Abstract
Protic pyrazoles (N-unsubstituted pyrazoles) have been versatile ligands in various fields, such as materials chemistry and homogeneous catalysis, owing to their proton-responsive nature. This review provides an overview of the reactivities of protic pyrazole complexes. The coordination chemistry of pincer-type 2,6-bis(1H-pyrazol-3-yl)pyridines is first surveyed as a class of compounds for which significant advances have made in the last decade. The stoichiometric reactivities of protic pyrazole complexes with inorganic nitrogenous compounds are then described, which possibly relates to the inorganic nitrogen cycle in nature. The last part of this article is devoted to outlining the catalytic application of protic pyrazole complexes, emphasizing the mechanistic aspect. The role of the NH group in the protic pyrazole ligand and resulting metal–ligand cooperation in these transformations are discussed.
1. Introduction
Pyrazole is an aromatic five-membered N-heterocycle containing a potentially Brønsted acidic NH group adjacent to a Schiff-base nitrogen atom. This amphiprotic character gives rise to the rich coordination chemistry of pyrazoles. Unlike aprotic N-heterocycles, such as pyridine, pyrazole can be deprotonated easily, and the resulting pyrazolate anion bridges two metal centers to form di- or polynuclear complexes in some cases. The NH group also provides a clue to integration of pyrazole units, making multidentate ligands, such as poly(pyrazolyl)borates []. The flexible ligand design based on the easy construction of the pyrazole ring [,,] and N-functionalization has led to the structural diversity of the pyrazole complexes [,,,] and their applications in various fields, including materials chemistry [], homogeneous catalysis [,], bioinorganic modeling [], supramolecular chemistry [,,], and medicinal chemistry [].
Coordination of a pyrazole to a Lewis acidic metal center renders the pyrazole NH proton more acidic []. The increased Brønsted acidity causes intra- and intermolecular hydrogen bonding as well as facile deprotonation that switches the coordination mode from a lone-pair donating L-type to a covalent X-type. These events should make an electronic impact on the complex. It is to be emphasized that the deprotonation in the position β to the metal may be coupled with ligand dissociation on the metal center. As illustrated in Scheme 1a, elimination of HX or outer sphere transfer of nucleophilic group X along with a proton from such a “β-protic” pyrazole complex would yield a coordinatively unsaturated pyrazolato complex. Bond cleavage of pronucleophile HX on this complex regenerates the pyrazole complex. The interconversion associated with change in the coordination mode of the pyrazole rather than the formal oxidation state of the metal would mediate various bond activation and transfer of HX. Such metal–ligand cooperative transformations have also been known for related proton-responsive ligands [,,,,,,,,,], exemplified in Scheme 1b. Nevertheless, difference in ligand acidity and relative positions to the metal center in the protic pyrazole complexes brings about their unique reactivities. Tamm and co-workers demonstrated that the pyrazolylborane 1 reacts with dihydrogen gas at room temperature to afford the zwitterionic pyrazolium borate 2 (Scheme 2) []. Heterocumulenes, such as carbon dioxide, also react with 1 to give the corresponding adduct 3, for example []. These reactions showcase the pyrazole-based bifunctional reactivities operating even in the field of transition-metal-free, frustrated Lewis pair chemistry.
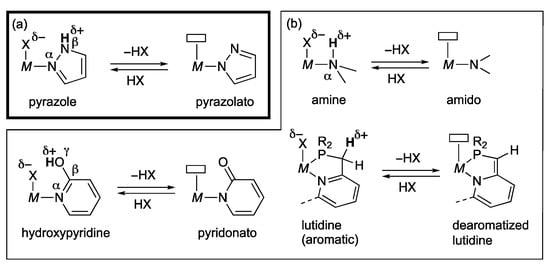
Scheme 1.
(a) Elimination and addition of pronucleophiles HX in pyrazole complexes. (b) Representative examples of related metal–ligand cooperative interconversion.
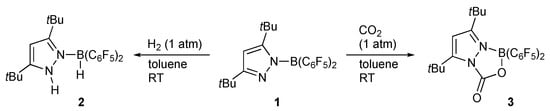
Scheme 2.
