


Endoplasmic Reticulum Stress and Mitochondrial Stress in Drug-Induced Liver Injury
Abstract
1. Introduction
2. Classification of Drugs Producing DILI and Their Associated Mechanisms in the Endoplasmic Reticulum and Mitochondrial Stress
2.1. DILI Classification According to the Liver Lesion and Drug Type
2.2. Endoplasmic Reticulum and Mitochondrial Stress in Drug-Induced Liver Injury
2.2.1. ERS and ERS-Related Molecules in the Development of DILI
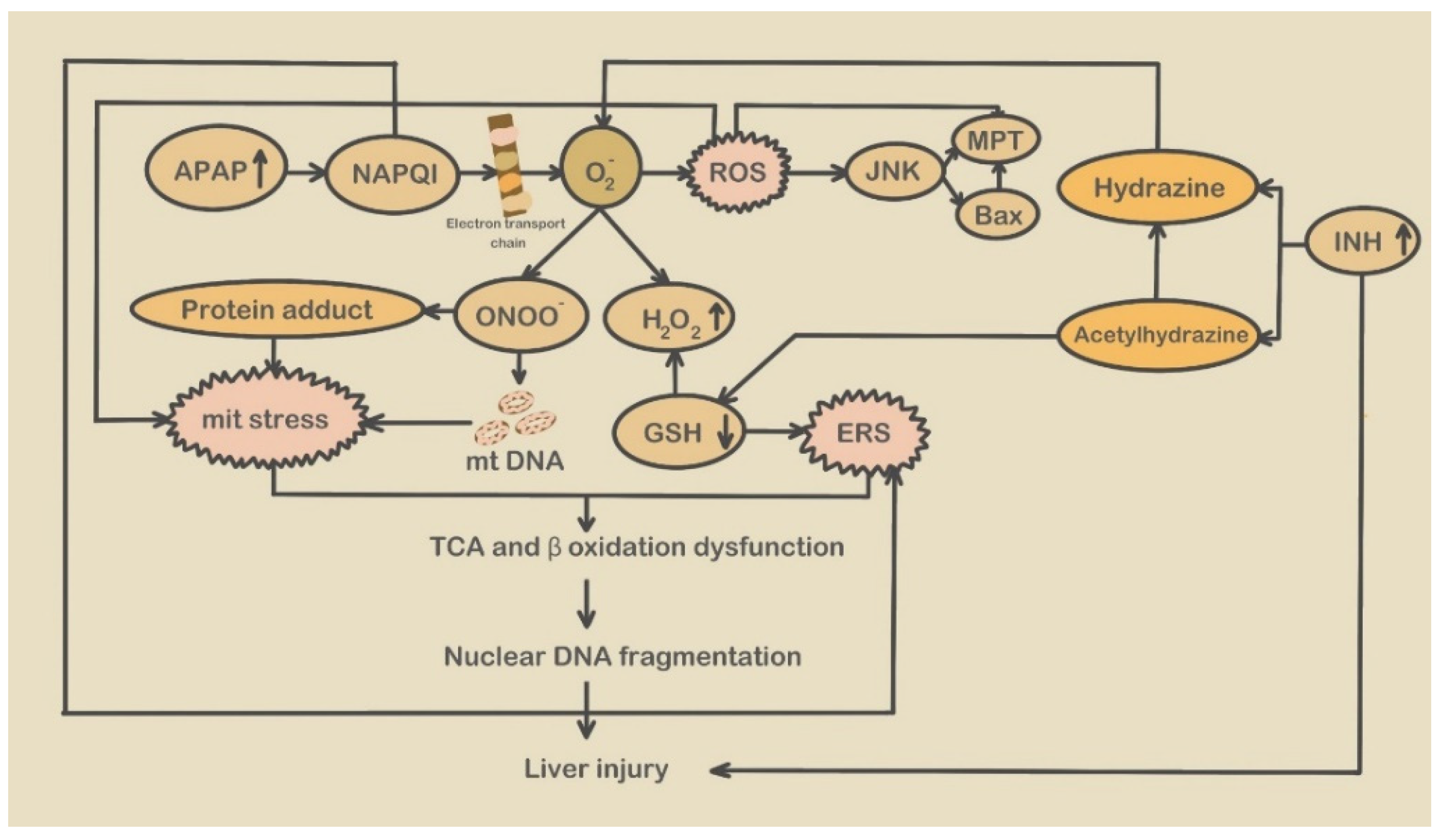
- APAP-induced endoplasmic reticulum stress
2.2.2. Mitochondrial Stress and Mitochondrial Stress-Related Molecules Participate in the Development of DILI
- APAP-induced mitochondrial stress
- Isoniazid-induced mitochondrial stress
- Valproic acid induced mitochondrial stress
3. ERS Triggers and Signaling Pathways in DILI
3.1. ERS Triggers
3.1.1. Abnormal Calcium Regulation
3.1.2. Lipid Metabolism Disorders
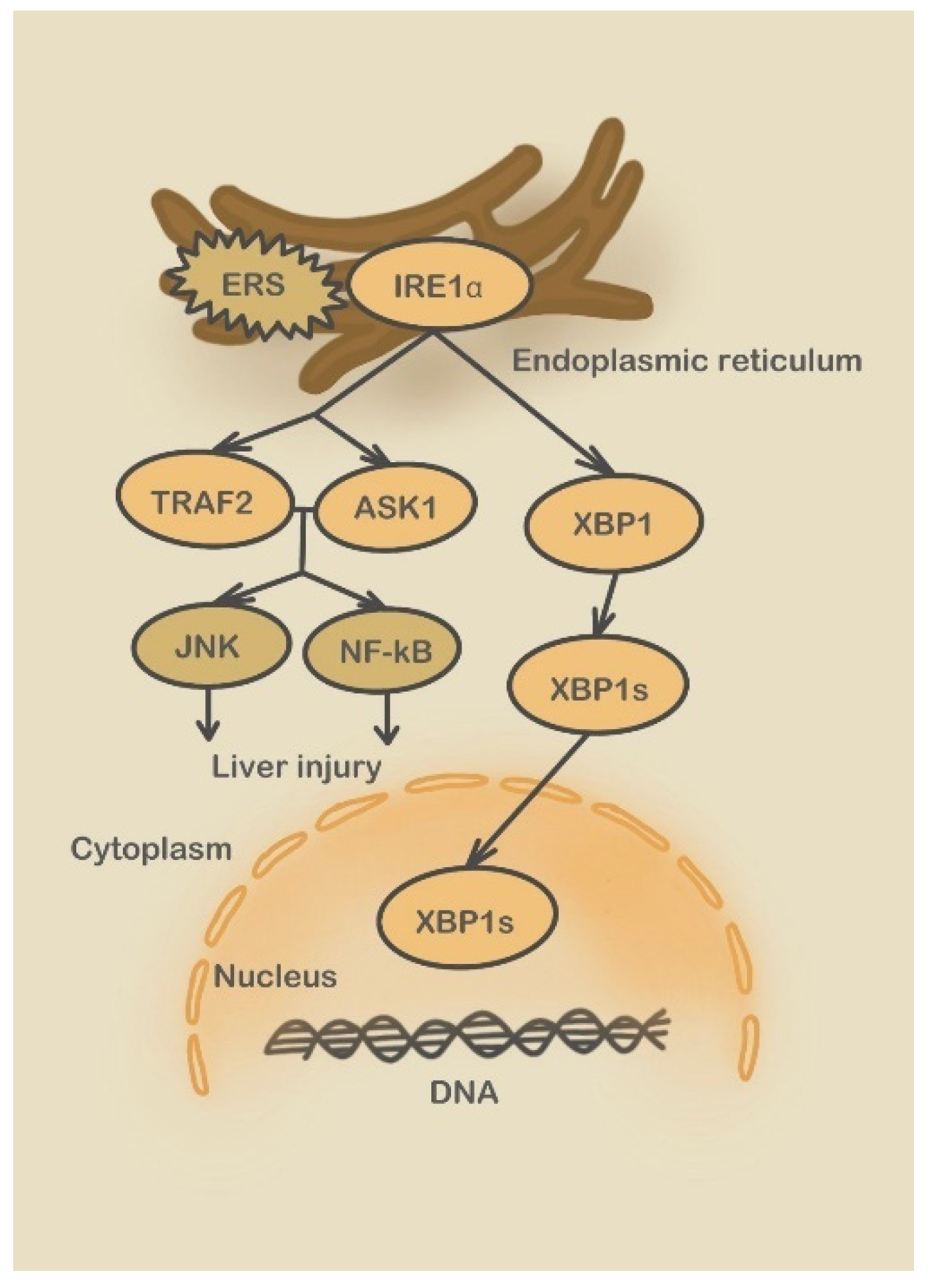
3.2. Endoplasmic Reticulum Stress Signaling Pathways Involved in DILI
3.2.1. Three UPR Pathways
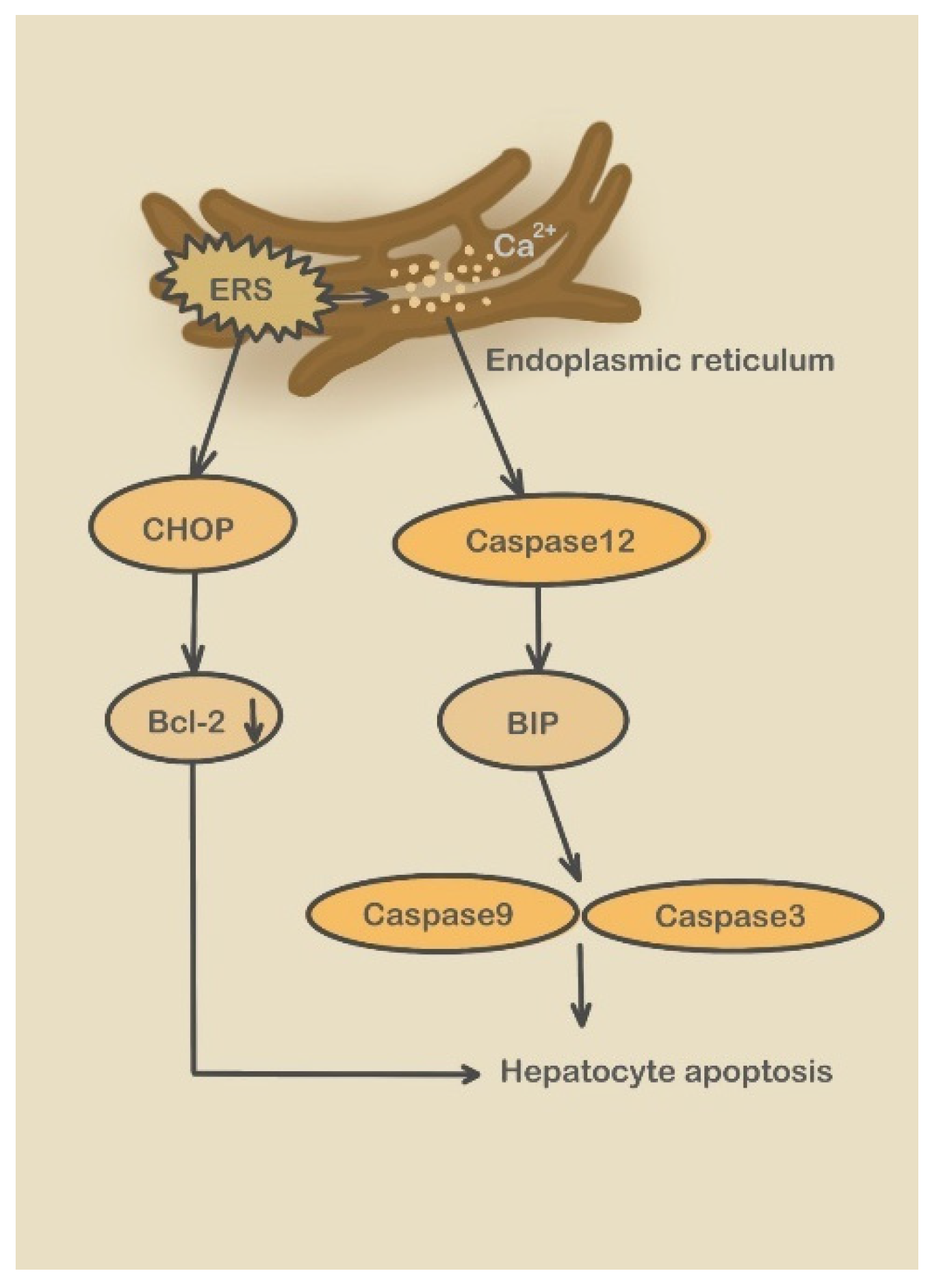
3.2.2. Caspase-12 Pathway
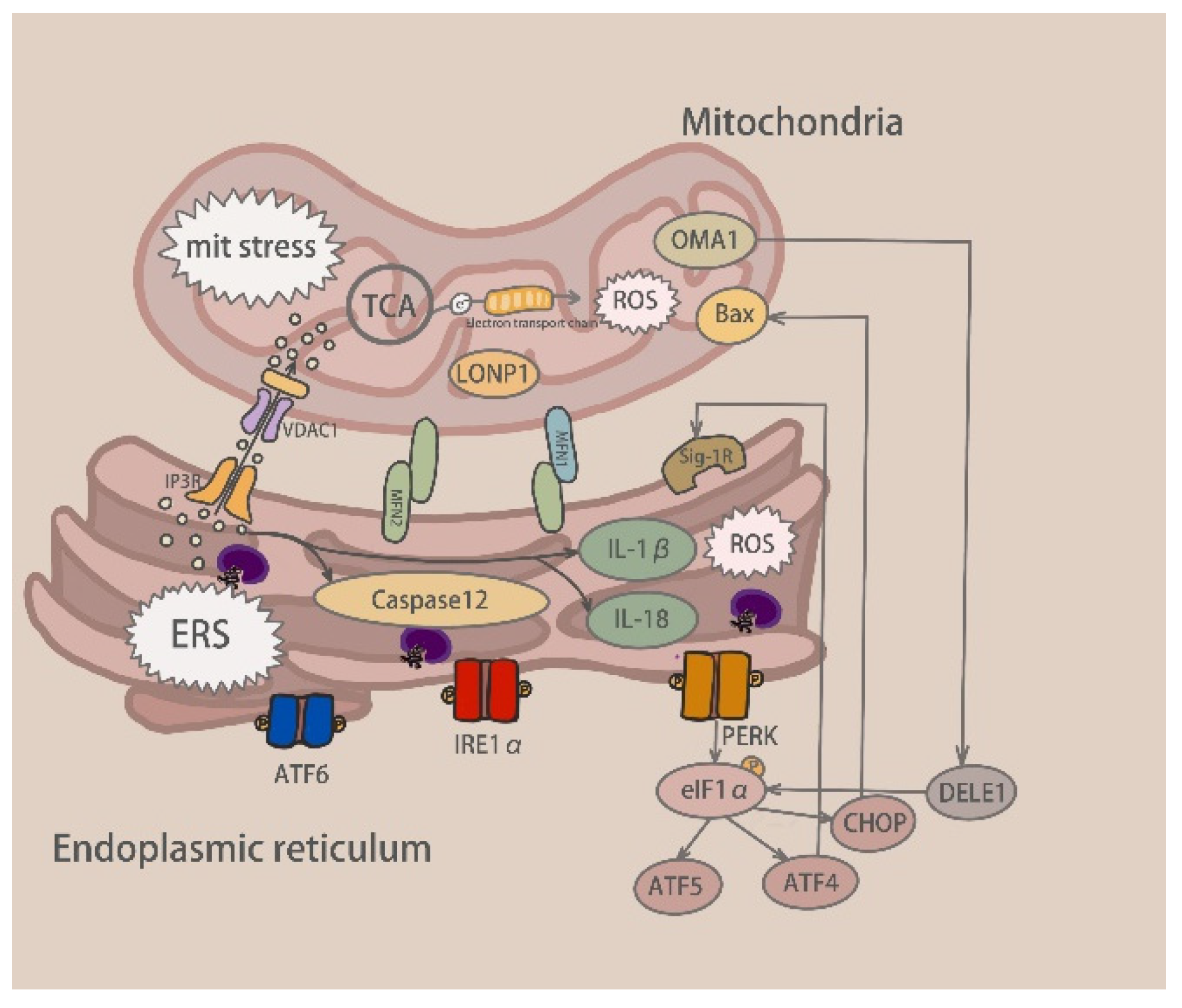
4. Mitochondrial Stress Signaling Channels Activated by Drug-Induced Liver Injury
5. Communication between Mitochondrial and Endoplasmic Reticulum Stress
5.1. Calcium Exchange
5.1.1. Contact Site between ER and Mitochondria-MAM
5.1.2. ER Receptor-Mediated Ca2+ Exchange
5.1.3. TRPM2 Involved in Ca2+ Exchange
5.2. Relationship between Mitochondrial and Endoplasmic Reticulum Stress
6. The Immunological Mechanisms of APAP-Induced Liver Injury
7. Pathways for the Treatment of a Drug-Induced Liver Injury
7.1. SPHK1–STARD1 Pathway
7.2. ATF4-ATF5-CHOP Pathway
7.3. TRPM2 Pathway
7.4. JNK Signaling Pathway
7.5. NF-κB Pathway
7.6. UPR Pathway
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Björnsson, H.; Björnsson, E. Drug-induced liver injury: Pathogenesis, epidemiology, clinical features, and practical management. Eur. J. Intern. Med. 2022, 97, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef]
- Ou, P.; Liu, X.; Tang, Z.; Hou, Z.; Liu, L.; Liu, J.; Zhou, S.; Fang, Z.; Sun, K.; Chen, Y.; et al. Gynura Segetum Related Hepatic Sinusoidal Obstruction Syndrome: A Liver Disease with High Mortality and Misdiagnosis Rate. Curr. Pharm. Des. 2019, 25, 3762–3768. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Dara, L. Cell death in drug-induced liver injury. Adv. Pharmacol. 2019, 85, 31–74. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Xu, D.; Zhu, J.; Ge, G.; Kong, X.; Zhou, Y. Herbal Therapy for the Treatment of Acetaminophen-Associated Liver Injury: Recent Advances and Future Perspectives. Front. Pharmacol. 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Kuang, W.; Hao, W.; Liang, J.; Wu, L.; Tang, C.; Wang, Y.; Liu, T. Antituberculosis Drugs (Rifampicin and Isoniazid) Induce Liver Injury by Regulating NLRP3 Inflammasomes. Mediat. Inflamm. 2021, 2021, 8086253. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Zhang, T.; Wang, Q.; Xie, W. Drug-Induced Liver Injury from Anti-Tuberculosis Treatment: A Retrospective Cohort Study. Med. Sci. Monit. 2020, 26, e920350. [Google Scholar] [CrossRef]
- Wang, N.; Wang, H.; Zhang, J.; Ji, X.; Su, H.; Liu, J.; Wang, J.; Zhao, W. Endogenous peroxynitrite activated fluorescent probe for revealing anti-tuberculosis drug induced hepatotoxicity. Chin. Chem. Lett. 2022, 33, 1584–1588. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 2022, 21, 115–140. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Lim, P.L. Mitochondrial abnormalities—A link to idiosyncratic drug hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Liang, X.; Liu, H.; Zhang, F.; Meng, W.; Hu, F. Mitochondrial stress protein HSP60 regulates ER stress-induced hepatic lipogenesis. J. Mol. Endocrinol. 2020, 64, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Zhang, J.; Sha, Y.; Wu, F.; Wen, S.; He, L.; Sheng, L.; You, Q.; Shi, M.; et al. SPHK1 deficiency protects mice from acetaminophen-induced ER stress and mitochondrial permeability transition. Cell Death Differ. 2020, 27, 1924–1937. [Google Scholar] [CrossRef]
- Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid ceramidase improves mitochondrial function and oxidative stress in Niemann-Pick type C disease by repressing STARD1 expression and mitochondrial cholesterol accumulation. Redox Biol. 2021, 45, 102052. [Google Scholar] [CrossRef] [PubMed]
- Kheradpezhouh, E.; Zhou, F.H.; Barritt, G.J.; Rychkov, G.Y. Oxidative stress promotes redistribution of TRPM2 channels to the plasma membrane in hepatocytes. Biochem. Biophys. Res. Commun. 2018, 503, 1891–1896. [Google Scholar] [CrossRef]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salinas, C.S.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, X.; Shao, C.; Ye, L.; Guo, J.; Peng, N.; Zhang, H.; Li, J.; Kong, Y.; You, H.; et al. A proposed pathologic sub-classification of drug-induced liver injury. Hepatol. Int. 2019, 13, 339–351. [Google Scholar] [CrossRef]
