
Computational Exploration of Licorice for Lead Compounds against Plasmodium vivax Duffy Binding Protein Utilizing Molecular Docking and Molecular Dynamic Simulation

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Structural Assessment of P. vivax DBP
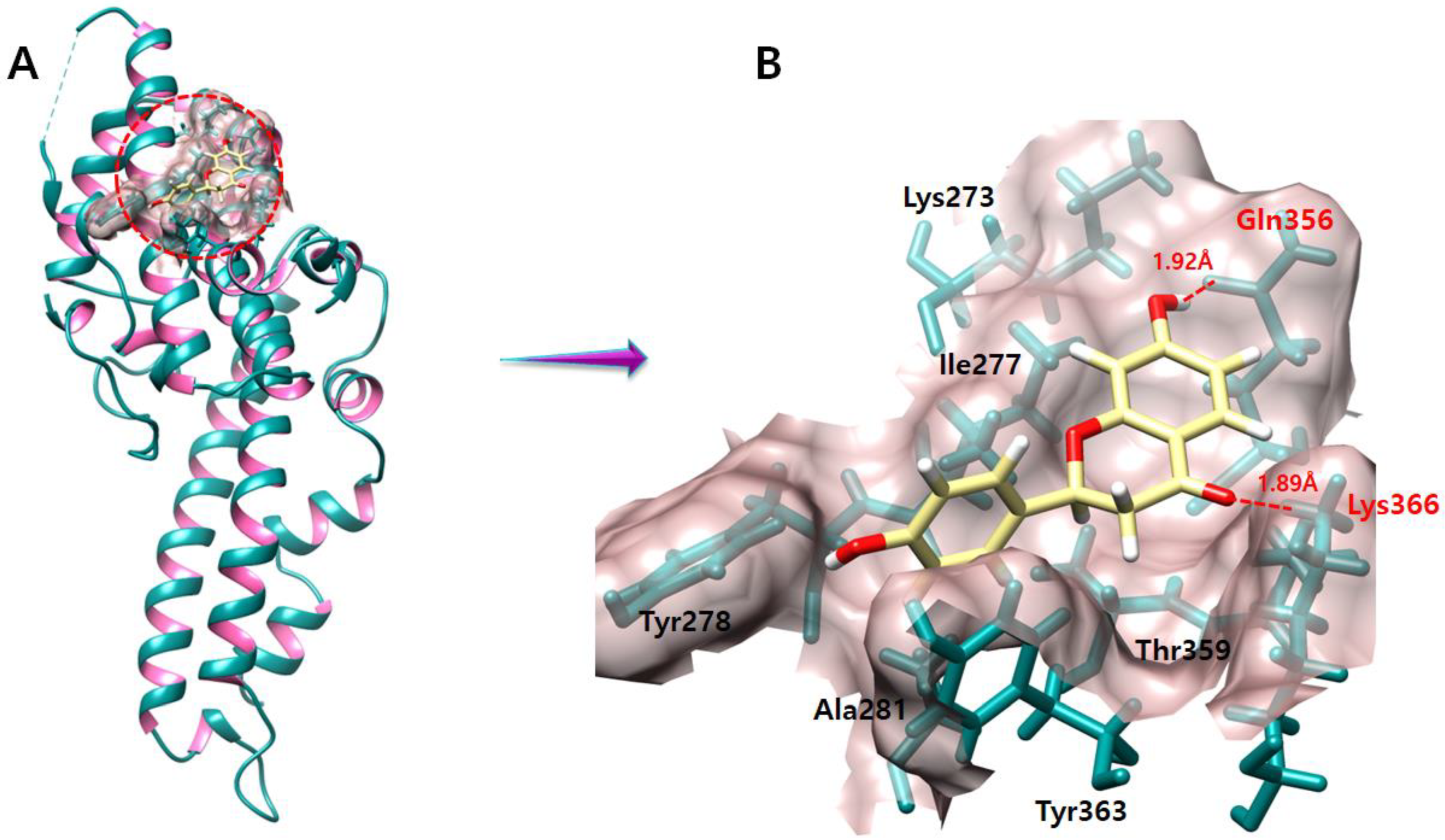
2.2. Binding Pocket Analysis
2.3. Licorice (Ligands) Preparation
2.4. Molecular Docking Analysis
2.5. Interaction Analyses of the Top Five Ligands against DBP
2.6. Licochalcone B
2.7. Echinatin
2.8. Licochalcone A
2.9. Licochalcone E
2.10. Liquiritigenin
2.11. Molecular Dynamic Simulation
2.12. RMSD Analysis
2.13. Binding Modes Analysis after the MD Simulation
2.14. MD Simulation Interaction Energy
3. Discussion
4. Methodology
4.1. Repossession of 4NUV from PDB
4.2. Binding Site Assessment of DBP
4.3. Licorice (Ligands) Preparation
4.4. Molecular Docking Analysis Using Discovery Studio
4.5. Molecular Dynamic Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Tizifa, T.A.; Kabaghe, A.N.; McCann, R.S.; Berg, H.V.D.; Van Vugt, M.; Phiri, K.S. Prevention Efforts for Malaria. Curr. Trop. Med. Rep. 2018, 5, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, C.J.; Tanomsing, N.; Nolder, D.; Oguike, M.; Jennison, C.; Pukrittayakamee, S.; Dolecek, C.; Hien, T.T.; Rosário, V.E.D.; Arez, A.P.; et al. Two Nonrecombining Sympatric Forms of the Human Malaria Parasite Plasmodium ovale Occur Globally. J. Infect. Dis. 2010, 201, 1544–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, T.H.; Hisam, S.; Lanza, M.; Jiram, A.I.; Ismail, N.; Rubio, J.M. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar. J. 2014, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Deane, L.M. Simian malaria in Brazil. Mem. Inst. Oswaldo Cruz 1992, 87, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Zalis, M.G.; de Pina-Costa, A.; Siqueira, A.M.; Júnior, C.B.; Silva, S.; Areas, A.L.L.; Pelajo-Machado, M.; de Alvarenga, D.A.M.; da Silva Santelli, A.C.F.; et al. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: A molecular epidemiological investigation. Lancet Glob. Health 2017, 5, e1038–e1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalremruata, A.; Magris, M.; Vivas-Martínez, S.; Koehler, M.; Esen, M.; Kempaiah, P.; Jeyaraj, S.; Perkins, D.J.; Mordmüller, B.; Metzger, W.G. Natural infection of Plasmodium brasilianum in humans: Man and monkey share quartan malaria parasites in the Venezuelan Amazon. Ebiomedicine 2015, 2, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, R.E.; Battle, K.E.; Mendis, K.N.; Smith, D.L.; Cibulskis, R.E.; Baird, J.K.; Hay, S.I. Global Epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 2016, 95, 15–34. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Sinha, A. Plasmodium vivax Duffy Binding Protein-Based Vaccine: A Distant Dream. Front. Cell. Infect. Microbiol. 2022, 12, 990. [Google Scholar] [CrossRef]
