

A Highly Efficient Workflow for Detecting and Identifying Sequence Variants in Therapeutic Proteins with a High Resolution LC-MS/MS Method


Abstract
1. Introduction
2. Results and Discussion
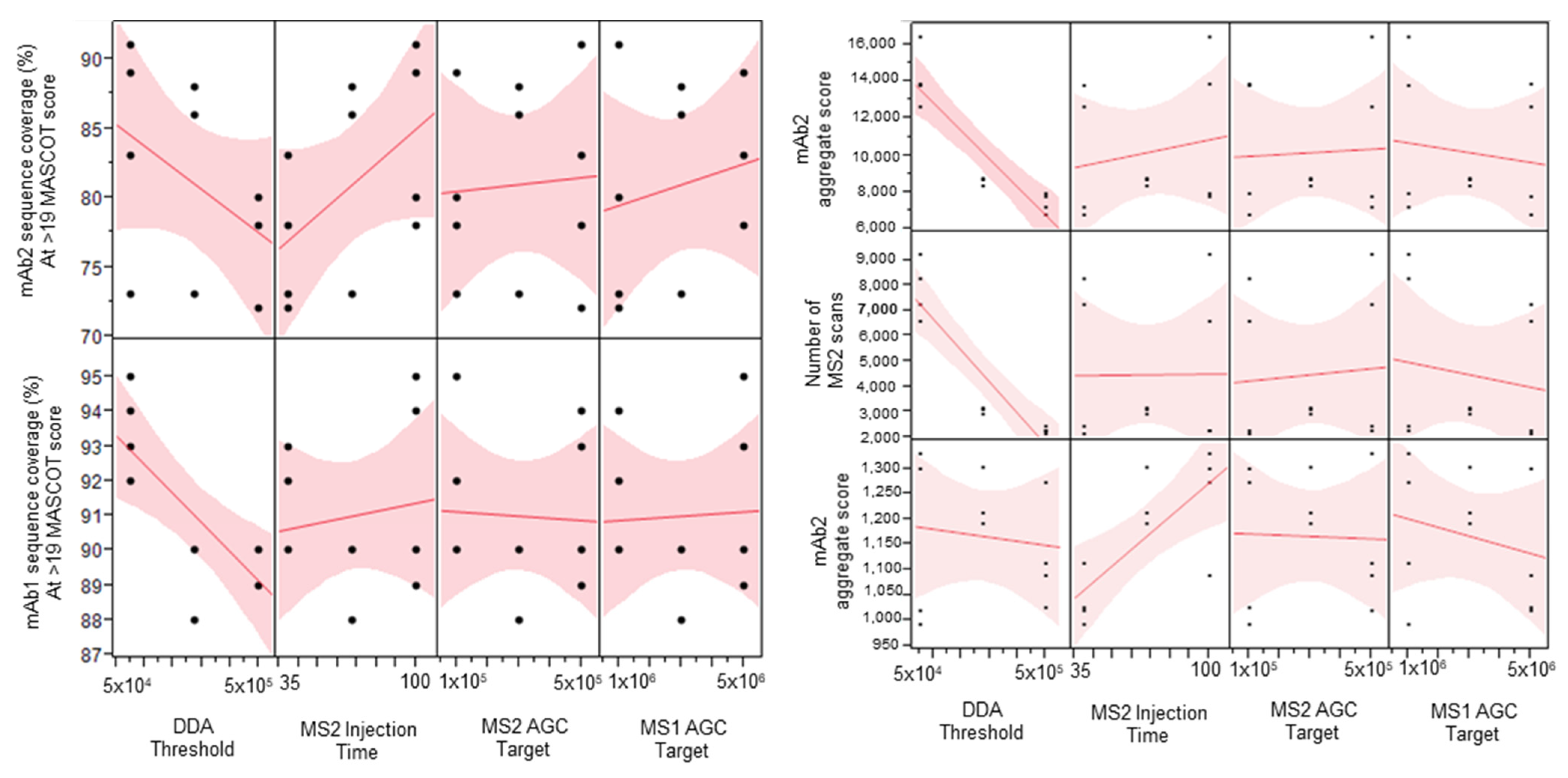
2.1. Parameter Screening DOE Study
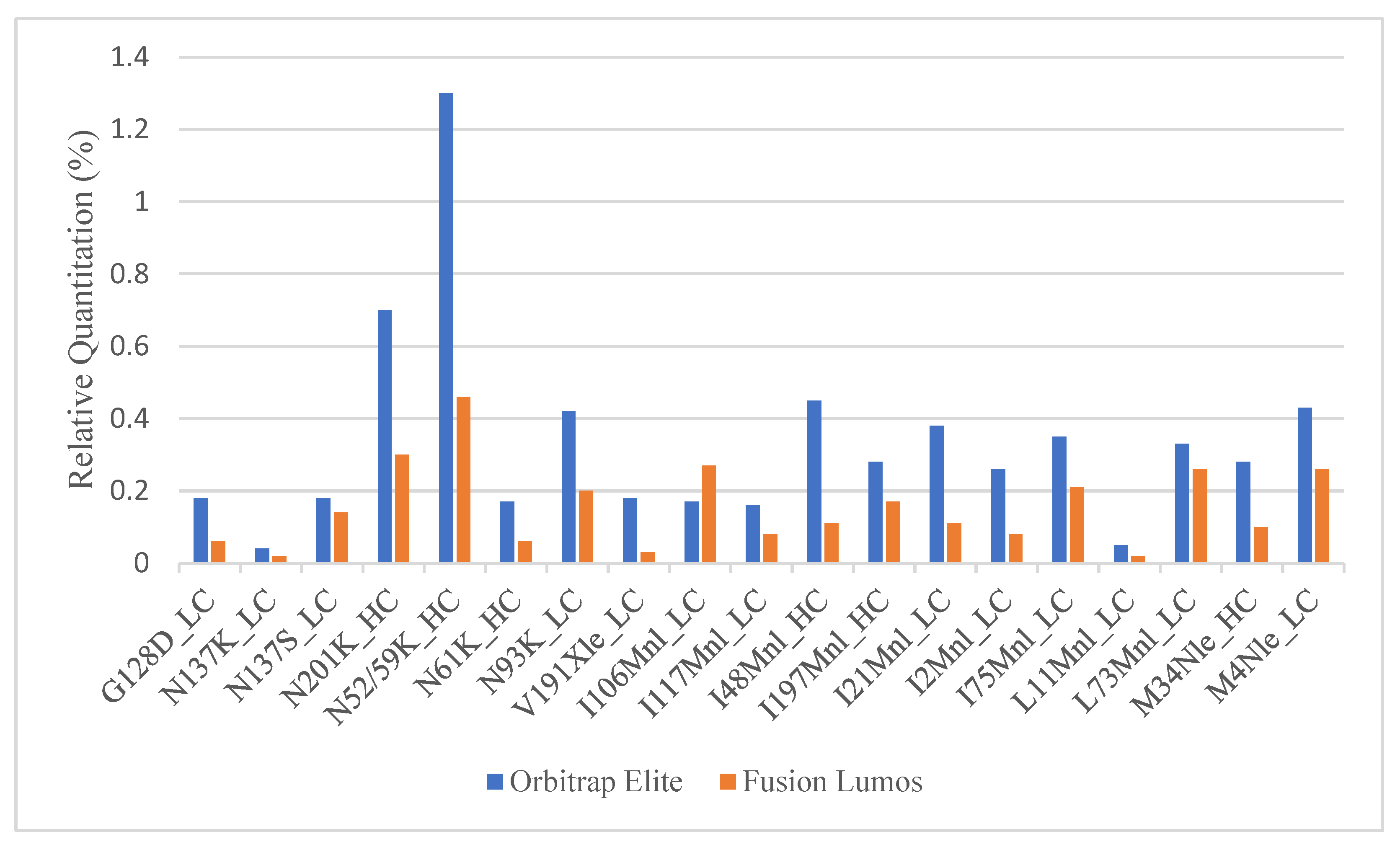
2.2. Fixing the Under-Quantitation in the Fusion Lumos
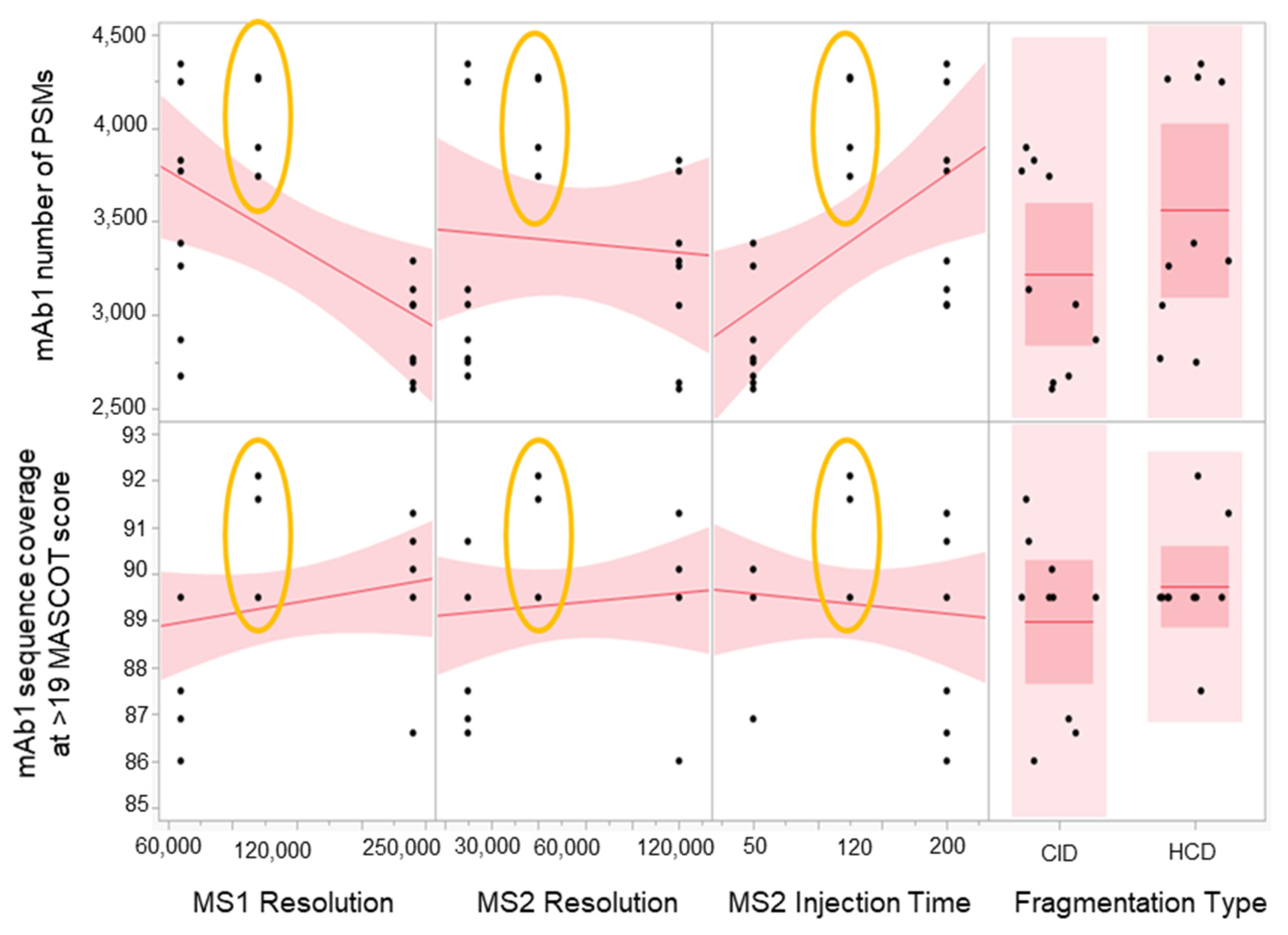
2.3. Method Robustness DOE
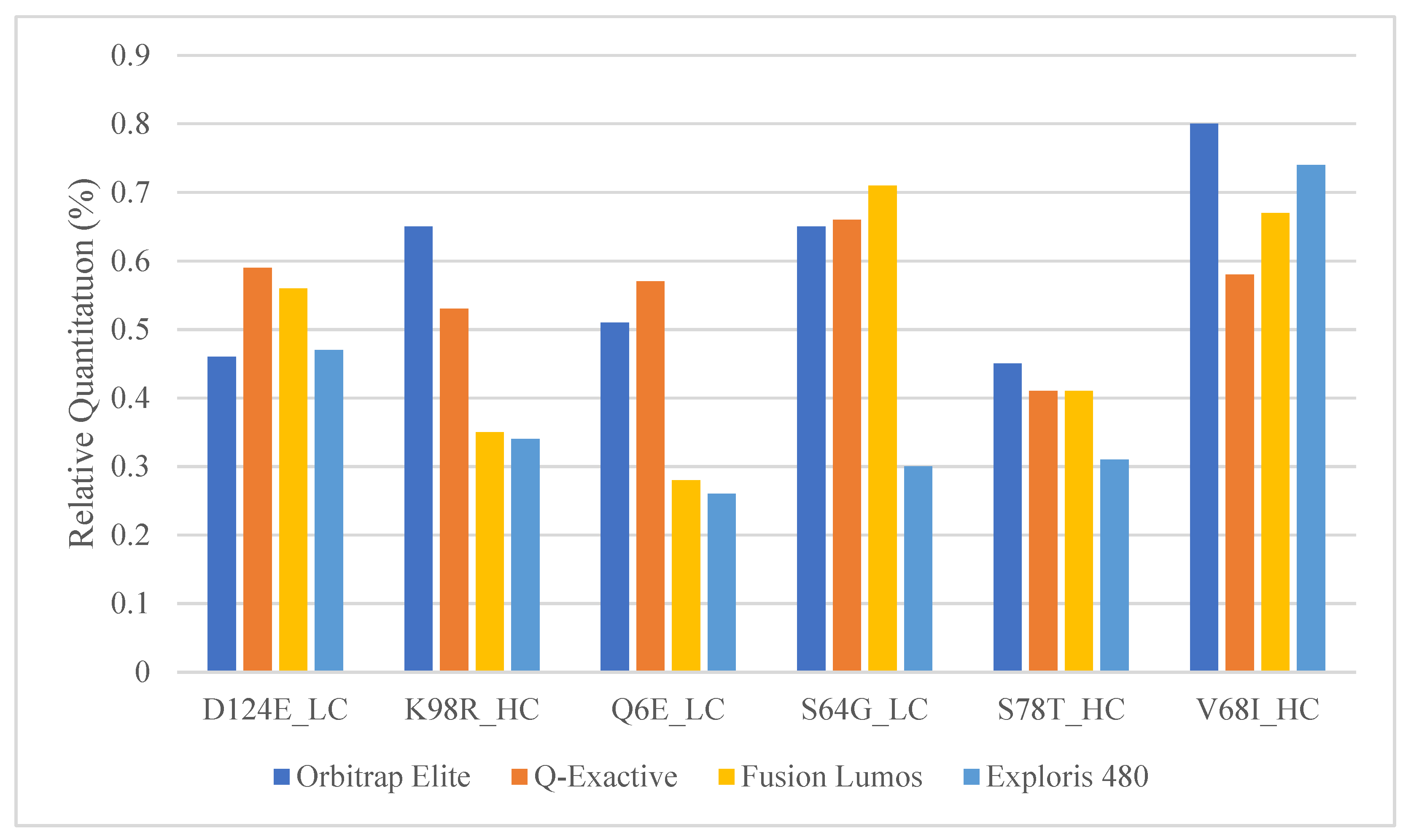
2.4. Comparison of Relative Quantitation between Commonly Used Orbitrap Platforms
2.5. PMI Byos SVA Parameter Selection
2.6. Direct Comparison of Low-Resolution vs. High-Resolution Methods
3. Material and Methods
3.1. Material
3.2. Methods
3.2.1. Peptide Map by Low-Resolution MS/MS
3.2.2. High Resolution MS Parameter Optimization and Robustness Testing
3.2.3. PMI Byos Parameter Selection
3.2.4. Peptide Map by High-Resolution LC-MS/MS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alt, N.; Zhang, T.Y.; Motchnik, P.; Taticek, R.; Quarmby, V.; Schlothauer, T.; Beck, H.; Emrich, T.; Harris, R.J. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals 2016, 44, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Trill, J.J.; Shatzman, A.R.; Ganguly, S. Production of Monoclonal-Antibodies in Cos and Cho Cells. Curr. Opin. Biotechnol. 1995, 6, 553–560. [Google Scholar] [CrossRef]
- O’Callaghan, P.M.; McLeod, J.; Pybus, L.P.; Lovelady, C.S.; Wilkinson, S.J.; Racher, A.J.; Porter, A.; James, D.C. Cell Line-Specific Control of Recombinant Monoclonal Antibody Production by CHO Cells. Biotechnol. Bioeng. 2010, 106, 938–951. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Vijayasankaran, N.; Shen, A.Y.