
Diastereoselective Synthesis of (–)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid via Morpholinone Derivatives



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
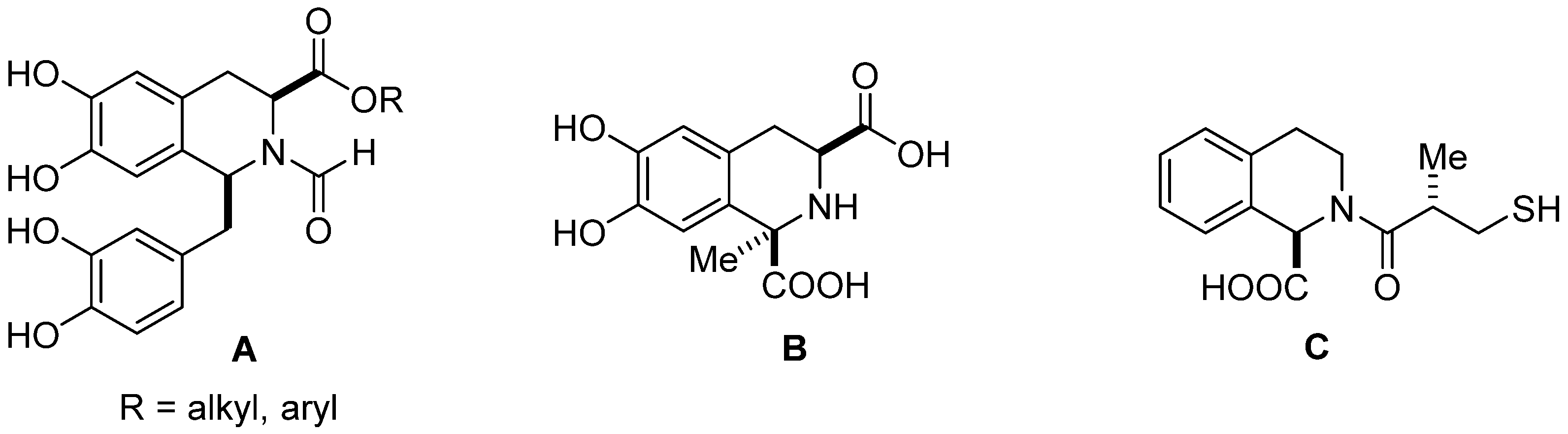
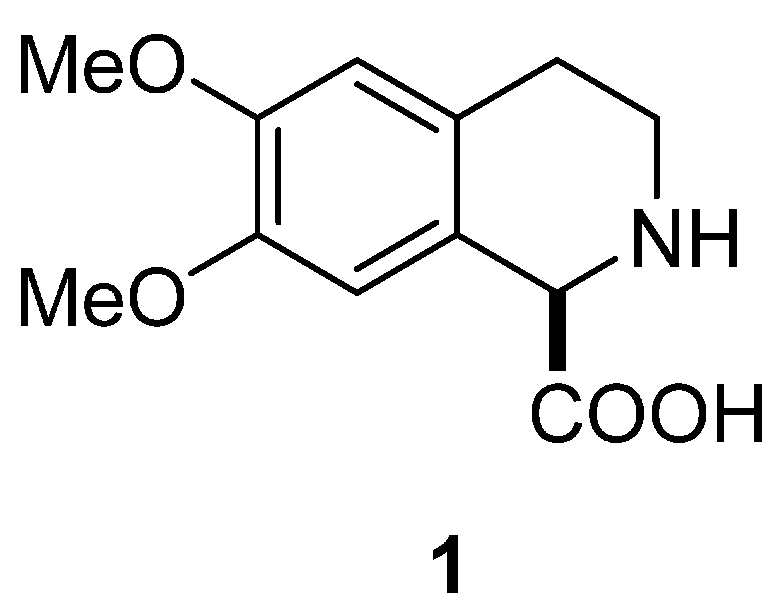
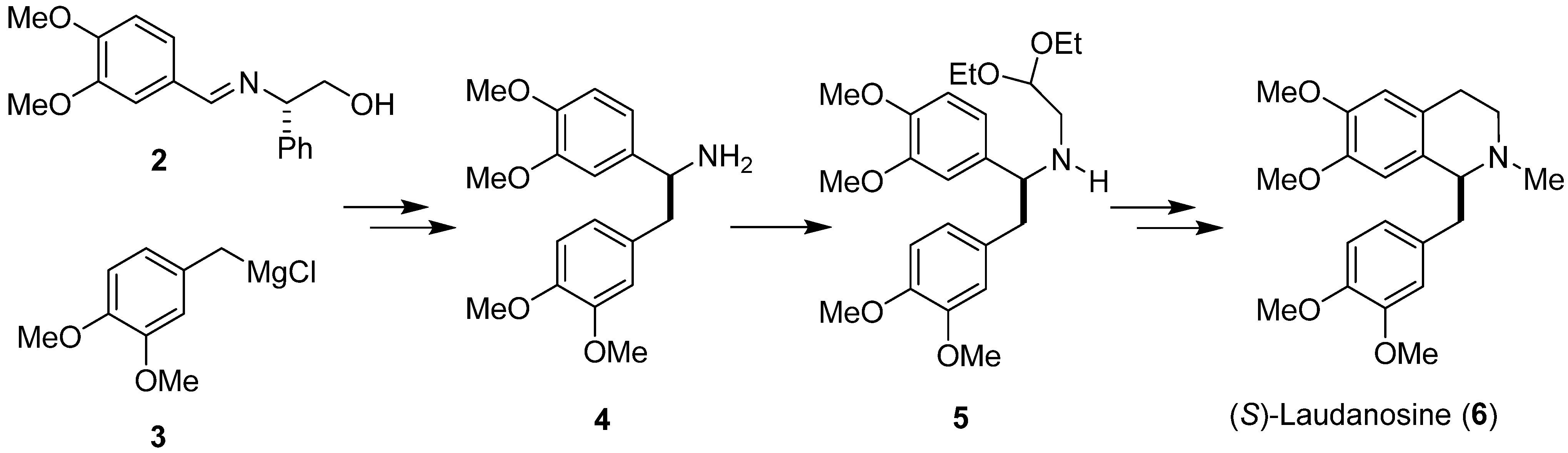
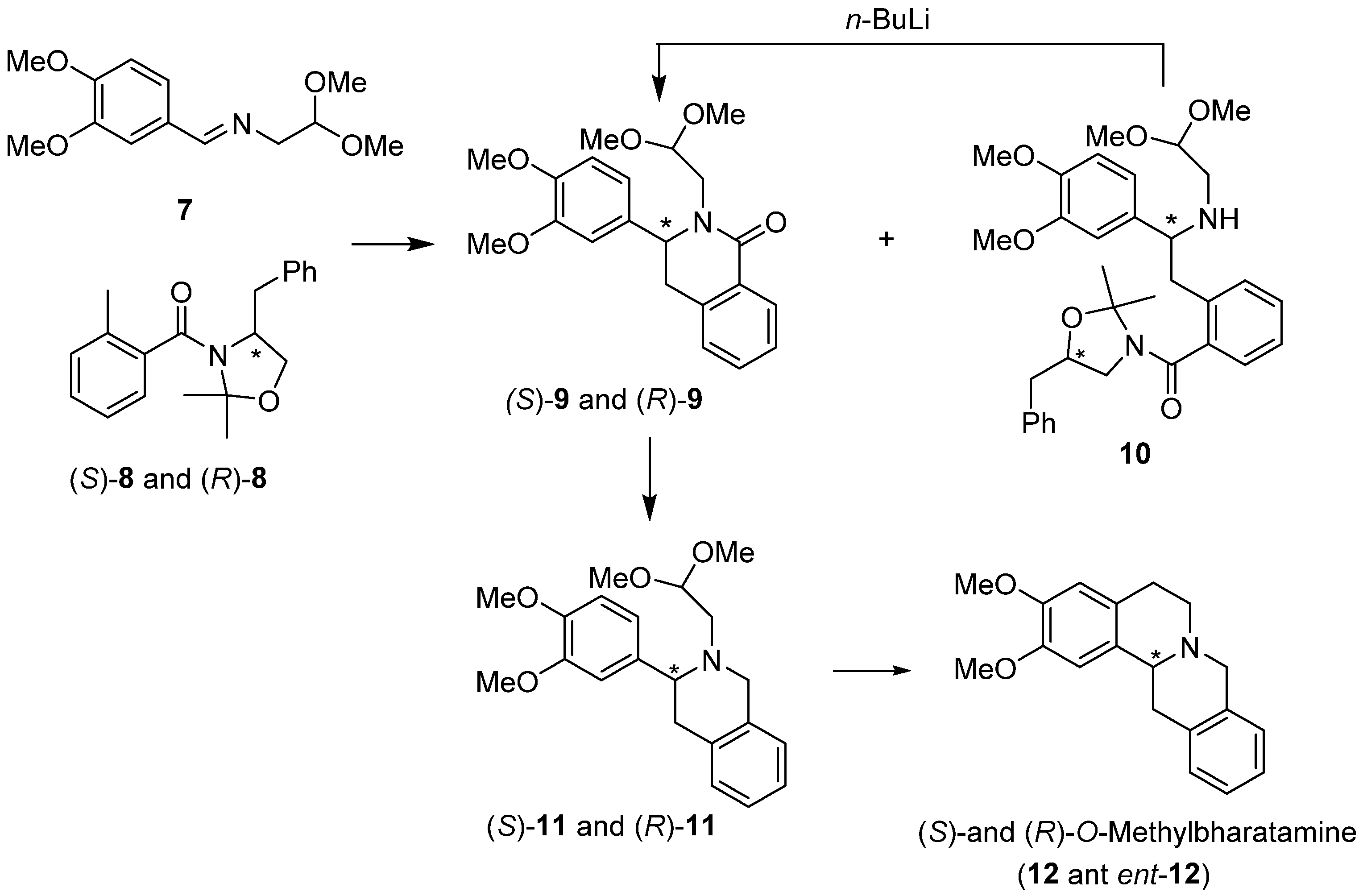
1. Introduction
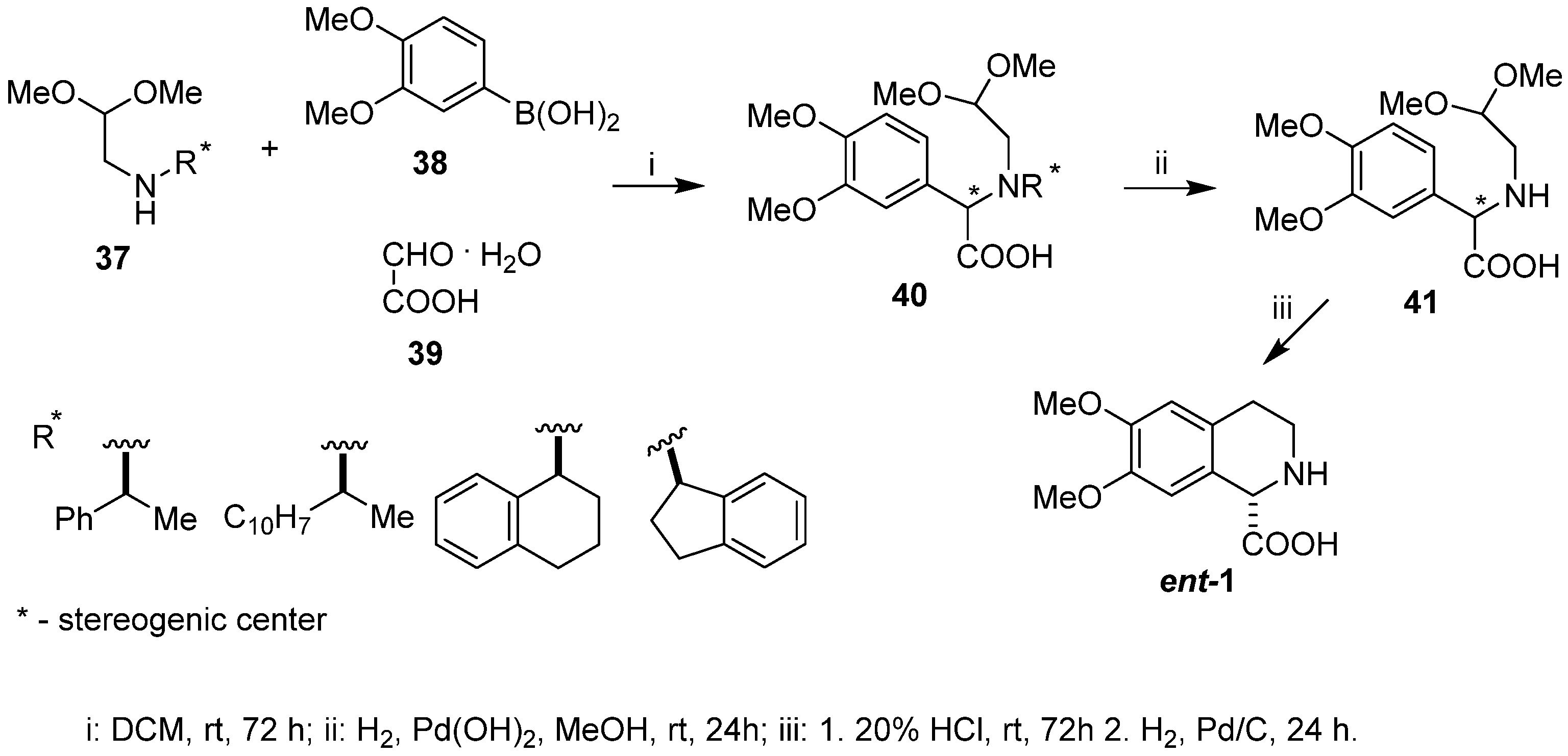
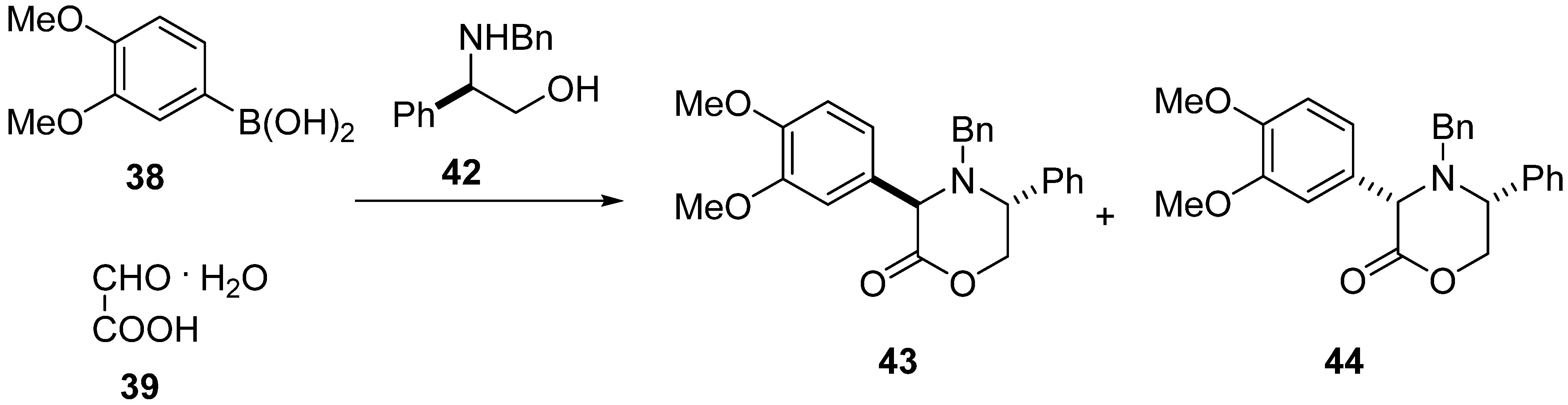
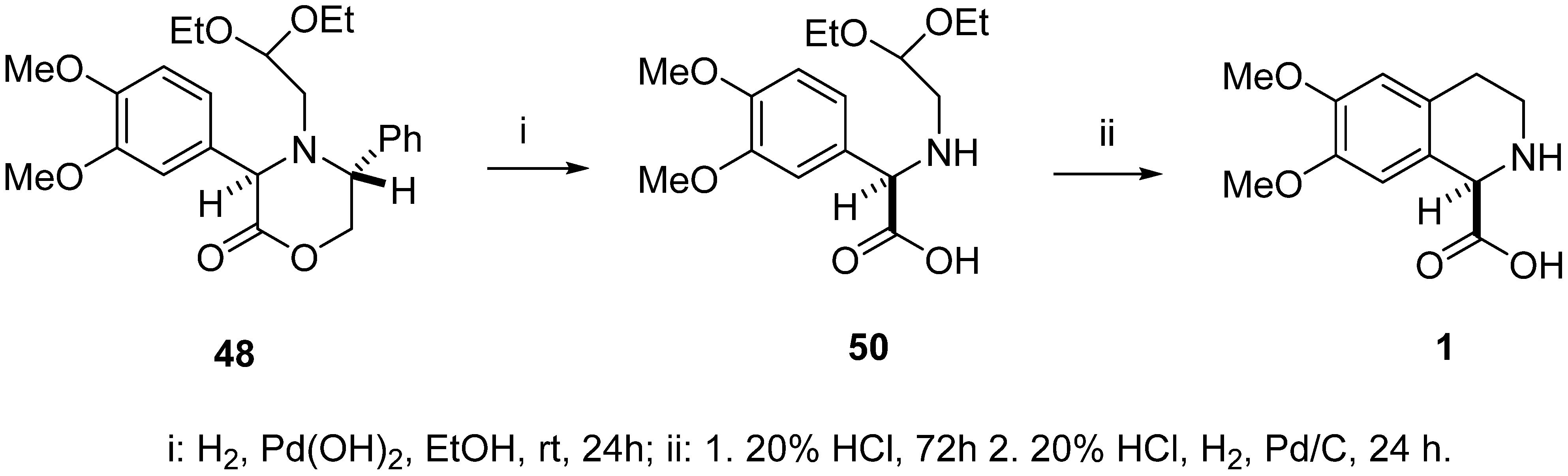
2. Results
3. Conclusions
4. Experimental Section
4.1. General
4.2. (R)-(–)-N-(2,2-Diethoxyethyl)-2-phenylglycinol (47)
4.3. Synthesis of (3R,5R) and (3S,5R)-4-(2,2-diethoxyethyl)-3-(3,4-dimethoxyphenyl)-5-phenyl-1,4-oxazin-2-ones (48) and (49)
4.4. (R)-(–)-N-(2,2-Diethoxyethyl)-3,4-dimethoxyphenylglycine (50)
4.5. (R)-(–)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid (1)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shang, X.-F.; Yang, C.-J.; Morris-Natschke, S.L.; Li, J.-C.; Yin, X.-D.; Liu, Y.-Q.; Guo, X.; Peng, J.-W.; Goto, M.; Zhang, J.-Y.; et al. Biologically active isoquinoline alkaloids covering 2014–2018. Med. Res. Rev. 2020, 40, 2212–2289. [Google Scholar] [CrossRef] [PubMed]
- Plazas, E.; Avila, M.M.C.; Munoz, D.R.; Cuca, S.L.E. Natural isoquinoline alkaloids: Pharmacological features and multi-target potential for complex diseases. Pharmacol. Res. 2022, 177, 106126. [Google Scholar] [CrossRef] [PubMed]