- Soeiro, M.D.N.C.; Vergoten, G.; Bailly, C. Mechanism of action of glycyrrhizin against Plasmodium falciparum. Mem. Inst. Oswaldo Cruz 2021, 116, e210084. [Google Scholar] [CrossRef]
- Adams, J.H.; Ntumngia, F.; Thomson-Luque, R.; Pires, C.V. The role of the human Duffy antigen receptor for chemokines in malaria susceptibility: Current opinions and future treatment prospects. J. Recept. Ligand Channel Res. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Salinas, N.D.; Ntumngia, F.B.; Adams, J.H.; Tolia, N.H. Structural Analysis of the Synthetic Duffy Binding Protein (DBP) Antigen DEKnull Relevant for Plasmodium vivax Malaria Vaccine Design. PLoS Negl. Trop. Dis. 2015, 9, e0003644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, A.; Majlesi, M.; Rafieian-Kopaei, M. Herbal versus synthetic drugs; beliefs and facts. J. Nephropharmacol. 2015, 4, 27–30. [Google Scholar] [PubMed]
- Siracusa, L.; Saija, A.; Cristani, M.; Cimino, F.; D’Arrigo, M.; Trombetta, D.; Rao, F.; Ruberto, G.J.F. Phytocomplexes from liquorice (Glycyrrhiza glabra L.) leaves—Chemical characterization and evaluation of their antioxidant, anti-genotoxic and anti-inflammatory activity. Fitoterapia 2011, 82, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, D.M.; Abd El-Alim, S.H.; Kassem, A.A. Chapter 6-Nanoemulsions: A New Approach for Enhancing Phytonutrient Efficacy. In Nanotechnology Applications in Food; Oprea, A.E., Grumezescu, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 107–127. [Google Scholar]
- Akbar, S. Glycyrrhiza glabra L. (Fabaceae/Leguminosae): (Syns.: G. glandulifera Waldst. & Kit.; G. hirsuta Pall.; G. pallida Boiss. & Noe; G. violacea Boiss. & Noe). In Handbook of 200 Medicinal Plants; Springer: Cham, Switzerland, 2020; pp. 963–980. [Google Scholar] [CrossRef] [Green Version]
- Rashidzadeh, H.; Mosavi, F.S.; Shafiee, T.; Adyani, S.M.; Eghlima, G.; Sanikhani, M.; Kheiry, A.; Amiri, M.; Tavakolizadeh, M.; Ramazani, A. Anti-Plasmodial Effects of Different Ecotypes of Glycyrrhiza glabra Traditionally Used for Malaria in Iran. Rev. Bras. Farm. 2023, 33, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Nasri, H. Cisplatin therapy and the problem of gender-related nephrotoxicity. J. Nephropharmacol. 2013, 2, 13–14. [Google Scholar]
- Talevi, A. Computer-Aided Drug Design: An Overview. Comput. Drug Discov. Des. 2018, 1762, 1–19. [Google Scholar] [CrossRef]
- Huang, H.-J.; Yu, H.W.; Chen, C.-Y.; Hsu, C.-H.; Chen, H.-Y.; Lee, K.-J.; Tsai, F.-J.; Chen, C.Y.-C. Current developments of computer-aided drug design. J. Taiwan Inst. Chem. Eng. 2010, 41, 623–635. [Google Scholar] [CrossRef]
