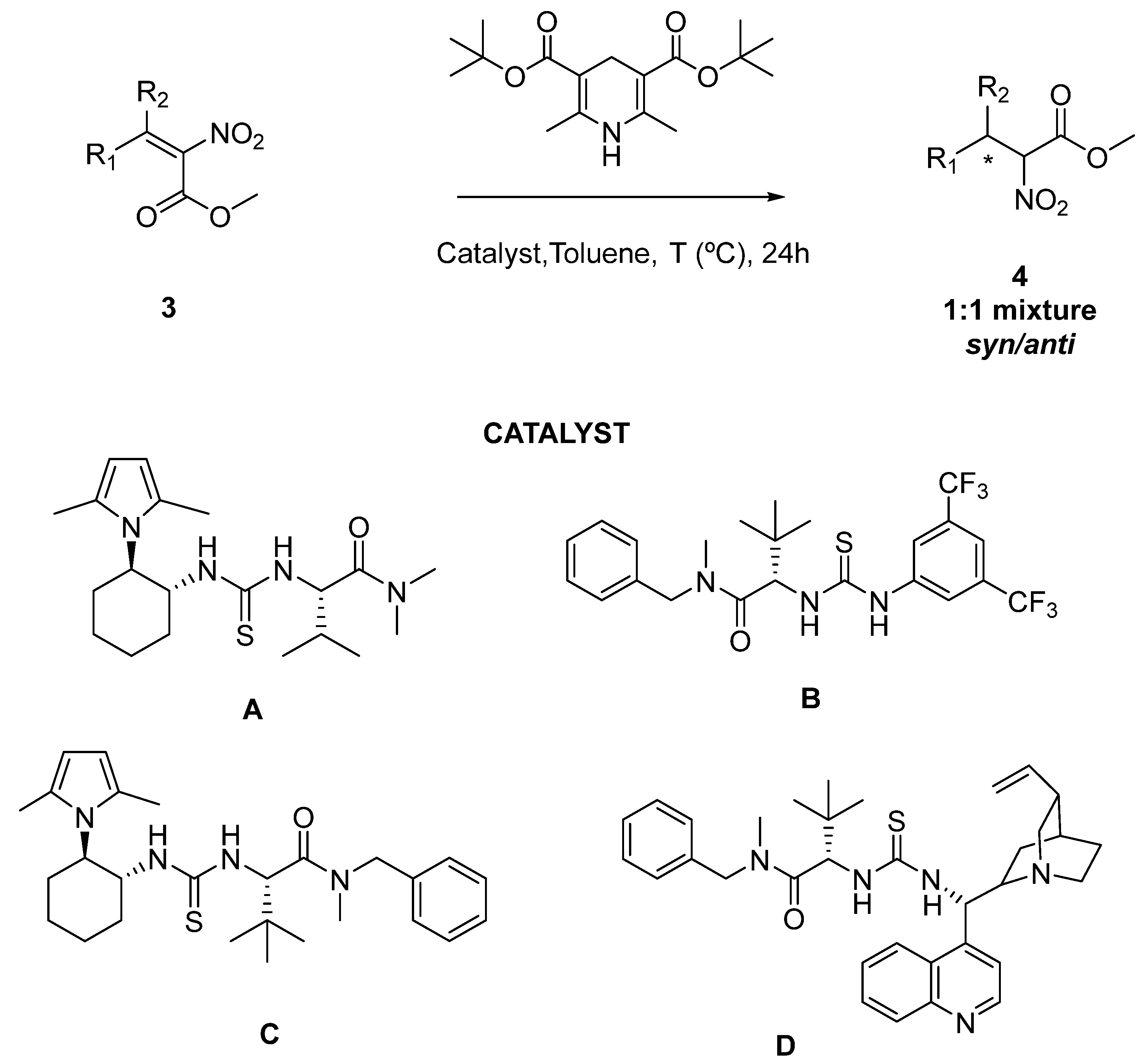
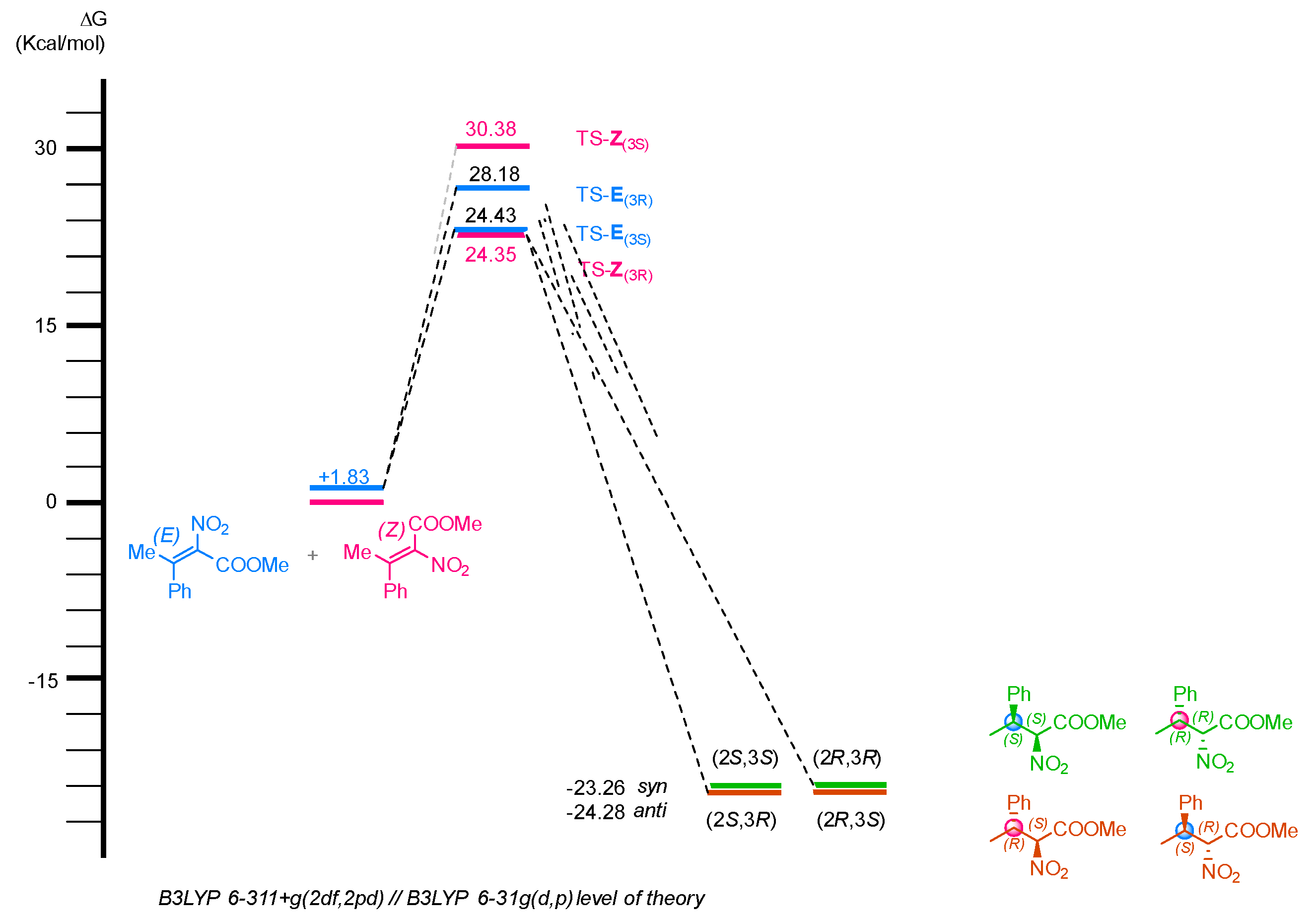
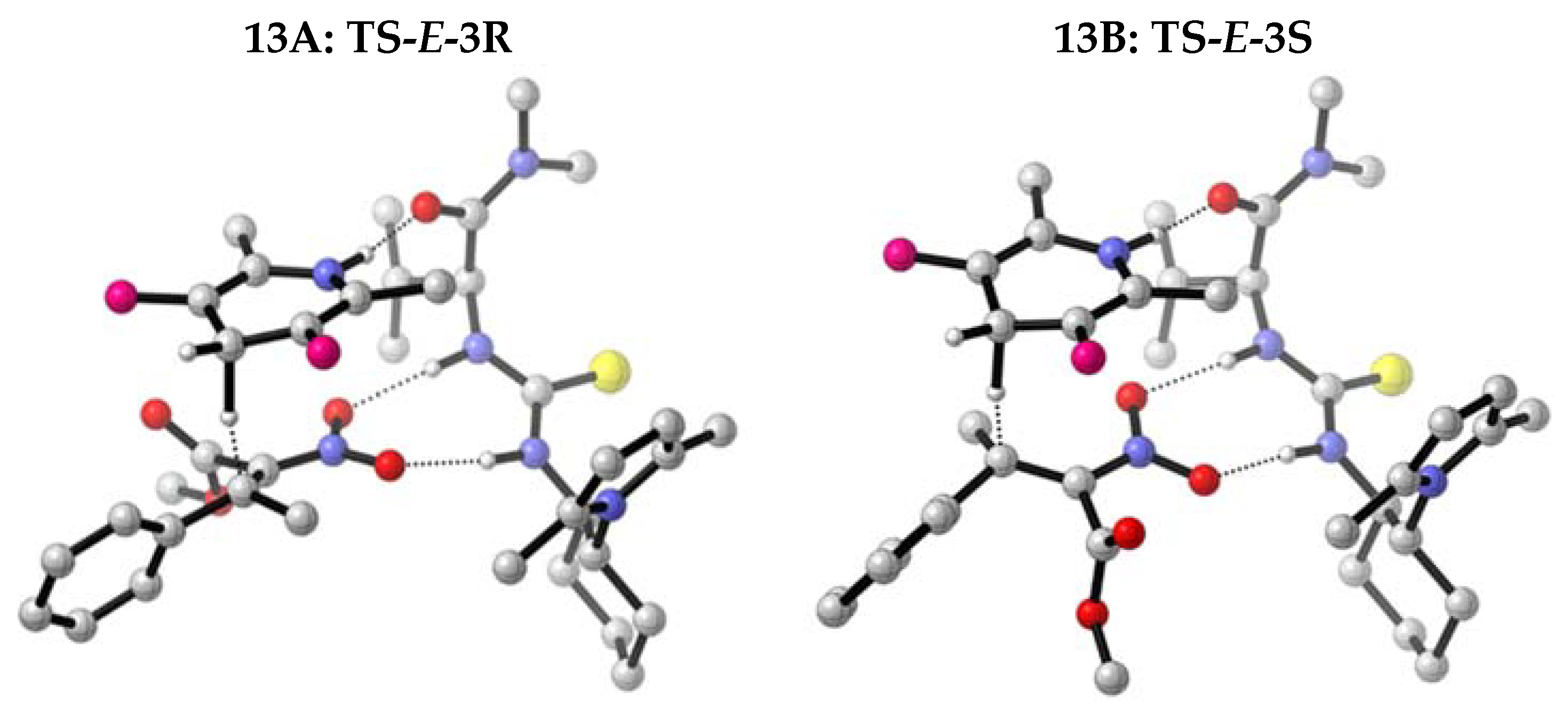
2.2. Enantioselective Reduction of Tetrasubstituted Nitroalkenes (3)
2.3. Determination of the Absolute Configuration of Nitroalkanes (4)
3. Materials and Methods
3.1. General Procedure for the Synthesis of Tetrasubstituted Nitroalkenes (3)
3.2. General Procedure for the Enantioselective Synthesis of Nitroalkanes (4)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Narancic, T.; Almahboub, S.A.; O’Connor, K.E. Unnatural amino acids: Production and biotechnological potential. World J. Microbiol. Biotechnol. 2019, 35, 67. [Google Scholar] [CrossRef]
- Osberger, T.J.; Rogness, D.C.; Kohrt, J.T.; Stepan, A.F.; White, M.C. Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 2016, 537, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Bisht, A.; Juyal, D. Unnatural amino acids (UAA’S): A trendy scaffold for pharmaceutical research. J. Drug Deliv. Ther. 2019, 9, 601–609. [Google Scholar]
- Ohfune, Y.; Shinada, T. Enantio- and Diastereoselective Construction of α,α-Disubstituted α-Amino Acids for the Synthesis of Biologically Active Compounds. Eur. J. Org. Chem. 2005, 2005, 5127–5143. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Xu, Y.; Yang, X.; Li, L. Improved synthesis of unnatural amino acids for peptide stapling. Tetrahedron Lett. 2017, 58, 24–28. [Google Scholar] [CrossRef]
- Vogt, H.; Bräse, S. Recent approaches towards the asymmetric synthesis of α,α-disubstituted α-amino acids. Org. Biomol. Chem. 2007, 5, 406–430. [Google Scholar] [CrossRef]
- Noboru, O. Chapter 2: Preparation of Nitro Compounds. In The Nitro Group in Organic Synthesis; Willey-VCH: Hoboken, NJ, USA, 2001; pp. 3–29. [Google Scholar]