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Wecksler, A.T.; Lundin, V.; Williams, A.J.; Veeravalli, K.; Reilly, D.E.; Grieco, S.-H. Bioprocess Development and Characterization of a 13C-Labeled Hybrid Bispecific Antibody Produced in Escherichia coli. Antibodies 2023, 12, 16. [Google Scholar] [CrossRef]
- Castineiras, T.S.; Williams, S.G.; Hitchcock, A.G.; Smith, D.C. E. coli strain engineering for the production of advanced biopharmaceutical products. FEMS Microbiol. Lett. 2018, 365, fny162. [Google Scholar]
- Carter, P.; Kelley, R.F.; Rodrigues, M.L.; Snedecor, B.; Covarrubias, M.; Velligan, M.D.; Wong, W.L.T.; Rowland, A.M.; Kotts, C.E.; Carver, M.E.; et al. High-Level Escherichia coli Expression and Production of a Bivalent Humanized Antibody Fragment. Bio/Technol. 1992, 10, 163–167. [Google Scholar] [CrossRef]
- Tang, D.M.; Gunson, J.; Tran, E.; Lam, C.; Shen, A.; Snedecor, B.; Barnard, G.; Misaghi, S. Expressing antigen binding fragments with high titers in a targeted integration CHO host by optimizing expression vector gene copy numbers and position: A case study. Biotechnol. Prog. 2022, 38, e3290. [Google Scholar] [CrossRef]
- Wei, B.C.; Han, G.H.; Tang, J.; Sandoval, W.; Zhang, Y.T. Native Hydrophobic Interaction Chromatography Hyphenated to Mass Spectrometry for Characterization of Monoclonal Antibody Minor Variants. Anal. Chem. 2019, 91, 15360–15364. [Google Scholar] [CrossRef] [PubMed]
- Brorson, K.; Jia, A.Y. Therapeutic monoclonal antibodies and consistent ends: Terminal heterogeneity, detection, and impact on quality. Curr. Opin. Biotechnol. 2014, 30, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.J.; Murnane, A.A.; Utter, S.L.; Wagner, K.L.; Cox, E.T.; Polastri, G.D.; Helder, J.C.; Sliwkowski, M.B. Assessing Genetic-Heterogeneity in Production Cell-Lines-Detection by Peptide-Mapping of a Low-Level Tyr to Gln Sequence Variant in a Recombinant Antibody. Bio/Technol. 1993, 11, 1293–1297. [Google Scholar] [CrossRef]