- Faheem; Kumar, B.K.; Chandra Sekhar, K.V.G.; Chander, S.; Kunjiappan, S.; Murugesan, S. Medicinal chemistry perpectives of 1,2,3,4-tetrahydroisoquinoline analogs — biological activities and SAR studies. RSC Adv. 2021, 11, 12254–12287. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, I.P.; Shah, P. Tetrahydroisoquinolines in therapeutics: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 17–36. [Google Scholar] [CrossRef]
- Faheem; Kumar, B.K.; Chandra Sekhar, K.V.G.; Chander, S.; Kunjiappan, S.; Murugesan, S. 1,2,3,4-Tetrahydroisoquinoline (THIQ) as privileged scaffold for anticancer de novo drug design. Expert Opin. Drug Discov. 2021, 16, 119–1147. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, N.; Manchanda, N.; Prasad, S.K.; Sharma, P.C.; Thakur, V.K.; Rahman, M.M.; Dhobi, M. Bioactivity and in Silico Studies of Isoquinoline and Related Alkaloids as Promising Antiviral Agents: An Insight. Biomolecules 2023, 13, 17. [Google Scholar] [CrossRef]
- Ju, S.; Qian, M.; Xu, G.; Yang, L.; Wu, J. Chemoenzymatic Approach to (S)-1,2,3,4-Tetrahydroisoquinoline Carboxylic Acids Employing D-Amino Acid Oxidase. Adv. Synth. Catal. 2019, 361, 3101–3199. [Google Scholar] [CrossRef]
- Ju, S.; Qian, M.; Li, J.; Xu, G.; Yang, L.; Wu, J. A biocatalytic redox cascade approach for one-pot deracemization of carboxyl-substituted tetrahydroisoquinolines by stereoinversion. Green Chem. 2019, 21, 5579–5585. [Google Scholar] [CrossRef]
- Liu, Z.; Gu, S.; Zhu, X.; Liu, M.; Cao, Z.; Qiu, P.; Li, S.; Liu, S.; Song, G. Discovery and optimization of new 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline derivatives as potent influenza virus PAN inhibitors. Eur. J. Med. Chem. 2022, 227, 113929. [Google Scholar] [CrossRef]