- Hoofnagle, J.H.; Björnsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef]
- Björnsson, E.; Talwalkar, J.; Treeprasertsuk, S.; Kamath, P.S.; Takahashi, N.; Sanderson, S.; Neuhauser, M.; Lindor, K. Drug-induced autoimmune hepatitis: Clinical characteristics and prognosis. Hepatology 2010, 51, 2040–2048. [Google Scholar] [CrossRef]
- Jadhav, S.; Russo, S.; Cottier, S.; Schneiter, R.; Cowart, A.; Greenberg, M.L. Valproate Induces the Unfolded Protein Response by Increasing Ceramide Levels. J. Biol. Chem. 2016, 291, 22253–22261. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Qi, J.; Sun, Y.; Gao, X.; Ma, J.; Zhao, S. An integrated RNA-Seq and network study reveals that valproate inhibited progesterone production in human granulosa cells. J. Steroid Biochem. Mol. Biol. 2021, 214, 105991. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Grummich, H.; Birkmann, J.; Wilhelm, M.; Holzgrabe, U.; Sörgel, F. Severe Drug-Induced Liver Injury as an Adverse Drug Event of Antibiotics: A Case Report and Review of the Literature. Chemotherapy 2017, 62, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Liu, F.; Zhao, C.; Fan, L.; Hu, H.; Yin, S. Combination of oxytetracycline and quinocetone synergistically induces hepatotoxicity via generation of reactive oxygen species and activation of mitochondrial pathway. Toxicol. Mech. Methods 2022, 32, 49–57. [Google Scholar] [CrossRef]
- Björnsson, E.S. Drug-induced liver injury due to antibiotics. Scand. J. Gastroenterol. 2017, 52, 617–623. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, A.K.; Faridi, K.; Jain, P. Cefazolin Induced Liver Injury and Hypoprothrombinemia. J. Clin. Exp. Hepatol. 2018, 8, 213–214. [Google Scholar] [CrossRef]
- Zoubek, M.E.; Lucena, M.I.; Andrade, R.J.; Stephens, C. Systematic review: Ibuprofen-induced liver injury. Aliment. Pharmacol. Ther. 2020, 51, 603–611. [Google Scholar] [CrossRef]
- Wen, C.; Zhuang, Z.; Song, H.; Tong, S.; Wang, X.; Lin, Y.; Zhan, H.; Chen, Z.; Hu, L. Metabolism of liver CYP450 and ultrastructural changes after long-term administration of aspirin and ibuprofen. Biomed. Pharmacother. 2018, 108, 208–215. [Google Scholar] [CrossRef]
- Jung, S.-H.; Lee, W.; Park, S.-H.; Lee, K.-Y.; Choi, Y.-J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.-S.; Hong, S.-S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef]
- Gunther, M.; Dopheide, J.A. Antipsychotic Safety in Liver Disease: A Narrative Review and Practical Guide for the Clinician. J. Acad. Consult.-Liaison Psychiatry 2023, 64, 73–82. [Google Scholar] [CrossRef]
- Vukotić, N.T.; Đorđević, J.; Pejić, S.; Đorđević, N.; Pajović, S.B. Antidepressants- and antipsychotics-induced hepatotoxicity. Arch. Toxicol. 2021, 95, 767–789. [Google Scholar] [CrossRef]
- Shah, J.; Muir, J.; Furfaro, D.; Beitler, J.R.; Dzierba, A.L. Use of N-Acetylcysteine for Clozapine-Induced Acute Liver Injury: A Case Report and Literature Review. J. Pharm. Pract. 2021, 8971900211034007. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S. Hepatotoxicity of statins and other lipid-lowering agents. Liver Int. 2017, 37, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Meunier, L.; Larrey, D. Chemotherapy-associated steatohepatitis. Ann. Hepatol. 2020, 19, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Alessandrino, F.; Qin, L.; Cruz, G.; Sahu, S.; Rosenthal, M.H.; Meyerhardt, J.A.; Shinagare, A.B. 5-Fluorouracil induced liver toxicity in patients with colorectal cancer: Role of computed tomography texture analysis as a potential biomarker. Abdom. Radiol. 2019, 44, 3099–3106. [Google Scholar] [CrossRef]
- Honda, S.; Tsujimoto, M.; Minegaki, T.; Mori, T.; Muraoka, J.; Nishiguchi, K. A case of idiosyncratic liver injury after oxaliplatin-induced thrombocytopenia. J. Clin. Pharm. Ther. 2020, 45, 373–375. [Google Scholar] [CrossRef]
- Kim, J.-H.; Nam, W.S.; Kim, S.J.; Kwon, O.K.; Seung, E.J.; Jo, J.J.; Shresha, R.; Lee, T.H.; Jeon, T.W.; Ki, S.H.; et al. Mechanism Investigation of Rifampicin-Induced Liver Injury Using Comparative Toxicoproteomics in Mice. Int. J. Mol. Sci. 2017, 18, 1417. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, Y.; Li, Y. Pyrazinamide enhances lipid peroxidation and antioxidant levels to induce liver injury in rat models through PI3k/Akt inhibition. Toxicol. Res. 2020, 9, 149–157. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Chen, K.-H.; Chen, Y.-L.; Lin, S.-W.; Liu, W.-D.M.; Wang, J.-T.M.; Hung, C.-C.M. Pyrazinamide related prolonged drug-induced liver injury: A case report. Medicine 2022, 101, e30955. [Google Scholar] [CrossRef]
