
Targeting mTOR for Anti-Aging and Anti-Cancer Therapy


Abstract
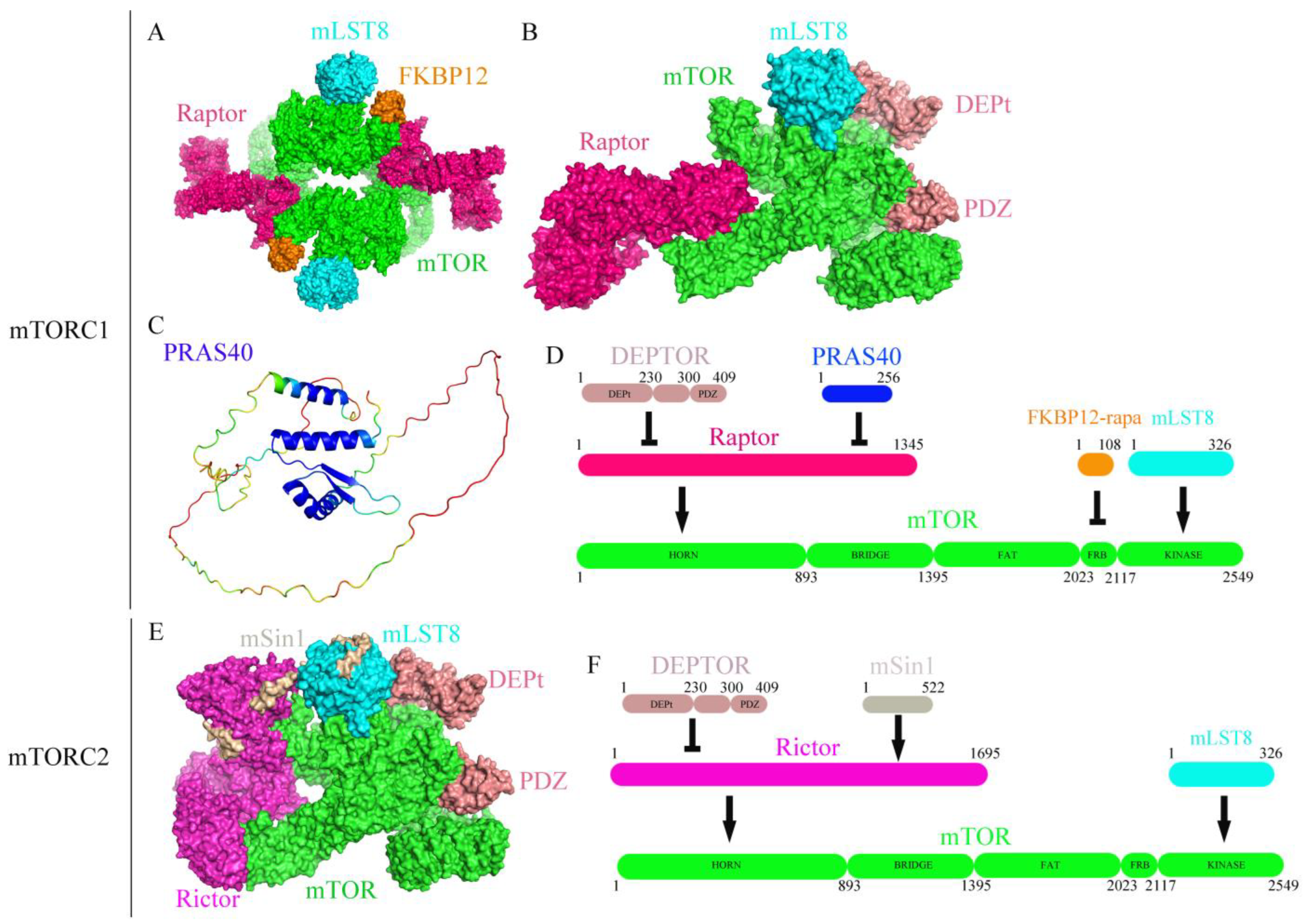
:1. Introduction
2. Anti-Aging by Targeting mTOR
3. The Complexity of Anti-Aging by Targeting mTOR
4. Cancer Therapy by Targeting mTOR
5. The Disadvantage of Cancer Therapy by Targeting mTOR
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Diaz, V.; Walsh, M.E.; Zhang, Y. Moderate lifelong overexpression of tuberous sclerosis complex 1 (TSC1) improves health and survival in mice. Sci. Rep. 2017, 7, 834. [Google Scholar] [CrossRef] [Green Version]
- Garratt, M.; Bower, B.; Garcia, G.G.; Miller, R.A. Sex differences in lifespan extension with acarbose and 17-alpha estradiol: Gonadal hormones underlie male-specific improvements in glucose tolerance and mTORC2 signaling. Aging Cell 2017, 16, 1256–1266. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Powers RW 3rd Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [Green Version]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73. [Google Scholar] [CrossRef]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes. Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e6. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Nelson, G.; Rabanal-Ruiz, Y.; Kucheryavenko, O.; Dunhill-Turner, N.A.; Chesterman, C.C.; Zahari, Q.; Zhang, T.; Conduit, S.E.; Mitchell, C.A.; et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol. 2017, 216, 1949–1957. [Google Scholar] [CrossRef] [Green Version]
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 2016, 8, 231–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Wu, B.; Fan, X.; Xu, G.; Ou, C.; Chen, M. YAP accelerates vascular senescence via blocking autophagic flux and activating mTOR. J. Cell Mol. Med. 2021, 25, 170–183. [Google Scholar] [CrossRef]
- Tarrago, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, H.E.; Cox, L.S. mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leontieva, O.V.; Blagosklonny, M.V. Gerosuppression by pan-mTOR inhibitors. Aging 2016, 8, 3535–3551. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is. Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Deng, Z.; Ma, Y.; Jin, J.; Qi, F.; Li, S.; Liu, C.; Lyu, F.J.; Zheng, Q. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int. J. Biol. Sci. 2020, 16, 2675–2691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sergin, I.; Evans, T.D.; Jeong, S.J.; Rodriguez-Velez, A.; Kapoor, D.; Chen, S.; Song, E.; Holloway, K.B.; Crowley, J.R.; et al. High-protein diets increase cardiovascular risk by activating macrophage mTOR to suppress mitophagy. Nat. Metab. 2020, 2, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, J.; Elia, A.; Carroll, V.A.; Moumen, A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget 2015, 6, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Hu, H.; Tong, X.; Li, L.; Liu, X.; Chen, M.; Yuan, H.; Xie, X.; Li, Q.; Zhang, Y.; et al. The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat. Cell Biol. 2018, 20, 320–331. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Blanchard, P.G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brule, S.; Turcotte, V.; Cote, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARgamma-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Le Bacquer, O.; Petroulakis, E.; Paglialunga, S.; Poulin, F.; Richard, D.; Cianflone, K.; Sonenberg, N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J. Clin. Investig. 