- Feeney, L.; Carvalhal, V.; Yu, X.C.; Chan, B.; Michels, D.A.; Wang, Y.J.; Shen, A.; Ressl, J.; Dusel, B.; Laird, M.W. Eliminating tyrosine sequence variants in CHO cell lines producing recombinant monoclonal antibodies. Biotechnol. Bioeng. 2013, 110, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Chen, Z.; Li, M.; Qiu, H.; Lawrence, S.; Bak, H.; Li, N. A general evidence-based sequence variant control limit for recombinant therapeutic protein development. mAbs 2020, 12, 1791399. [Google Scholar] [CrossRef]
- Frye, C.; Deshpande, R.; Estes, S.; Francissen, K.; Joly, J.; Lubiniecki, A.; Munro, T.; Russell, R.; Wang, T.; Anderson, K. Industry view on the relative importance of “clonality” of biopharmaceutical-producing cell lines. Biologicals 2016, 44, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.J.; Beal, K.M.; Brown, P.W.; DeGruttola, H.S.; Ly, M.; Wang, W.E.; Chu, C.H.; Dufield, R.L.; Casperson, G.F.; Carroll, J.A.; et al. Evolution of a comprehensive, orthogonal approach to sequence variant analysis for biotherapeutics. Mabs 2019, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.C.; Borisov, O.V.; Alvarez, M.; Michels, D.A.; Wang, Y.J.; Ling, V. Identification of Codon-Specific Serine to Asparagine Mistranslation in Recombinant Monoclonal Antibodies by High-Resolution Mass Spectrometry. Anal. Chem. 2009, 81, 9282–9290. [Google Scholar] [CrossRef]