- Chalasani, N.; Bonkovsky, H.L.; Stine, J.G.; Gu, J.; Barnhart, H.; Jacobsen, E.; Björnsson, E.; Fontana, R.J.; Kleiner, D.E.; Hoofnagle, J.H. Clinical characteristics of antiepileptic-induced liver injury in patients from the DILIN prospective study. J. Hepatol. 2022, 76, 832–840. [Google Scholar] [CrossRef]
- Ezhilarasan, D.; Mani, U. Valproic acid induced liver injury: An insight into molecular toxicological mechanism. Environ. Toxicol. Pharmacol. 2022, 95, 103967. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-L.; Jing, X.; Sun, J.-Y.; Hu, Y.-H.; Xu, Z.-J.; Ni, M.-M.; Chen, F.; Lu, X.-P.; Qiu, J.-C.; Wang, T. Valproic Acid and the Liver Injury in Patients with Epilepsy: An Update. Curr. Pharm. Des. 2019, 25, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Liu, Y.-T.; Zeng, X.-C.; Li, C.-P.; Ou-Yang, D.-S. The hepatotoxicity of Polygonum multiflorum: The emerging role of the immune-mediated liver injury. Acta Pharmacol. Sin. 2021, 42, 27–35. [Google Scholar] [CrossRef]
- Li, H.; Peng, Y.; Zheng, J. Dioscorea bulbifera L.-induced hepatotoxicity and involvement of metabolic activation of furanoterpenoids. Drug Metab. Rev. 2020, 52, 568–584. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Fan, L.; Peng, C.; Zhang, Q.; Wang, L.; Li, L.; Wang, J.; Zhang, D.; Peng, W.; Wu, C. Traditional Uses, Botany, Phytochemistry, Pharmacology, Pharmacokinetics and Toxicology of Xanthium strumarium L.: A Review. Molecules 2019, 24, 359. [Google Scholar] [CrossRef]
- Lambert, A.; Cordeanu, M.; Gaertner, S.; Nouri, S.; Alt, M.; Stephan, D. Rivaroxaban-induced liver injury: Results from a venous thromboembolism registry. Int. J. Cardiol. 2015, 191, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Licata, A.; Puccia, F.; Lombardo, V.; Serruto, A.; Minissale, M.G.; Morreale, I.; Giannitrapani, L.; Soresi, M.; Montalto, G.; Almasio, P.L. Rivaroxaban-induced hepatotoxicity: Review of the literature and report of new cases. Eur. J. Gastroenterol. Hepatol. 2018, 30, 226–232. [Google Scholar] [CrossRef]
- Song, A.B.; Rosovsky, R.P.; Connors, J.M.; Al-Samkari, H. Direct oral anticoagulants for treatment and prevention of venous thromboembolism in cancer patients. Vasc. Health Risk Manag. 2019, 15, 175–186. [Google Scholar] [CrossRef]
- Machlab, S.; Miquel, M.; Vergara, M.; Escoda, M.R.; Casas, M. Apixaban-induced liver injury. Rev. Esp. Enfermadades Dig. 2018, 111, 161–163. [Google Scholar] [CrossRef]
- Saha, M.; Sikder, P.; Saha, A.; Shah, S.; Sultana, S.; Emran, T.; Banik, A.; Islam, Z.; Islam, M.S.; Sharker, M.S.; et al. QbD Approach towards Robust Design Space for Flutamide/PiperineSelf-Emulsifying Drug Delivery System with Reduced Liver Injury. AAPS PharmSciTech 2022, 23, 62. [Google Scholar] [CrossRef]
- Thole, Z.; Salgueiro-Vázquez, E.; Revuelta, P.; Manso, G.; Hidalgo, A. Hepatotoxicity Induced by Antiandrogens: A Review of the Literature. Urol. Int. 2004, 73, 289–295. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; Mikus, G. Ketoconazole and Liver Injury: A Five-Year Update. Clin. Pharmacol. Drug Dev. 2019, 8, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Ogimura, E.; Nakagawa, T.; Deguchi, J.; Sekine, S.; Ito, K.; Bando, K. Troglitazone Inhibits Bile Acid Amidation: A Possible Risk Factor for Liver Injury. Toxicol. Sci. 2017, 158, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Mohamed, M.; Alhail, M.; Awwad, F.A.; Wahba, R.M.; Hassan, S.B.; Omar, K.; El Kassem, W.; Rouf, P.A. A case of probable esomeprazole-induced transient liver injury in a pregnant woman with hyperemesis. Clin. Pharmacol. Adv. Appl. 2016, 8, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, K.; Nakagawa, E.; Kurata, T.; Iwamoto, T.; Okuda, M. Altered Pharmacokinetics of Cimetidine Caused by Down-regulation of Renal Rat Organic Cation Transporter 2 (rOCT2) after Liver Ischemia-Reperfusion Injury. Drug Metab. Pharmacokinet. 2013, 28, 504–509. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.-X. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef]
- Ishitsuka, Y.; Kondo, Y.; Kadowaki, D. Toxicological Property of Acetaminophen: The Dark Side of a Safe Antipyretic/Analgesic Drug? Biol. Pharm. Bull. 2020, 43, 195–206. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol. Res. Perspect. 2016, 4, e00211. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Kardon, T.; Wunderlich, L.; Szarka, A.; Kiss, A.; Schaff, Z.; Bánhegyi, G.; Mandl, J. Acetaminophen induces ER dependent signaling in mouse liver. Arch. Biochem. Biophys. 2007, 459, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Kusama, H.; Kon, K.; Ikejima, K.; Arai, K.; Aoyama, T.; Uchiyama, A.; Yamashina, S.; Watanabe, S. Sodium 4-phenylbutyric acid prevents murine acetaminophen hepatotoxicity by minimizing endoplasmic reticulum stress. J. Gastroenterol. 2017, 52, 611–622. [Google Scholar] [CrossRef]