- Gabrielli, S.; Chiurchiù, E.; Palmieri, A. β-Nitroacrylates: A Versatile and Growing Class of Functionalized Nitroalkenes. Adv. Synth. Catal. 2019, 361, 630–651. [Google Scholar] [CrossRef]
- Wen, J.; Jiang, J.; Zhang, X. Rhodium-Catalyzed Asymmetric Hydrogenation of α,β-Unsaturated Carbonyl Compounds via Thiourea Hydrogen Bonding. Org. Lett. 2016, 18, 4451–4453. [Google Scholar] [CrossRef]
- Sharma, V.; Kelly, G.T.; Watanabe, C.M.H. Exploration of the Molecular Origin of the Azinomycin Epoxide: Timing of the Biosynthesis Revealed. Org. Lett. 2008, 10, 4815–4818. [Google Scholar] [CrossRef]
- Maity, S.; Manna, S.; Rana, S.; Naveen, T.; Mallick, A.; Maiti, D. Efficient and Stereoselective Nitration of Mono- and Disubstituted Olefins with AgNO2 and TEMPO. J. Am. Chem. Soc. 2013, 135, 3355–3358. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Pujol, M.D. Straightforward synthesis of nitroolefins by microwave- or ultrasound-assisted Henry reaction. Tetrahedron Lett. 2011, 52, 2629–2632. [Google Scholar] [CrossRef]
- Zhu, L.; Yan, P.; Zhang, L.; Chen, Z.; Zeng, X.; Zhong, G. TiCl4/DMAP mediated Z-selective knovenagel condensation of isatins with nitroacetates and related compounds. RSC Adv. 2017, 7, 51352–51358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Blazecka, P.G.; Mark, P.A.; Lovdahl, T.; Curran, T. Indium (III) mediated Markovnikov addition of malonates and β-ketoesters to terminal alkynes and the formation of Knoevenagel condensation products. Tetrahedron 2005, 61, 7807–7813. [Google Scholar] [CrossRef]
- Nakamura, M.; Fujimoto, T.; Endo, K.; Nakamura, E. Stereoselective Synthesis of Tetra-Substituted Olefins via Addition of Zinc Enolates to Unactivated Alkynes. Org. Lett. 2004, 6, 4837–4840. [Google Scholar] [CrossRef]