- Yang, Y.; Strahan, A.; Li, C.; Shen, A.; Liu, H.B.; Ouyang, J.; Katta, V.; Francissen, K.; Zhang, B.Y. Detecting low level sequence variants in recombinant monoclonal antibodies. Mabs 2010, 2, 285–298. [Google Scholar] [CrossRef]
- Li, W.Z.; Wypych, J.; Duff, R.J. Improved sequence variant analysis strategy by automated false positive removal. Mabs 2017, 9, 978–984. [Google Scholar] [CrossRef]
- Makarov, A.; Denisov, E.; Kholomeev, A.; Balschun, W.; Lange, O.; Strupat, K.; Horning, S. Performance Evaluation of a Hybrid Linear Ion Trap/Orbitrap Mass Spectrometer. Anal. Chem. 2006, 78, 2113–2120. [Google Scholar] [CrossRef]
- Second, T.P.; Blethrow, J.D.; Schwartz, J.C.; Merrihew, G.E.; MacCoss, M.J.; Swaney, D.L.; Russell, J.D.; Coon, J.J.; Zabrouskov, V. Dual-Pressure Linear Ion Trap Mass Spectrometer Improving the Analysis of Complex Protein Mixtures. Anal. Chem. 2009, 81, 7757–7765. [Google Scholar] [CrossRef]
- Kalli, A.; Smith, G.T.; Sweredoski, M.J.; Hess, S. Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: Focus on LTQ-Orbitrap mass analyzers. J. Proteome Res. 2013, 12, 3071–3086. [Google Scholar] [CrossRef]
- Michalski, A.; Damoc, E.; Lange, O.; Denisov, E.; Nolting, D.; Muller, M.; Viner, R.; Schwartz, J.; Remes, P.; Belford, M.; et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteom. 2012, 11, O111.013698. [Google Scholar] [CrossRef] [PubMed]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Martínez-Val, A.; Steigerwald, S.; Rüther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, K.; Op de Beeck, J.; Dürnberger, G.; Jacobs, P.; Mechtler, K. Ultrasensitive NanoLC-MS of Subnanogram Protein Samples Using Second Generation Micropillar Array LC Technology with Orbitrap Exploris 480 and FAIMS PRO. Anal. Chem. 2021, 93, 8704–8710. [Google Scholar] [CrossRef]