- Torres, S.; Baulies, A.; Insausti-Urkia, N.; Alarcón-Vila, C.; Fucho, R.; Solsona-Vilarrasa, E.; Núñez, S.T.; Robles, D.; Ribas, V.; Wakefield, L.; et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019, 157, 552–568. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Umbaugh, D.S.; Nguyen, N.T.; Jaeschke, H.; Ramachandran, A. Mitochondrial Membrane Potential Drives Early Change in Mitochondrial Morphology After Acetaminophen Exposure. Toxicol. Sci. 2021, 180, 186–195. [Google Scholar] [CrossRef]
- Han, D.; Dara, L.; Win, S.; Than, T.A.; Yuan, L.; Abbasi, S.Q.; Liu, Z.-X.; Kaplowitz, N. Regulation of drug-induced liver injury by signal transduction pathways: Critical role of mitochondria. Trends Pharmacol. Sci. 2013, 34, 243–253. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Du, K.; Akakpo, J.Y.; Umbaugh, D.S.; Jaeschke, H.; Ramachandran, A. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol. Lett. 2021, 338, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Mironova, G.D.; Pavlov, E.V. Mitochondrial Cyclosporine A-Independent Palmitate/Ca(2+)-Induced Permeability Transition Pore (PA-mPT Pore) and Its Role in Mitochondrial Function and Protection against Calcium Overload and Glutamate Toxicity. Cells 2021, 10, 125. [Google Scholar] [CrossRef]
- Vanova, K.H.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef]
- Gheena, S.; Ezhilarasan, D.; Harini, K.S.; Rajeshkumar, S. Syringic acid and silymarin concurrent administration inhibits sodium valproate-induced liver injury in rats. Environ. Toxicol. 2022, 37, 2143–2152. [Google Scholar] [CrossRef]
- Mishra, M.K.; Kukal, S.; Paul, P.R.; Bora, S.; Singh, A.; Kukreti, S.; Saso, L.; Muthusamy, K.; Hasija, Y.; Kukreti, R. Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile. Molecules 2021, 27, 104. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef]
- Zhou, H.-Y.; Sun, Y.-Y.; Chang, P.; Huang, H.-C. Curcumin Inhibits Cell Damage and Apoptosis Caused by Thapsigargin-Induced Endoplasmic Reticulum Stress Involving the Recovery of Mitochondrial Function Mediated by Mitofusin-2. Neurotox. Res. 2022, 40, 449–460. [Google Scholar] [CrossRef]
- Zhou, H.; Gurley, E.C.; Jarujaron, S.; Ding, H.; Fang, Y.; Xu, Z.; Pandak, W.M.; Hylemon, P.B. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 291, G1071–G1080. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Pandak, W.M.; Lyall, V.; Natarajan, R.; Hylemon, P.B. HIV Protease Inhibitors Activate the Unfolded Protein Response in Macrophages: Implication for Atherosclerosis and Cardiovascular Disease. Mol. Pharmacol. 2005, 68, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Smyrnias, I. The mitochondrial unfolded protein response and its diverse roles in cellular stress. Int. J. Biochem. Cell Biol. 2021, 133, 105934. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 Is a Transcription Factor Specializing in the Regulation of Quality Control Proteins in the Endoplasmic Reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Garcia de la Cadena, S.; Massieu, L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis 2016, 21, 763–777. [Google Scholar] [CrossRef]
- Shi, X.Y.; Li, J.T. Endoplasmic reticulum stress in regulation of hepatic fibrosis. Zhonghua Gan Zang Bing Za Zhi 2018, 26, 865–868. [Google Scholar]
- Zhang, P.; McGrath, B.; Li, S.A.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, R.D. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef]
- You, K.; Wang, L.; Chou, C.-H.; Liu, K.; Nakata, T.; Jaiswal, A.; Yao, J.; Lefkovith, A.; Omar, A.; Perrigoue, J.G.; et al. QRICH1 dictates the outcome of ER stress through transcriptional control of proteostasis. Science 2021, 371, eabb6896. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of Caspase-12, an Endoplastic Reticulum (ER) Resident Caspase, through Tumor Necrosis Factor Receptor-associated Factor 2-dependent Mechanism in Response to the ER Stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, S.; Zhou, L.; Seyhan, A.A.; Borrero, L.H.; Zhang, Y.; El-Deiry, W.S. Targeting the Integrated Stress Response in Cancer Therapy. Front. Pharmacol. 2021, 12, 747837. [Google Scholar] [CrossRef]