- Buevich, A.V.; Wu, Y.; Chan, T.M.; Stamford, A. An unusual contra-Michael addition of NaNO2–ceric ammonium nitrate to acrylic esters. Tetrahedron Lett. 2008, 49, 2132–2135. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef]
- Nolwenn, J.A.; Ozores, M.L.; List, B. Organocatalytic Asymmetric Transfer Hydrogenation of Nitroolefins. J. Am. Chem. Soc. 2007, 129, 8976–8977. [Google Scholar]
- Bernardi, L.; Fochi, M. A General Catalytic Enantioselective Transfer Hydrogenation Reaction of β,β-Disubstituted Nitroalkenes Promoted by a Simple Organocatalyst. Molecules 2016, 21, 1000. [Google Scholar] [CrossRef] [Green Version]
- Massolo, E.; Benaglia, M.; Orlandi, M.; Rossi, S.; Celentano, G. Enantioselective Organocatalytic Reduction of β-Trifluoromethyl Nitroalkenes: An Efficient Strategy for the Synthesis of Chiral β-Trifluoromethyl Amines. Chem. Eur. J. 2015, 21, 3589–3595. [Google Scholar] [CrossRef]
- Barrios-Rivera, J.; Xu, Y.; Willis, M.; Vyas, V.K. A diversity of recently reported methodology for asymmetric imine reduction. Org. Chem. Front. 2020, 7, 3312–3342. [Google Scholar] [CrossRef]
- Davis, F.A.; Liang, C.H.; Liu, H. Asymmetric Synthesis of β-Substituted α-Amino Acids Using 2H-Azirine-2-carboxylate Esters. Synthesis of 3,3-Disubstituted Aziridine-2-carboxylate Esters. J. Org. Chem. 1997, 62, 3796–3797. [Google Scholar] [CrossRef]
- Gagnot, G.; Hervin, V.; Coutant, E.P.; Desmons, S.; Baatallah, R.; Monnot, V.; Janin, Y.L. Synthesis of unnatural α-amino esters using ethyl nitroacetate and condensation or cycloaddition reactions. Beilstein J. Org. Chem. 2018, 14, 2846–2852. [Google Scholar] [CrossRef] [PubMed]