- Rajski, Ł.; Petromelidou, S.; Díaz-Galiano, F.J.; Ferrer, C.; Fernández-Alba, A.R. Improving the simultaneous target and non-target analysis LC-amenable pesticide residues using high speed Orbitrap mass spectrometry with combined multiple acquisition modes. Talanta 2021, 228, 122241. [Google Scholar] [CrossRef]
- Riter, L.S.; Vitek, O.; Gooding, K.M.; Hodge, B.D.; Julian, R.K., Jr. Statistical design of experiments as a tool in mass spectrometry. J. Mass Spectrom. 2005, 40, 565–579. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers. In Breakthroughs in Statistics: Methodology and Distribution; Kotz, S., Johnson, N.L., Eds.; Springer: New York, NY, USA, 1992; pp. 66–70. [Google Scholar]
- Huang, S.K.; Glancy, S.B.; Stansbury, W.F. Optimization of a commercial liquid chromatography/mass spectrometry interface by statistical experimental design. Rapid Commun. Mass Spectrom. 1993, 7, 722–724. [Google Scholar] [CrossRef]
- Cole, D.C.; Pagano, N.; Kelly, M.F.; Ellingboe, J. Use of Design of Experiments To Optimize High-Throughput Semipreparative LC and LC/MS Methods. J. Comb. Chem. 2004, 6, 78–82. [Google Scholar] [CrossRef]
- Seto, C.; Bateman, K.P.; Gunter, B. Development of generic liquid chromatography-mass spectrometry methods using experimental design. J. Am. Soc. Mass Spectrom. 2002, 13, 2–9. [Google Scholar] [CrossRef]
- Kil, Y.J.; Bern, M.; Crowell, K.; Kletter, D.; Bern, N.; Tang, W.; Carlson, E.; Becker, C. Towards a Comprehensive Bioinformatic Analysis of the NIST Reference mAb. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 3. Defining the Next Generation of Analytical and Biophysical Techniques; American Chemical Society: Washington, DC, USA, 2015; Volume 1202, pp. 395–414. [Google Scholar]