- Parrales-Macias, V.; Harfouche, A.; Ferrie, L.; Haik, S.; Michel, P.P.; Raisman-Vozari, R.; Figadere, B.; Bizat, N.; Maciuk, A. Effects of a New Natural Cathechol-O-methyl Transferase Inhibitor on Two In Vivo Models of Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 3303–3313. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, S.; Wu, K.; Zhang, W.; Ahmad, A.; Hao, Q.; Lei, X.; Zhang, H. Structure-guided optimization of D-captopril for discovery of potent NDM-1 inhibitors. Bioorg. Med. Chem. 2021, 29, 115902. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, M.; Rozwadowska, M.D. Asymmetric Synthesis of Isoquinoline Alkaloids. Chem. Rev. 2004, 104, 3341–3370. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.D. Asymmetric Synthesis of Isoquinoline Alkaloids: 2004–2016. Chem. Rev. 2016, 116, 12369–12465. [Google Scholar] [CrossRef]
- Shamma, M.; Moniot, L. Isoquinoline Alkaloids Research; Plenum Press: New York, NY, USA, 1978. [Google Scholar]
- Williams, R.M.; Hendrix, J.A. Asymmetric synthesis of aryl glycines. Chem. Rev. 1992, 92, 889–917. [Google Scholar] [CrossRef]
- Paal, T.A.; Liljeblad, A.; Kanerva, L.T.; Forro, E.; Fülöp, F. Directed (R)- or (S)-Selective Dynamic Kinetic Enzymatic Hydrolysis of 1,2,3,4-Tetrahydroisoquinoline-carboxylic Esters. Eur. J. Org. Chem. 2008, 31, 5269–5276. [Google Scholar] [CrossRef]