2007, 117, 387–396. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurez, V.; Dao, V.; Liu, A.; Pandeswara, S.; Gelfond, J.; Sun, L.; Bergman, M.; Orihuela, C.J.; Galvan, V.; Padron, A.; et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015, 14, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Morris, M.; Hockey, H.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizunuma, M.; Neumann-Haefelin, E.; Moroz, N.; Li, Y.; Blackwell, T.K. mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell 2014, 13, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Dominick, G.; Berryman, D.E.; List, E.O.; Kopchick, J.J.; Li, X.; Miller, R.A.; Garcia, G.G. Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology 2015, 156, 565–575. [Google Scholar] [CrossRef]
- Vodenik, B.; Rovira, J.; Campistol, J.M. Mammalian target of rapamycin and diabetes: What does the current evidence tell us? Transplant. Proc. 2009, 41, S31–S38. [Google Scholar] [CrossRef]
- Teutonico, A.; Schena, P.F.; Di Paolo, S. Glucose metabolism in renal transplant recipients: Effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J. Am. Soc. Nephrol. 2005, 16, 3128–3135. [Google Scholar] [CrossRef] [Green Version]
- Johnston, O.; Rose, C.L.; Webster, A.C.; Gill, J.S. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J. Am. Soc. Nephrol. 2008, 19, 1411–1418. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimbetov, D.; Davis, T.; Brook, A.J.; Cox, L.S.; Faragher, R.G.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T.; Haidinger, M.; Katholnig, K.; Kopecky, C.; Poglitsch, M.; Lassnig, C.; Rosner, M.; Zlabinger, G.J.; Hengstschlager, M.; Muller, M.; et al. Inhibition of mTOR blocks the anti-inflammatory effects of glucocorticoids in myeloid immune cells. Blood 2011, 117, 4273–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saemann, M.D.; Haidinger, M.; Hecking, M.; Horl, W.H.; Weichhart, T. The multifunctional role of mTOR in innate immunity: Implications for transplant immunity. Am. J. Transplant. 2009, 9, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, N.; Hall, M.N. TOR complex 2: A signaling pathway of its own. Trends Biochem. Sci. 2009, 34, 620–627. [Google Scholar] [CrossRef]
- Sparks, C.A.; Guertin, D.A. Targeting mTOR: Prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 2010, 29, 3733–3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulhati, P.; Cai, Q.; Li, J.; Liu, J.; Rychahou, P.G.; Qiu, S.; Lee, E.Y.; Silva, S.R.; Bowen, K.A.; Gao, T.; et al. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin. Cancer Res. 2009, 15, 7207–7216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007, 67, 11712–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Nardella, C.; Carracedo, A.; Alimonti, A.; Hobbs, R.M.; Clohessy, J.G.; Chen, Z.; Egia, A.; Fornari, A.; Fiorentino, M.; Loda, M.; et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci. Signal. 2009, 2, ra2. [Google Scholar] [CrossRef] [Green Version]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gaboury, L.A.; et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 2010, 107, 14134–14139. [Google Scholar] [CrossRef] [Green Version]