- Abdel-Nour, M.; Carneiro, L.A.M.; Downey, J.; Tsalikis, J.; Outlioua, A.; Prescott, D.; Da Costa, L.S.; Hovingh, E.S.; Farahvash, A.; Gaudet, R.G.; et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 2019, 365, eaaw4144. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Büttner, S. Mitochondrial lipids in neurodegeneration. Cell Tissue Res. 2016, 367, 125–140. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T.; et al. The ER Stress Sensor PERK Coordinates ER-Plasma Membrane Contact Site Formation through Interaction with Filamin-A and F-Actin Remodeling. Mol. Cell 2017, 65, 885–899.e6. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Médard, J.-J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138 Pt 4, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Ali, E.S.; Rychkov, G.; Barritt, G.J. Deranged hepatocyte intracellular Ca2+ homeostasis and the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma. Cell Calcium 2019, 82, 102057. [Google Scholar] [CrossRef]
- Ali, E.S.; Rychkov, G.Y.; Barritt, G.J. Targeting Ca(2+) Signaling in the Initiation, Promotion and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 2755. [Google Scholar] [CrossRef]
- Hofmann, J.; Otarashvili, G.; Meszaros, A.; Ebner, S.; Weissenbacher, A.; Cardini, B.; Oberhuber, R.; Resch, T.; Öfner, D.; Schneeberger, S.; et al. Restoring Mitochondrial Function While Avoiding Redox Stress: The Key to Preventing Ischemia/Reperfusion Injury in Machine Perfused Liver Grafts? Int. J. Mol. Sci. 2020, 21, 3132. [Google Scholar] [CrossRef]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Zheng, L.; Tuo, B. Role of Ca2+ channels in non-alcoholic fatty liver disease and their implications for therapeutic strategies (Review). Int. J. Mol. Med. 2022, 50, 113. [Google Scholar] [CrossRef]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol–disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free. Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef]
- Li, Q.; Chen, F.; Wang, F. The immunological mechanisms and therapeutic potential in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Cell Biosci. 2022, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, F.; Huang, X.; Lee, C.Z.W.; Ginhoux, F.; Yu, H. Recent Advances in Models of Immune-Mediated Drug-Induced Liver Injury. Front. Toxicol. 2021, 3, 605392. [Google Scholar] [CrossRef]
- Shoda, L.K.; Battista, C.; Siler, S.Q.; Pisetsky, D.S.; Watkins, P.B.; Howell, B.A. Mechanistic Modelling of Drug-Induced Liver Injury: Investigating the Role of Innate Immune Responses. Gene Regul. Syst. Biol. 2017, 11, 1177625017696074. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Natalini, A.; Antonangeli, F.; Mancuso, C.; Agostinetto, E.; Barisani, D.; Di Rosa, F.; Andrade, R.; Invernizzi, P. Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models. Int. J. Mol. Sci. 2021, 22, 4557. [Google Scholar] [CrossRef] [PubMed]
- Verginadis, I.I.; Avgousti, H.; Monslow, J.; Skoufos, G.; Chinga, F.; Kim, K.; Leli, N.M.; Karagounis, I.V.; Bell, B.I.; Velalopoulou, A.; et al. A stromal Integrated Stress Response activates perivascular cancer-associated fibroblasts to drive angiogenesis and tumour progression. Nat. Cell Biol. 2022, 24, 940–953. [Google Scholar] [CrossRef]
- Gho, J.W.-M.; Ip, W.-K.; Chan, K.Y.-Y.; Law, P.T.-Y.; Lai, P.B.-S.; Wong, N. Re-Expression of Transcription Factor ATF5 in Hepatocellular Carcinoma Induces G2-M Arrest. Cancer Res. 2008, 68, 6743–6751. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Zhang, T.; Huang, W.; Ma, Y. Down-regulation of TRPM2 attenuates hepatic ischemia/reperfusion injury through activation of autophagy and inhibition of NLRP3 inflammasome pathway. Int. Immunopharmacol. 2022, 104, 108443. [Google Scholar] [CrossRef]
- Kheradpezhouh, E.; Ma, L.; Morphett, A.; Barritt, G.J.; Rychkov, G.Y. TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. USA 2014, 111, 3176–3181. [Google Scholar] [CrossRef]
- Ali, E.S.; Rychkov, G.Y.; Barritt, G.J. TRPM2 Non-Selective Cation Channels in Liver Injury Mediated by Reactive Oxygen Species. Antioxidants 2021, 10, 1243. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, P.; Lin, H.; Jin, Z.; Zhao, S.; Zhang, Y.; Xu, Q.; Liu, Z.; Yang, W.; Zhang, L. The Discovery of Novel ACA Derivatives as Specific TRPM2 Inhibitors that Reduce Ischemic Injury Both In Vitro and In Vivo. J. Med. Chem. 2021, 64, 3976–3996. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Saito, C.; Lemasters, J.J.; Jaeschke, H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2010, 246, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Chen, S.; Qing, T.; Xuan, J.; Couch, L.; Yu, D.; Ning, B.; Shi, L.; Guo, L. Endoplasmic reticulum stress and MAPK signaling pathway activation underlie leflunomide-induced toxicity in HepG2 Cells. Toxicology 2017, 392, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Yu, L.-R.; Abdelmegeed, M.A.; Gao, Y.; Banerjee, A.; Song, B.-J. Critical role of c-jun N-terminal protein kinase in promoting mitochondrial dysfunction and acute liver injury. Redox Biol. 2015, 6, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Gores, G.J.; Hirsova, P.; Kirby, M.; Miles, L.; Jaeschke, A.; Kohli, R. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver Int. 2014, 34, 427–437. [Google Scholar] [CrossRef]