- Zhang, Z.; Shah, B.; Bondarenko, P.V. G/U and certain wobble position mismatches as possible main causes of amino acid misincorporations. Biochemistry 2013, 52, 8165–8176. [Google Scholar] [CrossRef]
- Michalski, A.; Neuhauser, N.; Cox, J.; Mann, M. A Systematic Investigation into the Nature of Tryptic HCD Spectra. J. Proteome Res. 2012, 11, 5479–5491. [Google Scholar] [CrossRef] [PubMed]
- Wilburn, D.B.; Richards, A.L.; Swaney, D.L.; Searle, B.C. CIDer: A Statistical Framework for Interpreting Differences in CID and HCD Fragmentation. J. Proteome Res. 2021, 20, 1951–1965. [Google Scholar] [CrossRef] [PubMed]
- Sadek, M.; Moore, B.N.; Yu, C.; Ruppe, N.; Abdun-Nabi, A.; Hao, Z.; Alvarez, M.; Dahotre, S.; Deperalta, G. A Robust Purity Method for Biotherapeutics Using New Peak Detection in an LC–MS-Based Multi-Attribute Method. J. Am. Soc. Mass Spectrom. 2023, 34, 484–492. [Google Scholar] [CrossRef]
- Hao, Z.; Moore, B.; Ren, C.; Sadek, M.; Macchi, F.; Yang, L.; Harris, J.; Yee, L.; Liu, E.; Tran, V.; et al. Multi-attribute method performance profile for quality control of monoclonal antibody therapeutics. J. Pharm. Biomed. Anal. 2021, 205, 114330. [Google Scholar] [CrossRef] [PubMed]
- Bern, M.; Cai, Y.; Goldberg, D. Lookup Peaks: A Hybrid of de Novo Sequencing and Database Search for Protein Identification by Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 1393–1400. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinform. 2012, 40, 13.20.1–13.20.14. [Google Scholar] [CrossRef]
- Niu, B.; Lu, Y.; Chen, X.; Xu, W. Using New Peak Detection to Solve Sequence Variants Analysis Challenges in Bioprocess Development. J. Am. Soc. Mass Spectrom. 2023, 34, 401–408. [Google Scholar] [CrossRef]