- Bułyszko, I.; Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.D. Synthesis of (+)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid, a Diastereoselective Approach. Eur. J. Org. Chem. 2015, 2015, 383–388. [Google Scholar] [CrossRef]
- Fritsch, P. Synthesen in der Isocumarin- und Isochinolinreihe. Ber. Dtsch. Chem. Ges. 1893, 26, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Bobbitt, J.M.; Moore, T.E. Synthesis of Isoquinolines. VIII. J. Org. Chem. 1968, 33, 2958–2959. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; Winter, D.P.; Kiely, J.M. Synthesis of Isoquinolines. IV. 4-benzylisoquinolines. J. Org. Chem. 1965, 30, 2459–2460. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; McNew Kiely, J.; Khanna, K.L.; Ebermann, R. Synthesis of Isoquinolines. III. A new Synthesis of 1,2,3,4-tetrahydroisoquinolines. J. Org. Chem. 1965, 30, 2247–2250. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; Roy, D.N.; Marchand, A.; Allen, C.W. Synthesis of Isoquinolines. VI. N-Alkyl-1,2,3,4-tetrahydroisoquinolines. J. Org. Chem. 1967, 32, 2225–2227. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; Steinfeld, A.S.; Weisgraber, K.H.; Dutta, S. Synthesis of Isoquinolines. X. 1-Alkyl-1,2,3,4-tetrahydroisoquinolines. J. Org. Chem. 1969, 34, 2478–2479. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; Sih, J.C. Synthesis of Isoquinolines. VII. 4-hydroxy-1,2,3,4-tetrahydroisoquinolines. J. Org. Chem. 1968, 33, 856–858. [Google Scholar] [CrossRef]