- Dowling, R.J.; Topisirovic, I.; Alain, T.; Bidinosti, M.; Fonseca, B.D.; Petroulakis, E.; Wang, X.; Larsson, O.; Selvaraj, A.; Liu, Y.; et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010, 328, 1172–1176. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, M.M.; Young, C.D.; Wang, S.; Sobolik, T.; Sanchez, V.M.; Hicks, D.J.; Cook, R.S.; Brantley-Sieders, D.M. mTOR Directs Breast Morphogenesis through the PKC-alpha-Rac1 Signaling Axis. PLoS Genet. 2015, 11, e1005291. [Google Scholar] [CrossRef] [Green Version]
- Morrison Joly, M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; Sarbassov, D.D.; Muller, W.J.; et al. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast Cancers. Cancer Res. 2016, 76, 4752–4764. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, W.; Li, C.; Zhang, J.; Su, Z. Chemotherapy in idiopathic pulmonary fibrosis and small-cell lung cancer with poor lung function. BMC Pulm. Med. 2021, 21, 122. [Google Scholar] [CrossRef]
- Woodcock, H.V.; Eley, J.D.; Guillotin, D.; Platé, M.; Nanthakumar, C.B.; Martufi, M.; Peace, S.; Joberty, G.; Poeckel, D.; Good, R.B.; et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Junpaparp, P.; Sharma, B.; Samiappan, A.; Rhee, J.H.; Young, K.R. Everolimus-induced severe pulmonary toxicity with diffuse alveolar hemorrhage. Ann. Am. Thorac. Soc. 2013, 10, 727–729. [Google Scholar] [CrossRef]
- Malouf, M.A.; Hopkins, P.; Snell, G.; Glanville, A.R. Everolimus in IPF Study Investigators. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 2011, 16, 776–783. [Google Scholar] [CrossRef]
- Platé, M.; Guillotin, D.; Chambers, R.C. The promise of mTOR as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2020, 29, 200269. [Google Scholar] [CrossRef]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phung, T.L.; Ziv, K.; Dabydeen, D.; Eyiah-Mensah, G.; Riveros, M.; Perruzzi, C.; Sun, J.; Monahan-Earley, R.A.; Shiojima, I.; Nagy, J.A.; et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 2006, 10, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, E.T.; Cao, C.; Niermann, K.; Mu, Y.; Zeng, F.; Hallahan, D.E.; Lu, B. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 2005, 24, 5414–5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokas, E.; Im, J.H.; Hill, S.; Yameen, S.; Stratford, M.; Beech, J.; Hackl, W.; Maira, S.M.; Bernhard, E.J.; McKenna, W.G.; et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res. 2012, 72, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [Green Version]
- Akbari Dilmaghani, N.; Safaroghli-Azar, A.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTORC signaling axis in head and neck squamous cell carcinoma: Possibilities for therapeutic interventions either as single agents or in combination with conventional therapies. IUBMB Life 2021, 73, 618–642. [Google Scholar] [CrossRef]