- Min, R.W.M.; Aung, F.W.M.; Liu, B.; Arya, A.; Win, S. Mechanism and Therapeutic Targets of c-Jun-N-Terminal Kinases Activation in Nonalcoholic Fatty Liver Disease. Biomedicines 2022, 10, 2035. [Google Scholar] [CrossRef]
- Umbaugh, D.S.; Soder, R.P.; Nguyen, N.T.; Adelusi, O.; Robarts, D.R.; Woolbright, B.; Duan, L.; Abhyankar, S.; Dawn, B.; Apte, U.; et al. Human Wharton’s Jelly-derived mesenchymal stem cells prevent acetaminophen-induced liver injury in a mouse model unlike human dermal fibroblasts. Arch. Toxicol. 2022, 96, 3315–3329. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.-K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Wu, H.; Deng, R.; Wang, Y. Therapeutic Potential of SphK1 Inhibitors Based on Abnormal Expression of SphK1 in Inflammatory Immune Related-Diseases. Front. Pharmacol. 2021, 12, 733387. [Google Scholar] [CrossRef]
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.-G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Pulkoski-Gross, M.J.; Uys, J.D.; Orr-Gandy, K.A.; Coant, N.; Bialkowska, A.B.; Szulc, Z.M.; Bai, A.; Bielawska, A.; Townsend, D.M.; Hannun, Y.A.; et al. Novel sphingosine kinase-1 inhibitor, LCL351, reduces immune responses in murine DSS-induced colitis. Prostaglandins Other Lipid Mediat. 2017, 130, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Hengst, J.A.; Hegde, S.; Paulson, R.F.; Yun, J.K. Development of SKI-349, a dual-targeted inhibitor of sphingosine kinase and microtubule polymerization. Bioorganic Med. Chem. Lett. 2020, 30, 127453. [Google Scholar] [CrossRef]
- Alshaker, H.; Srivats, S.; Monteil, D.; Wang, Q.; Low, C.M.R.; Pchejetski, D. Field template-based design and biological evaluation of new sphingosine kinase 1 inhibitors. Breast Cancer Res. Treat. 2018, 172, 33–43. [Google Scholar] [CrossRef]
- Childress, E.S.; Kharel, Y.; Brown, A.M.; Bevan, D.R.; Lynch, K.R.; Santos, W.L. Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2 Selective Inhibitors: Design, Synthesis, and in Vivo Activity. J. Med. Chem. 2017, 60, 3933–3957. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Insuasty, D.; Lima, S.; Gutiérrez, L.; Nogueras, M.; Marchal, A.; Abonia, R.; Andújar, S.; Spiegel, S.; Cobo, J.; et al. Design of new quinolin-2-one-pyrimidine hybrids as sphingosine kinases inhibitors. Bioorganic Chem. 2020, 94, 103414. [Google Scholar] [CrossRef]
- Chen, H.-J.; Yang, H.-R.; Zhi, Y.; Yao, Q.-Q.; Liu, B. Evaluation of pyrrolidine-based analog of jaspine B as potential SphK1 inhibitors against rheumatoid arthritis. Bioorganic Med. Chem. Lett. 2021, 34, 127754. [Google Scholar] [CrossRef]
- Sun, D.; Wang, S. Sphingosine kinases are involved in the regulation of all-trans retinoic acid sensitivity of K562 chronic myeloid leukemia cells. Oncol. Lett. 2021, 22, 581. [Google Scholar] [CrossRef]
- Sah, R.K.; Pati, S.; Saini, M.; Singh, S. Erythrocyte sphingosine kinase regulates intraerythrocytic development of Plasmodium falciparum. Sci. Rep. 2021, 11, 1257. [Google Scholar] [CrossRef]
- Hafizi, R.; Imeri, F.; Wenger, R.H.; Huwiler, A. S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2α Stabilization. Int. J. Mol. Sci. 2021, 22, 9467. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Macrophage Sphingosine 1-Phosphate Receptor 2 Blockade Attenuates Liver Inflammation and Fibrogenesis Triggered by NLRP3 Inflammasome. Front. Immunol. 2020, 11, 1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Y.; Sun, X.-J.; Wang, C.; Chen, S.-F.; Li, Z.-Y.; Chen, M.; Little, M.A.; Zhao, M.-H. Sphingosine-1-phosphate receptor modulator FTY720 attenuates experimental myeloperoxidase-ANCA vasculitis in a T cell-dependent manner. Clin. Sci. 2020, 134, 1475–1489. [Google Scholar] [CrossRef]
- Aye, I.L.; Waddell, B.J.; Mark, P.J.; Keelan, J.A. Oxysterols exert proinflammatory effects in placental trophoblasts via TLR4-dependent, cholesterol-sensitive activation of NF-κB. Mol. Hum. Reprod. 2012, 18, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Liu, B.; Cheng, Y.; Qian, H.; Yang, H.; Li, X.; Yang, G.; Zheng, X.; Shen, F. SN50 attenuates alveolar hypercoagulation and fibrinolysis inhibition in acute respiratory distress syndrome mice through inhibiting NF-κB p65 translocation. Respir. Res. 2020, 21, 130. [Google Scholar] [CrossRef]
- Yan, Y.; Qian, H.; Cao, Y.; Zhu, T. Nuclear factor-κB inhibitor Bay11-7082 inhibits gastric cancer cell proliferation by inhibiting Gli1 expression. Oncol. Lett. 2021, 21, 301. [Google Scholar] [CrossRef]
- Pao, H.-P.; Liao, W.-I.; Wu, S.-Y.; Hung, K.-Y.; Huang, K.-L.; Chu, S.-J. PG490-88, a derivative of triptolide, suppresses ischemia/reperfusion-induced lung damage by maintaining tight junction barriers and targeting multiple signaling pathways. Int. Immunopharmacol. 2019, 68, 17–29. [Google Scholar] [CrossRef]