- Petasis, N.A. Expanding Roles for Organoboron Compounds—Versatile and Valuable Molecules for Synthetic, Biological, and Medicinal Chemistry. Aust. J. Chem. 2007, 60, 795–798. [Google Scholar] [CrossRef]
- Candeias, N.R.; Montalbano, F.; Cal, P.M.S.D.; Gois, P.M.P. Boronic Acids and Esters in the Petasis-Borono Mannich Multicomponent Reaction. Chem. Rev. 2010, 110, 6169–6193. [Google Scholar] [CrossRef]
- Wu, P.; Givskov, M.; Nielsen, T.E. Reactivity and Synthetic Applications of Multicomponent Petasis Reaction. Chem. Rev. 2019, 119, 11245–11290. [Google Scholar] [CrossRef] [Green Version]
- Batey, R.A. Nucleophilic Addition Reactions of Aryl and Alkenylboronic Acids and Their Derivatives to Imines and Iminium Ions. In Boronic Acids; Hall, D.G., Ed.; Wiley VCH: Weinheim, Germany, 2005; pp. 279–304. [Google Scholar]
- Pyne, S.G.; Tang, M. The Boronic Acid Mannich Reaction. Org. React. 2014, 83, 211–498. [Google Scholar] [CrossRef]
- Pomeranz, C. Über eine neue Isochinolinsynthese. Monatsh. Chem. 1893, 14, 116–119. [Google Scholar] [CrossRef]
- Schlittler, E.; Müller, J. Über die Konstitution des Isothebaines. Helv. Chim. Acta 1948, 31, 1119–1132. [Google Scholar] [CrossRef]
- Birch, A.J.; Jackson, A.H.; Shannon, P.V.R. A new modification of the Pomeranz-Fritsch isoquinoline synthesis. J. Chem. Soc. Perkin Trans. 1974, 1, 2185–2190. [Google Scholar] [CrossRef]