Bifunctional reactions of a pyrazolylborane as a frustrated Lewis pair.
In this review, we outline the reactivities of the β-protic pyrazole complexes of transition metals. The proton-responsive nature and catalytic application of this class of complexes were surveyed about a decade ago [,]. This article updates these earlier reviews by focusing on their stoichiometric and catalytic reactivities. Special attention will be paid to the mechanistic aspects in the discussion on the catalysis. Protic pyrazoles are also known as modules in spin-crossover materials [,,,,] and phosphorescent complexes [], which are not covered in this review. The readers can also refer to papers [,,] to learn the design and synthesis of specific classes of protic pyrazole ligands.
2. Reactions of Protic Pyrazole Complexes
2.1. Pincer-Type Complexes Bearing Protic Pyrazole Arms
Chelation has been a rational strategy to ensure the coordination of pyrazoles for metal–ligand cooperative reactivities [,]. During the last decade, significant advances have been made particularly in the coordination chemistry of 2,6-bis(1H-pyrazol-3-yl)pyridines (RLH2; R represents the substituent at the 5-position of the pyrazole ring), which place the two protic pyrazole groups at trans positions rigorously, owing to the rigid pincer-type framework []. This class of compounds have long been used as a ligand in iron(II) complexes showing thermal and photochemical spin-state transitions [,,,,]. The protic pyrazole groups therein greatly affect the spin-crossover properties of the complexes through hydrogen bonding interaction with surrounding counteranion and solvent. In this section, we describe the transition metal RLH2 complexes and related pincer-type pyrazole complexes, focusing on their stoichiometric reactivities, which originate mostly from the protic pyrazole units.
2.1.1. Bis(1H-pyrazol-3-yl)pyridine Complexes
Ruthenium and Osmium
In 2010, Thiel and co-workers reported that deprotonation of the nBuLH2-ligated ruthenium(II) complex 4a under carbon monoxide leads to the formation of the bis(pyrazolato) carbonyl complex 5 (Scheme 3) []. The result shows the diprotic nature of the pincer-type complex 4a. The nBuL complex 5 is also obtained by the dehydrogenative coordination of nBuLH2 to [RuH2(CO)(PPh3)3].
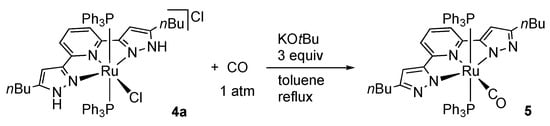
Scheme 3.
Dehydrochlorination of a protic pincer-type ruthenium complex in the presence of carbon monoxide.
Soon after that, our group demonstrated that the deprotonation can be done stepwise and reversible at least in the first step []. Thus, the ruthenium(II) complex 4b having a tert-butyl substituted pincer ligand, tBuLH2, undergoes reversible deprotonation by equimolar amount of a base to afford the pyrazole–pyrazolato complex 6 (Scheme 4). The partial deprotonation of the pincer ligand is established by the 1H NMR spectrum of 6, showing the D2O-exchangeable NH resonance at δ 10.17 with only 1H intensity as well as inequivalence of the two pyrazole arms. The single crystal X-ray analysis allows the detailed structural comparison between the pyrazole and deprotonated pyrazolato rings. The deprotonated pyrazole group lacks neighboring hydrogen bond acceptors and features a smaller NαNβC angle (106.8(4)°) due to the increased s character of the lone pair electrons on the deprotonated nitrogen atom [,]. Complete deprotonation of the pyrazole arms is achieved by an additional base in methanol, giving the bis(pyrazolato) methanol complex 7. In the crystal of 7, a hydrogen bonding network involving a co-crystalized methanol is observed (inset). The methanol ligand in 7 is replaced by molecular nitrogen and oxygen to yield the dinitrogen complex 8 and a side-on peroxo complex, respectively.