- Harel, T.; Rozen, S. Transforming Natural Amino Acids into α-Alkyl-Substituted Amino Acids with the Help of the HOF·CH3CN Complex. J. Org. Chem. 2007, 72, 6500–6503. [Google Scholar] [CrossRef]
- Metz, A.E.; Kozlowski, M.C. 2-Aryl-2-nitroacetates as Central Precursors to Aryl Nitromethanes, α-Ketoesters, and α-Amino Acids. J. Org. Chem. 2013, 78, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostmann, T.; Molloy, J.J.; Bussmann, K.; Gilmour, R. Light-Enabled Enantiodivergence: Stereospecific Reduction of Activated Alkenes Using a Single Organocatalyst Enantiomer. Org. Lett. 2019, 21, 10164–10168. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/citation/ (accessed on 13 December 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | R2 | T | Product | E/Z Ratio a | Yield b |
|---|---|---|---|---|---|---|
| 1 | Me | Ph | RT | 2a | 80:20 | 52% |
| 2 | CF3 | Ph | RT | 2b | 75:25 | 79% |
| 3 | Me | tBu | 66 °C | 2c | 90:10 | 14% |
| 4 | iPr | Ph | 66 °C | 2d | 67:33 | 33% |
| 5 | Me | 4-BrPh | RT | 2e | 65:35 | 76% |
| 6 | Me | 4-MePh | 66 °C | 2f | 75:25 | 86% |
| Entry | R1 | R2 | Acrylate | Nitroacrylate | Yield a | Isomers Ratio |
|---|---|---|---|---|---|---|
| 1 | Me | Ph | 2a | 3a | 46% | 60/40 |
| 2 | CF3 | Ph | 2b | - | b | - |
| 3 | Me | tBu | 2c | 3c | --- c | - |
| 4 | iPr | Ph | 2d | 3d | 25% | 99/1 |
| 5 | Me | 4-BrPh | 2e | 3e | 47% | 62/38 |
| 6 | Me | 4-MePh | 2f | 3f | 48% | 57/43 |
| Entry | T a | Nitroalkene | Nitroalkane | Catalyst | Yield d 4 | ee% Syn–ee% Anti e |
|---|---|---|---|---|---|---|
| 1a | 60 °C | 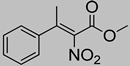 3a–Z | 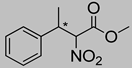 4a | A | trace | n.d. |
| 2b | 60 °C |  3a–E | 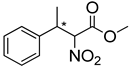 4a | A | 51% | 67-63 |
| 3c | 60 °C | 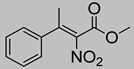 3a (E+Z) | 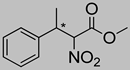 4a | A | 53% | 63–52 |