- Anakabe, E.; Carrillo, L.; Badia, D.; Vicario, J.L.; Villegas, M. Stereoselective Synthesis of Aporphinane Alkaloids Using a Hypervalent Iodine(III) Reagent-Promoted Oxidative Nonphenolic Biaryl Coupling Reaction. Total Synthesis of (S)-(+)-Glaucine. Synthesis 2004, 7, 1093–1101. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Dreas, A.; Rozwadowska, M.D. Synthesis of (S)-(–)- and (R)-(+)-O-methylbharatamine using a diastereoselective Pomeranz-Fritsch-Bobbitt methodology. Tetrahedron Asymmetry 2005, 16, 2954–2958. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Dreas, A. The Asymmetric Synthesis of (R)-(+)- and (S)-(–)-O-Methylbharatamine. Tetrahedron Asymmetry 2004, 15, 2561–2567. [Google Scholar] [CrossRef]
- Grajewska, A.; Rozwadowska, M.D. Diastereoselective Pomeranz–Fritsch–Bobbitt Synthesis of (S)-(–)-O-Methylbharatamine using (S)-N-tert-Butanesulfinimine as a Substrate. Tetrahedron Asymmetry 2007, 18, 2910–2914. [Google Scholar] [CrossRef]
- Boudou, M.; Enders, D. Asymmetric Synthesis of Tetrahydropalmatine via Tandem 1,2-Addition/Cyclization. J. Org. Chem. 2005, 70, 9486–9494. [Google Scholar] [CrossRef]
- Kościołowicz, A.; Rozwadowska, M.D. Diastereoselective Pomeranz–Fritsch–Bobbitt Synthesis of (S)-(–)-Salsolidine using (R)-N-tert-Butanesulfinylimine as a Substrate. Tetrahedron Asymmetry 2006, 17, 1444–1448. [Google Scholar] [CrossRef]
- Grajewska, A.; Rozwadowska, M.D. Total Synthesis of (R)-(+)-Salsolidine by Hydride Addition to (R)-N-tert-Butanesulfinyl Ketimine. Tetrahedron Asymmetry 2007, 18, 557–561. [Google Scholar] [CrossRef]