- Batchelor, J.D.; Malpede, B.M.; Omattage, N.S.; DeKoster, G.T.; Henzler-Wildman, K.; Tolia, N.H. Red Blood Cell Invasion by Plasmodium vivax: Structural Basis for DBP Engagement of DARC. PLoS Pathog. 2014, 10, e1003869. [Google Scholar] [CrossRef]
- Yang, R.; Wang, L.-Q.; Yuan, B.-C.; Liu, Y. The Pharmacological Activities of Licorice. Planta Med. 2015, 81, 1654–1669. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Liu, M.; Qin, H.; Lin, H.; An, X.; Shi, Z.; Song, L.; Yang, X.; Fan, H.; Tong, Y. Artemether, Artesunate, Arteannuin B, Echinatin, Licochalcone B and andrographolide effectively inhibit SARS-CoV-2 and related viruses in vitro. Front. Cell. Infect. Microbiol. 2021, 11, 680127. [Google Scholar] [CrossRef]
- Cao, Y.; Lei, E.; Wang, X.; Qi, X.; Li, L.; Ren, J.; Yang, J.; Wang, S. Licochalcone A inhibits enterovirus A71 replication in vitro and in vivo. Antivir. Res. 2021, 195, 105091. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Kakkar, R. Synthetic methods and biological applications of retrochalcones isolated from the root of Glycyrrhiza species: A review. Results Chem. 2021, 3, 100216. [Google Scholar] [CrossRef]
- Wang, L.; Yang, R.; Yuan, B.; Liu, Y.; Liu, C. The antiviral and antimicrobial activities of licorice, a widely-used Chinese herb. Acta Pharm. Sin. B 2015, 5, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.N.; Seo, J.H.; Lee, M.H.; Kim, C.; Kim, E.; Yoon, G.; Cho, S.S.; Cho, Y.S.; Choi, H.W.; Shim, J.H. Licochalcone C induced apoptosis in human oral squamous cell carcinoma cells by regulation of the JAK2/STAT3 signaling pathway. J. Cell. Biochem. 2018, 119, 10118–10130. [Google Scholar]
- Traboulsi, H.; Cloutier, A.; Boyapelly, K.; Bonin, M.-A.; Marsault, É.; Cantin, A.M.; Richter, M.V. The Flavonoid Isoliquiritigenin Reduces Lung Inflammation and Mouse Morbidity during Influenza Virus Infection. Antimicrob. Agents Chemother. 2015, 59, 6317–6327. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M.B.P.P. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. 2018, 32, 2323–2339. [Google Scholar] [CrossRef]
- Adianti, M.; Aoki, C.; Komoto, M.; Deng, L.; Shoji, I.; Wahyuni, T.S.; Lusida, M.I.; Fuchino, H.; Kawahara, N.; Hotta, H. Anti-hepatitis C virus compounds obtained from Glycyrrhiza uralensis and other Glycyrrhiza species. Microbiol. Immunol. 2014, 58, 180–187. [Google Scholar] [CrossRef]
- Kim, H.J.; Lim, S.S.; Park, I.S.; Lim, J.S.; Seo, J.Y.; Kim, J.-S. Neuroprotective effects of dehydroglyasperin C through activation of heme oxygenase-1 in mouse hippocampal cells. J. Agric. Food Chem. 2012, 60, 5583–5589. [Google Scholar] [CrossRef]
- Seo, J.-H.; Choi, H.W.; Oh, H.-N.; Lee, M.-H.; Kim, E.; Yoon, G.; Cho, S.-S.; Park, S.-M.; Cho, Y.S.; Chae, J.; et al. Licochalcone D directly targets JAK2 to induced apoptosis in human oral squamous cell carcinoma. J. Cell. Physiol. 2018, 234, 1780–1793. [Google Scholar] [CrossRef]