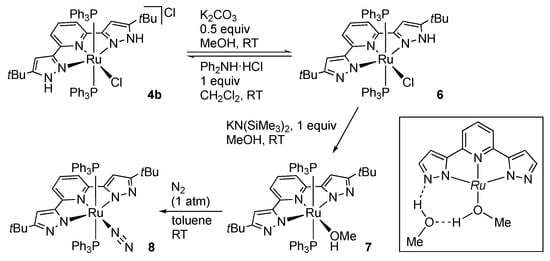
Scheme 4.
Stepwise dehydrochlorination of the protic pincer-type ruthenium complex 4b.
Some osmium complexes having RLH2 or its deprotonated form are known [,]. Substitution reactions of [OsCl2(H2O)(tBuLH2)] by bio-relevant molecules are reported [].
Rhodium and Iridium
Goldberg and co-workers reported the reactivities of the iridium(I) complex [Ir(tBuLH2)(coe)2]PF6 (9; coe = cyclooctene) bearing labile coe ligands (Scheme 5) []. The coe ligands are easily displaced by carbon monoxide, giving the bis(carbonyl) complex 11 featuring κ2-coordination of the tBuLH2 ligand. When 11 is dissolved in acetone-d6 under N2, a monocarbonyl species 10 having equivalent pyrazole arms is observed, suggesting an equilibrium between 10 and 11. Addition of 4-tert-butylpyridine (tBuPy) to 11 results in an oxidative addition of one of the two pyrazole NH groups to afford the hydridoiridium(III) pyrazolato complex 12. Co-crystallization of an additional pyridine, which is engaged in hydrogen bonding with the pyrazole arm of the tBuLH ligand, has been confirmed by X-ray analysis. Deprotonation of 12 affords the bis(pyrazolato) complex [IrH(CO)(tBuPy)(tBuL)]. Triphenylphosphine also reacts with the carbonyl complex 11 with oxidative addition of the pyrazole arm; however, the isolated product is [IrH(CO)(PPh3)2(tBuLH)]PF6, having a κ2-bound tBuLH ligand. Interestingly, thermolysis of 12 leads to dinuclear reductive elimination of H2 to give the diiridium(II) complex 13. Complex 13 is also formed by treatment of 12 with (Ph3C)PF6. Deprotonation of the dicationic complex 13 yields the bis(pyrazolato) complex [{Ir(CO)(tBuPy)(tBuL)}2]. Isolation of the iridium complexes ranging from Ir(I) to Ir(III) indicates the electronic flexibility of the RLHn (n = 0–2) ligands.
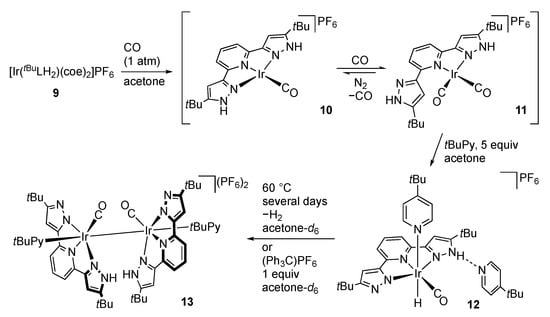
Scheme 5.
Synthesis and reactions of tBuLH2 iridium complexes. coe = cyclooctene; tBuPy = 4-tert-butylpyridine.
On the other hand, Bogojeski and Bugarčić [] and we [] reported the synthesis and crystal structures of the trichlorido rhodium(III) and iridium(III) complexes [MCl3(tBuLH2)] (M = Rh, Ir), respectively. The kinetics of the substitution reactions of the rhodium complex with small biomolecules, such as amino acids, is studied [].
Platinum
Goldberg and co-workers [] demonstrated that reaction of the chlorido complex 14 with methyllithium results in methylation of the platinum center, along with deprotonation of the tBuLH2 ligand to afford the methyl complex 15 with a dangling lithium cation on one of the two pyrazolato arms (Scheme 6). In the solution of THF-d8, the pincer ligand in 15 is symmetric, and the lithium cation is removed by the treatment with bis(triphenylphosphine)iminium (PPN) chloride to give the anionic complex 16. The methyl bis(pyrazolato) complex 16 undergoes three-step protonation with a pyridinium tetrafluoroborate. While the first protonation product is insoluble and remains uncharacterized, following protonation yields the cationic methyl complex 17 and acetonitrile complex 18 sequentially. Thus, the first site of protonation is the pyrazolato ligand rather than the Pt–Me bond. Methane release from [PtMe(tBuLH2)]Cl, a chloride salt of 17, is not observed in C6D6 until 180 °C, indicating that the high energy barrier for the intramolecular proton transfer to the methyl ligand.