- Głuszyńska, A.; Rozwadowska, M.D. Enantioselective Addition of Methyllithium to a Prochiral Imine–the Substrate in the Pomeranz–Fritsch–Bobbitt Synthesis of Tetrahydroisoquinoline Derivatives Mediated by Chiral Monooxazolines. Tetrahedron Asymmetry 2004, 15, 3289–3295. [Google Scholar] [CrossRef]
- Głuszyńska, A.; Rozwadowska, M.D. Enantioselective modification of the Pomeranz–Fritsch–Bobbitt synthesis of tetrahydroisoquinoline alkaloids: Synthesis of (–)-salsolidine and (–)-carnegine. Tetrahedron Asymmetry 2000, 11, 2359–2366. [Google Scholar] [CrossRef]
- Ji, X.; Huang, Z.; Lumb, J.-P. Synthesis of 1,2-Dihydroisoquinolines by a Modified Pomeranz-Fritsch Cyclization. J. Org. Chem. 2020, 85, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, H.; Okitsu, T.; Sawama, Y.; Murata, N.; Li, R.; Kita, Y. Reaction of the Acetals with TESOTf-Base Combination; Speculation of the Intermediates and Efficient Mixed Acetal Formation. J. Am. Chem. Soc. 2006, 128, 5930–5938. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Ji, X.; Lumb, J.-P. Total Synthesis of (S)-Cularine via Nucleophilic Substitution on a Catechol. Org. Lett. 2021, 23, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.; Wiatrowska, I. A concise synthesis of tetrahydroisoquinoline-1-carboxylic acids using a Petasis reaction and Pomeranz-Fritsch-Bobbitt cyclization sequence. Tetrahedron 2012, 68, 3092–3097. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.D. Synthesis of Calycotomine and N-Methylcalycotomine using a Petasis reaction—Pomeranz-Fritsch-Bobbitt cyclizaton sequence. Heterocycles 2012, 86, 1119–1127. [Google Scholar] [CrossRef]