- Hosseinzadeh, H.; Nassiri-Asl, M. Pharmacological effects of Glycyrrhiza spp. and its bioactive constituents: Update and review. Phytother. Res. 2015, 29, 1868–1886. [Google Scholar] [CrossRef]
- Huang, W.; Tang, S.; Qiao, X.; Ma, W.; Ji, S.; Wang, K.; Ye, M.; Yu, S. Isoangustone A induces apoptosis in SW480 human colorectal adenocarcinoma cells by disrupting mitochondrial functions. Fitoterapia 2014, 94, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Patel, V.; Parmar, B. Discovery of some antiviral natural products to fight against novel coronavirus (SARS-CoV-2) using an in silico approach. Comb. Chem. High Throughput Screen. 2021, 24, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Kırmızıbekmez, H.; Uysal, G.B.; Masullo, M.; Demirci, F.; Bağcı, Y.; Kan, Y.; Piacente, S. Prenylated polyphenolic compounds from Glycyrrhiza iconica and their antimicrobial and antioxidant activities. Fitoterapia 2015, 103, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Gazzani, G.; Daglia, M.; Papetti, A. Food components with anticaries activity. Curr. Opin. Biotechnol. 2012, 23, 153–159. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, L.; Heber, D.; Shi, W.; Lu, Q.-Y. Antibacterial Compounds from Glycyrrhiza u ralensis. J. Nat. Prod. 2006, 69, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltina, L.A.; Kondratenko, R.M.; Plyasunova, O.A.; Pokrovskii, A.G.; Khalilov, L.M.; Galin, F.Z.; Tolstikov, G.A. Synthesis and anti-HIV activity of triterpene conjugates of α-d-glucosamine. Pharm. Chem. J. 2008, 42, 64–67. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lee, Y.; Lee, H.J.; Kwon, Y.-S.; Chun, W. In silico screening of GABA aminotransferase inhibitors from the constituents of Valeriana officinalis by molecular docking and molecular dynamics simulation study. J. Mol. Model. 2020, 26, 1–13. [Google Scholar] [CrossRef]
- DJA Studio. Discovery Studio; Dassault Systemes BIOVIA: San Diego, CA, USA, 2008. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Breznik, M.; Ge, Y.; Bluck, J.P.; Briem, H.; Hahn, D.F.; Christ, C.D.; Mortier, J.; Mobley, D.L.; Meier, K. Prioritizing Small Sets of Molecules for Synthesis through in-silico Tools: A Comparison of Common Ranking Methods. Chemmedchem 2022, 18, e202200425. [Google Scholar] [CrossRef]
- Abraham, M.J.; Gready, J.E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J. Comput. Chem. 2011, 32, 2031–2040. [Google Scholar] [CrossRef]