| 4c | 100 °C |  3a (E+Z) |  4a | A | 37% | 27–22 |
| 5c | 25 °C | 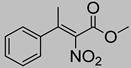 3a (E+Z) | 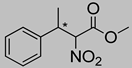 4a | A | 15% | n.d. |
| 6c | 60 °C | 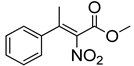 3a (E+Z) |  4a | B | 69% | 33–32 |
| 7 | 60 °C | 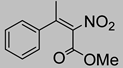 3a (E+Z) | 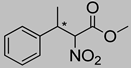 4a | C | 19% | n.d. |
| 8 | 60 °C | 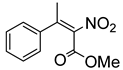 3a (E+Z) | 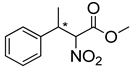 4a | D | 10% | n.d |
| 9 | 60 °C | 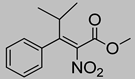 3d | 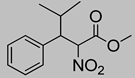 4d | A | 32% | 12–13 |
| 10 | 60 °C | 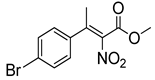 3e | 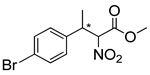 4e | A | 60% | 24–4 |
| 11 | 60 °C |  3f | 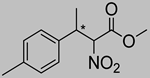 4f | A | 51% | 50–46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, P.C.; Rossi, S.; Sanz, M.; Vasile, F.; Benaglia, M. Synthesis of Tetrasubstituted Nitroalkenes and Preliminary Studies of Their Enantioselective Organocatalytic Reduction. Molecules 2023, 28, 3156. https://doi.org/10.3390/molecules28073156
González PC, Rossi S, Sanz M, Vasile F, Benaglia M. Synthesis of Tetrasubstituted Nitroalkenes and Preliminary Studies of Their Enantioselective Organocatalytic Reduction. Molecules. 2023; 28(7):3156. https://doi.org/10.3390/molecules28073156
Chicago/Turabian StyleGonzález, Patricia Camarero, Sergio Rossi, Miguel Sanz, Francesca Vasile, and Maurizio Benaglia. 2023. "Synthesis of Tetrasubstituted Nitroalkenes and Preliminary Studies of Their Enantioselective Organocatalytic Reduction" Molecules 28, no. 7: 3156. https://doi.org/10.3390/molecules28073156