- Grajewska, A.; Chrzanowska, M.; Adamska, W. Synthesis of a new substituted 7,12-dihyro-6,12-methanodibenzo[c,f]azocine-5-carboxylic acids containing a tetracyclic tetrahydroisoquinoline core structure. Beilstein J. Org. Chem. 2021, 17, 2511–2519. [Google Scholar] [CrossRef]
- Grajewska, A.; Rozwadowska, M.D.; Gzella, A. Synthesis and stereochemistry assignment of (3R,5R)- and (3S,5R)-4-benzyl-3-(3,4-dimethoxyphenyl)-5-phenyl-1,4-oxazin-2-ones. J. Mol. Struct. 2019, 1194, 204–210. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.D. Diastereoselective Synthesis of (–)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid via Morpholinone Derivatives. Molecules 2023, 28, 3200. https://doi.org/10.3390/molecules28073200
Chrzanowska M, Grajewska A, Rozwadowska MD. Diastereoselective Synthesis of (–)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid via Morpholinone Derivatives. Molecules. 2023; 28(7):3200. https://doi.org/10.3390/molecules28073200
Chicago/Turabian StyleChrzanowska, Maria, Agnieszka Grajewska, and Maria D. Rozwadowska. 2023. "Diastereoselective Synthesis of (–)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid via Morpholinone Derivatives" Molecules 28, no. 7: 3200. https://doi.org/10.3390/molecules28073200
APA StyleChrzanowska, M., Grajewska, A., & Rozwadowska, M. D. (2023). Diastereoselective Synthesis of (–)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid via Morpholinone Derivatives. Molecules, 28(7), 3200. https://doi.org/10.3390/molecules28073200