- Ge, Y.; Hahn, D.F.; Mobley, D.L. A Benchmark of Electrostatic Method Performance in Relative Binding Free Energy Calculations. J. Chem. Inf. Model. 2021, 61, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhang, R.; Jiang, H.; Zhang, H.; Luo, C. Computer-Aided Drug Design in Epigenetics. Front. Chem. 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Bai, Y.; Cui, J.; Zong, Z.; Gao, Y.; Zheng, Z. Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules 2022, 27, 5710. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful Applications of Computer Aided Drug Discovery: Moving Drugs from Concept to the Clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Hassan, M.; Yasir, M.; Shahzadi, S.; Kloczkowski, A. Exploration of Potential Ewing Sarcoma Drugs from FDA-Approved Pharmaceuticals through Computational Drug Repositioning, Pharmacogenomics, Molecular Docking, and MD Simulation Studies. ACS Omega 2022, 7, 19243–19260. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Hassan, M.; Kloczkowski, A.; Chun, W. Exploration of Flavonoids as Lead Compounds against Ewing Sarcoma through Molecular Docking, Pharmacogenomics Analysis, and Molecular Dynamics Simulations. Molecules 2023, 28, 414. [Google Scholar] [CrossRef]
- Ziegler, H.L.; Hansen, H.S.; Stærk, D.; Christensen, S.B.; Haägerstrand, H.; Jaroszewski, J.W. The Antiparasitic Compound Licochalcone A Is a Potent Echinocytogenic Agent That Modifies the Erythrocyte Membrane in the Concentration Range Where Antiplasmodial Activity Is Observed. Antimicrob. Agents Chemother. 2004, 48, 4067–4071. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.; Abbasi, M.A.; Aziz ur, R.; Siddiqui, S.Z.; Hussain, G.; Shah, S.A.A.; Shahid, M.; Seo, S.-Y. Exploration of synthetic multifunctional amides as new therapeutic agents for Alzheimer’s disease through enzyme inhibition, chemoinformatic properties, molecular docking and dynamic simulation insights. J. Theor. Biol. 2018, 458, 169–183. [Google Scholar] [CrossRef]
- Sharma, M.; Kohli, D.; Chaturvedi, S.; Sharma, S. Molecular modelling studies of some substitued 2-butylbenzimidazoles angiotensin ii receptor a ntagonists as antihypertensive agents. Dig. J. Nanomater. Biostruct. 2009, 4, 843–856. [Google Scholar]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr No | Ligand Name | Activity | Reference |
|---|---|---|---|
| 1 | Licochalcone B | Antitumor, anti-inflammatory, antiviral | [21,22,23] |
| 2 | Echinatin | Antiviral | [22,24] |
| 3 | Licochalcone A | Antiviral, antimicrobial, immunoregulatory | [21,25] |
| 4 | Licochalcone E | Antiviral, antimicrobial | [25] |
| 5 | Liquiritigenin | Antiviral, antimicrobial, immunoregulatory | [21,25] |
| 6 | Licochalcone C | Anticancer | [26] |
| 7 | Isoliquiritigenin | Antiviral | [27] |
| 8 | Glabridin | Antiviral | [28,29] |
| 9 | Dehydroglyasperin C | Hepatoprotective | [30] |
| 10 | Licochalcone D | Antiviral, antioxidant | [31] |
| 11 | Glycygenzofuran | Diabetes mellitus, obesity | [32] |