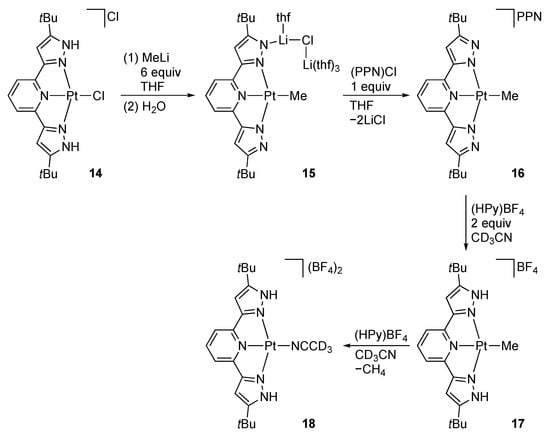
Scheme 6.
Reactions of the bis(pyrazolato) pincer-type platinum complex 14. PPN = (Ph3P)2N+; Hpy = 2,6-dimethoxypyridinium cation.
In this connection, uncharged bis(pyrazolato) phosphine complexes [Pt(PPh3)(RL)] are also synthesized []. The complexes display green phosphorescence in solution and in the solid state; however, the reactivities are unknown.
First-Row Transition Metals
The 3d transition metal complexes of RLH2 have been known much earlier []. Most of the studies were, however, limited to their structural determination, except for the spin-crossover properties of the iron(II) complexes with the Fe:ligand ratio of 1:2 until recently. In 2013, we synthesized a 1:1 complex 19 of iron(II) and tBuLH2 and uncovered the reactivity []. The paramagnetic dichlorido complex 19 is converted to the diamagnetic phosphine complex 20a with the aid of sodium triflate. Complex 20a catalyzes disproportionation of hydrazine (vide infra). We later obtained the cobalt and manganese analogues [MCl2(tBuLH2)] (M = Co (21b), Mn) and explored the ligand substitution reactions of 19 and 21b, as summarized in Scheme 7 []. The iron and cobalt complexes, 19 and 21b, are converted into the high-spin, triflato complexes 22, upon treatment with silver triflate in acetonitrile. The iron complex 22a reacts with dioxygen to give the oxido-bridged diiron(III) complex 23 [,]. The two pincer ligands in 23 are almost perpendicular, and the protic pyrazole arms make a hydrogen bond with the triflato ligand on the opposite iron center. The low-spin phosphine and carbonyl complexes, 20 and 24, can further be derivatized by sequential ligand replacement of the triflato complexes of 22. The ammine complex [Fe(NH3)(PMe3)2(tBuLH2)](OTf)2 is obtained similarly [].
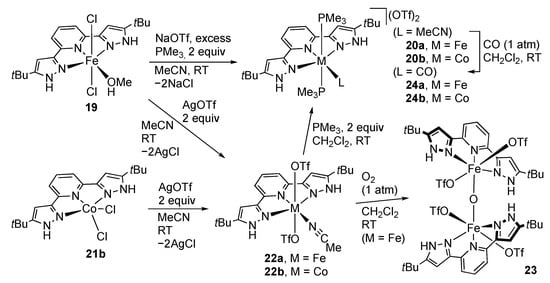
Scheme 7.
Ligand substitution of iron and cobalt complexes of tBuLH2.
Following that, Caulton and co-workers reported dehydrochlorination of the iron(II) and cobalt(II) complexes 21 with two equiv of a lithium silylamide in THF (Scheme 8) [,]. Full dehydrochlorination is, however, not achieved, and the products are the monochlorido-bridged, anionic complexes 25. For cobalt, the reactions in diethyl ether or toluene yield single crystals of unexpected polynuclear complexes, apparently caused by partial dissociation of the tBuLH2. Complexes 25 can be regarded as lithium chloride adducts of dimers of the expected two-fold dehydrochlorination products MtBuL with coordinative unsaturation.