- Montgomery, D.C. Design and Analysis of Experiments; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS/MS Injection Time (ms) | mAb1 Aggregate Score | mAb1 Sequence Coverage | mAb2 Aggregate Score | mAb2 Sequence Coverage |
|---|---|---|---|---|
| 100 | 1662 | 88% | 14,924 | 90% |
| 150 | 2222 | 89% | 25,304 | 93% |
| 200 | 2302 | 89% | 31,381 | 93% |
| 250 | 2557 | 89% | 34,722 | 94% |
| Molecule Name | High Resolution | Low Resolution | % Reduction | |
|---|---|---|---|---|
| mAb3 | Unique Peptides | 83 | 346 | 76% |
| Number of true SVs | 5 | 5 | ||
| Sequence Coverage | 100 | 100 | ||
| mAb4 | Unique Peptides | 43 | 194 | 78% |
| Number of true SVs | 1 | 1 | ||
| Sequence Coverage | 100 | 100 | ||
| mAb5 | Unique Peptides | 30 | 298 | 90% |
| Number of true SVs | 0 | 0 | ||
| Sequence Coverage | 100 | 99 | ||
| FP1 1 | Unique Peptides | 49 | 458 | 89% |
| Number of true SVs | 15 | 15 | ||
| Sequence Coverage | 91 2 | 92 2 |
| Molecule | Sequence Variant | High Resolution | Low Resolution | ||
|---|---|---|---|---|---|
| Rel. Quant (%) | Number of PSMs | Rel. Quant (%) | Number of PSMs | ||
| mAb3 | D1E_LC | 0.3 | 9 | 0.2 | 18 |
| D17E_LC | 0.3 | 10 | 0.3 | 3 | |
| G128E_LC | 0.3 | 6 | 0.2 | 18 | |
| I48Mnl 1_LC | 0.2 | 4 | 0.2 | 8 | |
| I70Mnl 1_HC | 0.2 | 7 | 0.2 | 12 | |
| mAb4 | T20I_LC | 0.9 | 3 | 0.9 | 9 |
| mAb5 | N/A 2 | N/A | N/A | N/A | N/A |
| FP1 3 | Y58F | 0.7 | 18 | 0.8 | 20 |
| Y76F | 0.7 | 2 | 0.8 | 2 | |
| Y80F | 1 | 2 | 1.2 | 2 | |
| Y129F | 0.8 | 32 | 0.8 | 16 | |
| Y153F | 1 | 20 | 1.5 | 10 | |
| Y171F | 1 | 18 | 1.5 | 10 | |
| Y187F | 0.9 | 8 | 1.2 | 10 | |
| Y216F | 0.6 | 8 | 0.7 | 12 | |
| Y276F | 5 | 4 | 8.8 | 8 | |