| 12 | Glabrol | Hepatoprotective | [25] |
| 13 | Isoangustone A | Adenocarcinoma | [33] |
| 14 | Glycyrol | Antiviral | [29] |
| 15 | Licoricidin | Antiviral | [34] |
| 16 | Licorisoflavan A | Antimicrobial | [35] |
| 17 | Glycyrrhizol A | Anticaries, antimicrobial | [36,37] |
| 18 | 18β Glycyrrhetinic acid | Antiviral, antimicrobial | [25] |
| 19 | 11Deoxyglycyrrhetic acid | Antiviral | [38] |
| No | Ligand Name | CDocker Energy | CDocker Interaction Energy |
|---|---|---|---|
| 1 | Licochalcone B | −40.646 | −47.6473 |
| 2 | Echinatin | −36.4715 | −44.7142 |
| 3 | Licochalcone A | −33.3302 | −52.3068 |
| 4 | Licochalcone E | −30.6573 | −54.8755 |
| 5 | Liquiritigenin | −22.072 | −27.4566 |
| 6 | Licochalcone C | −20.0702 | −52.0653 |
| 7 | Isoliquiritigenin | −14.967 | −26.2369 |
| 8 | Glabridin | −11.7479 | −31.5265 |
| 9 | Dehydroglyasperin C | −5.66293 | −48.4455 |
| 10 | Licochalcone D | −1.45512 | −33.7997 |
| 11 | Glycygenzofuran | 6.02888 | −34.8505 |
| 12 | Glabrol | 12.2899 | −37.0889 |
| 13 | Isoangustone A | 12.5396 | −35.0905 |
| 14 | Glycerol | 18.4799 | −32.3047 |
| 15 | Licoricidin | 20.9019 | −36.0512 |
| 16 | Licorisoflavan A | 25.2122 | −35.4716 |
| 17 | Glycyrrhizol A | 35.3237 | −33.7042 |
| 18 | 18β Glycyrrhetinic acid | 43.0477 | −38.4474 |
| 19 | 11Deoxyglycyrrhetic acid | 51.5132 | −38.6611 |
| Ligand Name | Total Energy | Average Energy | ||
|---|---|---|---|---|
| R1 | R2 | R3 | ||
| Licochalcone B | –105.8469 | –116.8744 | –99.7185 | –107.4799 |
| Echinatin | –102.3061 | –143.673 | –113.2652 | –119.7481 |
| Licochalcone A | –112.8598 | –104.1913 | –100.7909 | –105.9473 |
| Liquiritigenin | –84.437 | –92.9508 | –93.1907 | –90.1928 |
| Licochalcone E | –110.7497 | –101.6259 | –81.5792 | –97.9849 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Chun, W. Computational Exploration of Licorice for Lead Compounds against Plasmodium vivax Duffy Binding Protein Utilizing Molecular Docking and Molecular Dynamic Simulation. Molecules 2023, 28, 3358. https://doi.org/10.3390/molecules28083358
Yasir M, Park J, Han E-T, Park WS, Han J-H, Kwon Y-S, Lee H-J, Chun W. Computational Exploration of Licorice for Lead Compounds against Plasmodium vivax Duffy Binding Protein Utilizing Molecular Docking and Molecular Dynamic Simulation. Molecules. 2023; 28(8):3358. https://doi.org/10.3390/molecules28083358
Chicago/Turabian StyleYasir, Muhammad, Jinyoung Park, Eun-Taek Han, Won Sun Park, Jin-Hee Han, Yong-Soo Kwon, Hee-Jae Lee, and Wanjoo Chun. 2023. "Computational Exploration of Licorice for Lead Compounds against Plasmodium vivax Duffy Binding Protein Utilizing Molecular Docking and Molecular Dynamic Simulation" Molecules 28, no. 8: 3358. https://doi.org/10.3390/molecules28083358