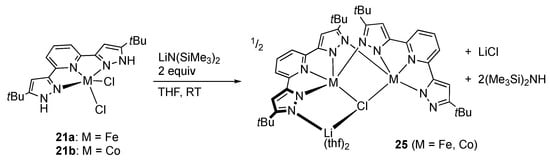
Scheme 8.
Dehydrochlorination of the iron and cobalt complexes of tBuLH2, 21.
In contrast, treatment of the iron complex 21a in the presence of two-electron donor ligands results in complete dissociation of the chloride ligands to give the bis(pyrazolato) complexes, such as 26–28 (Scheme 9) []. The 4-dimethylaminopyridine (DMAP) complex 26 is paramagnetic, while the isocyanide complex 27 as well as the diphosphine complex 28 obtained similarly is diamagnetic. The DMAP complex 26 is further converted to the oxido-bridged Fe(III)2 complex 29 upon treatment with silver triflate, although the oxidant remains unclear []. In 29, the Lewis-basic pyrazolate arms are bridged by the silver cation. On the other hand, chloride abstraction of 21a with two equiv of NaBArF4 (ArF = C6H3(CF3)2-3,5) in THF leads to the formation of the dicationic THF complex 30 [], which, however, is found to decompose into [Fe(tBuLH2)2]2+ with ligand redistribution during recrystallization from dichloromethane.
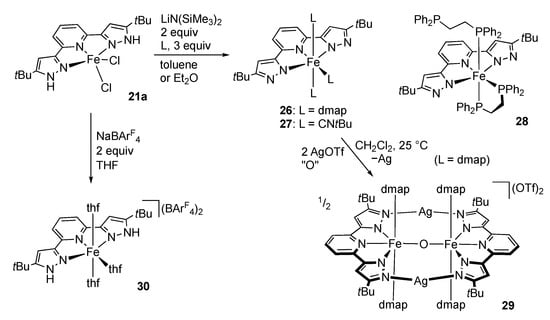
Scheme 9.
Dehydrochlorination of the tBuLH2 iron complex 21a in the presence of donor ligands. dmap = 4-dimethylaminopyridine.
As in the case of the iron analogue 21a, dehydrochlorination of the cobalt(II) complex 21b in the presence of triethylphosphine affords the bis(phosphine) complex 31 (Scheme 10) []. Subsequent treatment with nitrous oxide results in oxidation of the phosphine ligand to yield the (phosphine oxide)-bridged dinuclear complex 32. This complex catalyzes oxidation of PEt3 and PPh3 with nitrous oxide or O2, owing to the interconversion between 31 and 32. Meanwhile, the ligand basicity of 31 was suggested by the formation of the cobalt(III) complex 33 with each pyrazolato arm binding to a silver center []. Even cooperation of Lewis acid–Brønsted base centers is proposed for the oxidation of 31 in dichloromethane to give the cobalt(III) chlorido complex 34 (Scheme 11) [].
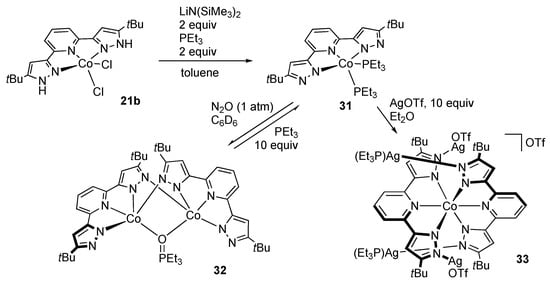
Scheme 10.
Dehydrochlorination and subsequent reactions of the tBuLH2 cobalt complex 21b.
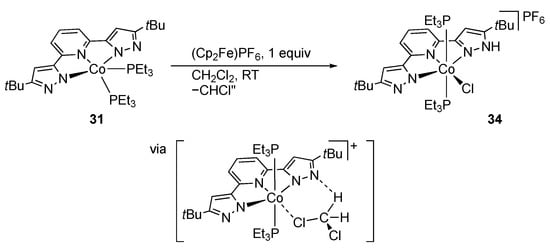
Scheme 11.