- El-Hashim, A.Z.; Renno, W.M.; Abduo, H.T.; Jaffal, S.M.; Akhtar, S.; Benter, I.F. Effect of inhibition of the ubiquitin-proteasome-system and IκB kinase on airway inflammation and hyperresponsiveness in a murine model of asthma. Int. J. Immunopathol. Pharmacol. 2011, 24, 33–42. [Google Scholar] [CrossRef]
- Li, S.-Z.; Zhang, H.-H.; Zhang, J.-N.; Zhang, Z.-Y.; Zhang, X.-F.; Du, R.-L. ALLN hinders HCT116 tumor growth through Bax-dependent apoptosis. Biochem. Biophys. Res. Commun. 2013, 437, 325–330. [Google Scholar] [CrossRef]
- Kelly-Laubscher, R.; Somers, S.; Lacerda, L.; Lecour, S. Role of nuclear factor kappa-B in TNF-induced cytoprotection. Cardiovasc. J. Afr. 2022, 33, 1–7. [Google Scholar] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-Methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a Potent and Selective First-in-Class Inhibitor of Protein Kinase R (PKR)-like Endoplasmic Reticulum Kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.-S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.-Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Andrews, K.L.; Beckmann, H.; Bellon, S.F.; Beltran, P.J.; Booker, S.; Chen, H.; Chung, Y.-A.; D’Angelo, N.D.; Dao, J.; et al. Discovery of 1H-Pyrazol-3(2H)-ones as Potent and Selective Inhibitors of Protein Kinase R-like Endoplasmic Reticulum Kinase (PERK). J. Med. Chem. 2015, 58, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Perera, B.G.K.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schürer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.C.; Vidadala, V.N.; Potter, Z.E.; Papa, F.R.; Backes, B.J.; Maly, D.J. Development of a Chemical Toolset for Studying the Paralog-Specific Function of IRE1. ACS Chem. Biol. 2019, 14, 2595–2605. [Google Scholar] [CrossRef]
- Lebeau, P.; Byun, J.H.; Yousof, T.; Austin, R.C. Pharmacologic inhibition of S1P attenuates ATF6 expression, causes ER stress and contributes to apoptotic cell death. Toxicol. Appl. Pharmacol. 2018, 349, 1–7. [Google Scholar] [CrossRef]
- Torres, S.E.; Gallagher, C.M.; Plate, L.; Gupta, M.; Liem, C.R.; Guo, X.; Tian, R.; Stroud, R.M.; Kampmann, M.; Weissman, J.S.; et al. Ceapins block the unfolded protein response sensor ATF6alpha by inducing a neomorphic inter-organelle tether. eLife 2019, 8, e46595. [Google Scholar] [CrossRef]
- Ha, T.K.; Hansen, A.H.; Kildegaard, H.F.; Lee, G.M. BiP Inducer X: An ER Stress Inhibitor for Enhancing Recombinant Antibody Production in CHO Cell Culture. Biotechnol. J. 2019, 14, e1900130. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class | Compound | Drug Target | Damage Type | References |
|---|---|---|---|---|
| Antibiotics | Clindamycin | Hepatocyte; Bile duct epithelial | Hepatitis; Hepatic failure | [23] |
| Oxytetracycline | Hepatocyte; Vascular injury | Fatty degeneration | [24] | |
| Cefazolin | Hepatocyte | Hepatitis | [25,26] | |
| Nitrofurantoin | Hepatocyte | Hepatitis; Jaundice; Necrosis; Fibrosis | [25] | |
| Non-steroidal anti-inflammatory drugs | Acetaminophen | Hepatocyte | Hepatonecrosis | [4,5] |
| Ibuprofen | Hepatocyte; Bile duct epithelial; Vascular injury | Hepatocyte damage; Hepatic failure; Acute hepatitis | [27,28] | |
| Aspirin | Hepatocyte; Bile duct epithelial; Vascular injury | Hepatic enzymes elevations; Jaundice; Acute hepatitis | [28] | |
| Diclofenac | Hepatocyte | Hepatitis; Jaundice; Hepatauxe | [29] | |
| Antipsychotics | Risperidone | Hepatocyte; Bile duct epithelial | Hepatitis | [30,31] |
| Clozapine | Hepatocyte; Bile duct epithelial | Fulminant liver failure | [32] | |
| Antidepressant | Amitriptyline | Hepatocyte; Bile duct epithelial | Hepatitis; Fulminant liver failure or death | [31] |
| Trazodone | ||||
| Agomelatine | ||||
| Bupropion | ||||
| Iproniazid | ||||
| Duloxetine | ||||
| Imipramine | ||||
| Nefazodone | ||||
| Tianeptine | ||||
| Phenelzine | ||||
| Lipid-lowering drugs | Atorvastatin | Hepatocyte; Bile duct epithelial; Vascular injury | Hepatitis; Jaundice | [33] |
| Chemotherapeutics | Fluorouracil | Hepatocyte | Hepatitis; Steatosis | [34,35] |
| Irinotecan | Hepatocyte | Hepatitis; Steatosis | [34] | |
| Oxaliplatin | Hepatocyte; Vascular injury | Hepatic sinusoidal obstruction syndrome | [36] | |
| Antituberculosis drugs | Isoniazid | Hepatocyte; Bile duct epithelial | Hepatitis; Necrosis | [6,7,8] |
| Rifampicin | Hepatocyte; Bile duct epithelial | Hepatitis; Necrosis | [6,7,37] | |
| Pyrazinamide (PZA) | Hepatocyte; Vascular injury | Hepatitis; Necrosis | [38,39] | |
| Antiepileptic drugs | Lamotrigine | Hepatocyte | Hepatic enzymes elevations; Death; Necrosis | [40] |