- Sager, R.A.; Backe, S.J.; Ahanin, E.; Smith, G.; Nsouli, I.; Woodford, M.R.; Bratslavsky, G.; Bourboulia, D.; Mollapour, M. Therapeutic potential of CDK4/6 inhibitors in renal cell carcinoma. Nat. Rev. Urol. 2022, 19, 305–320. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Haxhinasto, S.; Mathis, D.; Benoist, C. The AKT–mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 2008, 205, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, H.; de la Rosa, G.M.; Howard, O.M.; Chen, X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int. Immunopharmacol. 2007, 7, 1819–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Towards DNA-damage induced autophagy: A Boolean model of p53-induced cell fate mechanisms. DNA Repair. (Amst) 2020, 96, 102971. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Qu, X. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes. Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes. Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Farhan, M.A.; Carmine-Simmen, K.; Lewis, J.D.; Moore, R.B.; Murray, A.G. Endothelial Cell mTOR Complex-2 Regulates Sprouting Angiogenesis. PLoS ONE 2015, 10, e0135245. [Google Scholar] [CrossRef]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009, 69, 6232–6240. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E Survival Pathways by Rapamycin-Mediated Mammalian Target of Rapamycin Inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Vinals, F.; Chambard, J.C.; Pouyssegur, J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J. Biol. Chem. 1999, 274, 26776–26782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Cohen, R.B.; Laird, A.D.; Macé, S.; Engelman, J.A.; Ruiz-Soto, R.; Rockich, K.; Xu, J.; Shapiro, G.I.; Martinez, P.; et al. Phase I safety and pharmacokinetic study of the PI3K/mTOR inhibitor SAR245409 (XL765) in combination with erlotinib in patients with advanced solid tumors. J. Thorac. Oncol. 2014, 9, 316–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Berger, S.L. Senescence Elicits Stemness: A Surprising Mechanism for Cancer Relapse. Cell Metab. 2018, 27, 710–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, T.; Koiwa, M.; Yonemura, A.; Miyake, K.; Kariya, R.; Kubota, S.; Yokomizo-Nakano, T.; Yasuda-Yoshihara, N.; Uchihara, T.; Itoyama, R.; et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep. 2021, 34, 108779. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug | Population | Phase | Status | Trail Identifier |
|---|---|---|---|---|
| Temsirolimus + Er | Advanced HNSCC | Phase II | Terminated | NCT01009203 |
| Temsirolimus + Cet | R/M HNSCC | Phase II | Completed | NCT01256385 |
| Temsirolimus + Cis, Cet | R/M HNSCC | Phase I/II | Terminated | NCT01015664 |
| Temsirolimus + Pac, Car | R/M HNSCC | Phase I/II | Completed | NCT01016769 |
| Everolimus + Cis Rad | HNSCC patients | Phase I | Terminated | NCT01057277 |
| Everolimus + Cis IMRT | HNSCC patients | Phase I | Completed | NCT00858663 |
| Everolimus + Er Rad | R/M HNSCC | Phase I | Withdrawn | NCT01332279 |
| Everolimus + Doc, Cis | HNSCC patients | Phase I | Completed | NCT00935961 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, W.; Wu, G. Targeting mTOR for Anti-Aging and Anti-Cancer Therapy. Molecules 2023, 28, 3157. https://doi.org/10.3390/molecules28073157
Fu W, Wu G. Targeting mTOR for Anti-Aging and Anti-Cancer Therapy. Molecules. 2023; 28(7):3157. https://doi.org/10.3390/molecules28073157
Chicago/Turabian StyleFu, Wencheng, and Geng Wu. 2023. "Targeting mTOR for Anti-Aging and Anti-Cancer Therapy" Molecules 28, no. 7: 3157. https://doi.org/10.3390/molecules28073157
APA StyleFu, W., & Wu, G. (2023). Targeting mTOR for Anti-Aging and Anti-Cancer Therapy. Molecules, 28(7), 3157. https://doi.org/10.3390/molecules28073157