| Y280F | 0.7 | 12 | 0.8 | 16 | |
| Y312F | 0.3 | 20 | 0.2 | 112 | |
| Y317F | 0.9 | 18 | 1.1 | 152 | |
| Y326F | 0.7 | 20 | 0.9 | 23 | |
| Y329F | 0.8 | 12 | 0.8 | 9 | |
| Y336F | 0.5 | 10 | 0.6 | 10 | |
| Parameter | Units | Minimum (−) | Maximum (+) | Midpoint (0) |
|---|---|---|---|---|
| DDA Intensity Threshold | ions | 50,000 | 500,000 | 275,000 |
| MS/MS Injection Time | ms | 100 | 35 | 67.5 |
| MS/MS AGC Target | ions | 100,000 | 500,000 | 300,000 |
| MS1 AGC Target | ions | 1,000,000 | 5,000,000 | 3,000,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadang, L.; Tam, C.Y.J.; Moore, B.N.; Fichtl, J.; Yang, F. A Highly Efficient Workflow for Detecting and Identifying Sequence Variants in Therapeutic Proteins with a High Resolution LC-MS/MS Method. Molecules 2023, 28, 3392. https://doi.org/10.3390/molecules28083392
Cadang L, Tam CYJ, Moore BN, Fichtl J, Yang F. A Highly Efficient Workflow for Detecting and Identifying Sequence Variants in Therapeutic Proteins with a High Resolution LC-MS/MS Method. Molecules. 2023; 28(8):3392. https://doi.org/10.3390/molecules28083392
Chicago/Turabian StyleCadang, Lance, Chi Yan Janet Tam, Benjamin Nathan Moore, Juergen Fichtl, and Feng Yang. 2023. "A Highly Efficient Workflow for Detecting and Identifying Sequence Variants in Therapeutic Proteins with a High Resolution LC-MS/MS Method" Molecules 28, no. 8: 3392. https://doi.org/10.3390/molecules28083392
APA StyleCadang, L., Tam, C. Y. J., Moore, B. N., Fichtl, J., & Yang, F. (2023). A Highly Efficient Workflow for Detecting and Identifying Sequence Variants in Therapeutic Proteins with a High Resolution LC-MS/MS Method. Molecules, 28(8), 3392. https://doi.org/10.3390/molecules28083392