| Valproic acid (VPA) | Hepatocyte | Hepatitis; | [41,42] | |
| Traditional Chinese medicine (TCM) | HeShouWu | Hepatocyte | Hepatitis; Jaundice; Necrosis | [43] |
| JuSanQi | Hepatocyte; Vascular injury | Hepatitis; Hepatic sinusoidal obstruction syndrome | [3] | |
| Dioscorea bulbifera L. | Hepatocyte; Bile duct epithelial; Vascular injury | Hepatitis | [44] | |
| CangErZi | Hepatocyte; Vascular injury | Hepatitis | [45] | |
| Oral anticoagulants | Rivaroxaban | Hepatocyte; Bile duct epithelial; Vascular injury | Hepatitis; Hepatic enzymes elevations; Jaundice | [46,47] |
| Apixaban | Hepatocyte; Bile duct epithelial; | Hepatitis | [48,49] | |
| Antiandrogen drug | Flutamide | Hepatocyte | Hepatitis; Jaundice; ALF | [50,51] |
| Antimicrobial | Ketoconazole | Hepatocyte | Liver cirrhosis; Jaundice | [52] |
| Hypoglycemic agents | Troglitazone * | Hepatocyte | ALF | [53] |
| Acid-inhibitory drugs | Esomeprazole | Hepatocyte | Hepatic enzymes elevations | [54] |
| Cimetidine | Hepatocyte | Jaundice; ALF | [55] |
| Pathway | Class | Compound | Comments | Target | References |
|---|---|---|---|---|---|
| SPHK1-STARD pathway | SPHK1 inhibitors | PF543 | Sphingosine kinase inhibitor | SPHK1 | [14,131] |
| SK1-I | Enhances autophagy and cancer cell death | SPHK1 | [132,133] | ||
| LCL351 | Reduces the expression of pro-inflammatory cytokine | SPHK1 | [134] | ||
| SKI-178/349 | High selectivity and low toxicity | ATP- binding site | [135] | ||
| SK-F | Without significant systemic toxicity | SPHK1 | [136] | ||
| SLC4011540 | High cell permeability | SPHK1 | [137] | ||
| 11b | Selective inhibition of SPHK1 | SPHK1 | [138] | ||
| CHJ01 | Anti-inflammatory | SPHK1 | [139] | ||
| SK1-II | Up-regulates Cer level | SPHK1 | [140] | ||
| DMS | Causes severe hemolysis in mice | SPHK1 | [141] | ||
| S1P inhibitors | NIBR0213 | Reduces peripheral blood lymphocyte counts | S1P1 | [142] | |
| JTE013 | Inhibits inflammasome priming and inflammatory cytokine | S1P2 | [143] | ||
| FTY720 | Reduces and inhibits T cells | S1P3 | [144] | ||
| TRPM2 pathway | TRPM2 inhibitors | Curcumin | Prevents ROS-induced liver injury | TRPM2 | [120] |
| Compound A23 | Highly active and selective | TRPM2 | [121] | ||
| JNK pathway | JNK inhibitor | SP600125 | Inhibits mitochondrial stress | JNK1/2/3 | [123] |
| LEF | Inhibits JNK1/2 phosphorylation | JNK1/2 | [124] | ||
| BI-78D3 | Binds D-domain of JIP1 | D-domain of JIP1 | [125] | ||
| ASK1 inhibitor | GS-444217 | Noneffective in late-phase clinical trials | ASK1 | [126] | |
| NF-κB pathway | IKK inhibitors | Parthenolide | Specific IkB inhibitor | IkB | [145] |
| NF-κB inhibitors | SN50 | NF-κB inhibitory peptides for permeable cells | NF-κB | [146] | |
| BAY11-7082 | Prevents IkBα phosphorylation | NF-κB | [147] | ||
| Immunosuppressive agents | PG490 | Target gene transcription inhibitor | P65 | [148] | |
| Protease inhibitors | MG-132 | Inhibits phosphorylation of IkBα | IkBα | [149] | |
| Antioxidants | ALLN | Prevents IkBα degradation | IkBα | [150] | |
| PDTC | Prevents IkB release | IkB | [151] | ||
| UPR pathway | PERK inhibitors | GSK2606414 | RIPK inhibitor; KIT inhibitor | EIF2AK3 | [152,153] |
| GSK2656157 | RIPK inhibitor | ATP- binding site | [154,155] | ||
| PERK-IN-2 | Low renal clearance | ATP- binding site | [152] | ||
| PERK-IN-3 | High renal clearance | ATP- binding site | [152] | ||
| AMG PERK 44 | GCN2 inhibitor | PERK | [156] | ||
| IRE1 inhibitors | Compound 3 | Compound 3 disfavors IRE1 oligomerization | STING | [157,158] | |
| Compound 15 | EGFR inhibitor | EGFR | [157,158] | ||
| ATF6 inhibitors | PF-429242 | Inhibits SREBP signaling | S1P | [159] | |
| Ceapin | Does not affect ABCD3 function | Tethers ATF6 cytosolic domain and ABCD3 | [160] | ||
| Bip inducer | BIX | ERS inhibitor | Bip | [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, S.; Pan, Y.; Zhang, Q.; You, T.; Yue, T.; Zhang, Y.; Wang, M. Endoplasmic Reticulum Stress and Mitochondrial Stress in Drug-Induced Liver Injury. Molecules 2023, 28, 3160. https://doi.org/10.3390/molecules28073160
Pu S, Pan Y, Zhang Q, You T, Yue T, Zhang Y, Wang M. Endoplasmic Reticulum Stress and Mitochondrial Stress in Drug-Induced Liver Injury. Molecules. 2023; 28(7):3160. https://doi.org/10.3390/molecules28073160
Chicago/Turabian StylePu, Sisi, Yangyang Pan, Qian Zhang, Ting You, Tao Yue, Yuxing Zhang, and Meng Wang. 2023. "Endoplasmic Reticulum Stress and Mitochondrial Stress in Drug-Induced Liver Injury" Molecules 28, no. 7: 3160. https://doi.org/10.3390/molecules28073160
APA StylePu, S., Pan, Y., Zhang, Q., You, T., Yue, T., Zhang, Y., & Wang, M. (2023). Endoplasmic Reticulum Stress and Mitochondrial Stress in Drug-Induced Liver Injury. Molecules, 28(7), 3160. https://doi.org/10.3390/molecules28073160