Oxidation of a tBuLH2 cobalt complex in dichloromethane.
Synthesis of the elusive, coligand-free bis(pyrazolato) complexes MRL has been achieved for chromium(II) by redox-neutral, salt metathesis reaction of K2tBuL [] with CrCl2 or treatment of a chromium(II) silylamide and tBuLH2 []. The primary product was claimed to be the bis(pyrazolato)-type dimer 35, which subsequently converts into the tetramer 36 during recrystallization (Scheme 12). Owing to the coordinative unsaturation, 35 reacts with four equiv of DMAP to give the mononuclear bis(dmap) complex 37 with a square-pyramidal geometry. In contrast, addition of chloride anion to 35 affords the anionic monochlorido complex 38, maintaining the pyrazolato-bridged dichromium(II) core.
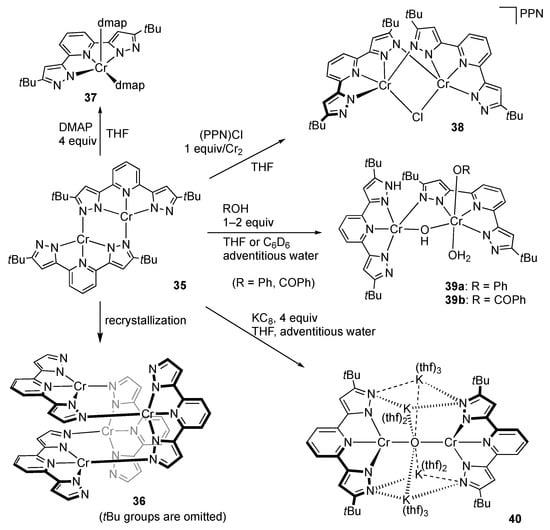
Scheme 12.
Reactions of the pyrazolato-bridged dichromium complex 35. PPN = (Ph3P)2N+; DMAP = 4-dimethylaminopyridine.
Cooperative reactivities of the Lewis acidic metal center and Brønsted basic pyrazolato nitrogen atoms in 35 are also reported. Treatment of 35 with phenol does not result in simple redox-neutral acid–base reaction to give a phenoxido–pyrazole complex; instead, the Cr(II)Cr(III) mixed-valent phenoxido complex 39a is obtained (Scheme 12) []. The oxidation has been explained by evolution of dihydrogen gas, which, however, is not detected in the reaction mixture. Benzoic acid similarly reacts with 35 to yield the corresponding benzoate complex 39b. On the other hand, treatment of 35 with an equimolar amount of 2-naphthol (NapOH) results in partial dissociation of the pincer ligand to afford the trinuclear Cr(II)Cr(III)2 complex [{(tBuLH2)Cr(ONap)(µ2-ONap)2}2Cr](Cl)2, wherein the two chloride counteranions are accommodated between the pyrazole rings through hydrogen bonding [].
Chemical reduction of 35 with four equiv of KC8 suggests that the pincer-type bis(1H-pyrazol-3-yl)pyridine ligand is redox non-innocent. The reaction eventually yields the oxido-bridged dichromium complex [K4(thf)10][Cr2tBuL2(µ2-O)] (40), possibly after reactions with adventitious water and dihydrogen evolution as postulated in the previous reactions (Scheme 12) []. The X-ray analysis of 40 revealed that the C–C bond distances around the 4-position of the pyridine ring (1.420(10)–1.439(10) Å) are much longer than the distances of the C2–C3 and C5–C6 bonds (1.352(9)–1.367(8) Å), indicating reduction in the pincer ligand tBuL with an unpaired electron at the 4-position of the pyridine ring. The DFT calculation also supports the description of the oxidation state of 40 as Cr(II)(tBuL•−) rather than Cr(I)tBuL.
In addition, reduction of 35 with two equiv of KC8 as well as oxidation of 35 with ferrocenium cation is examined []. Recrystallization of the reaction products again results in uptake of adventitious water to produce oxido-bridged tri- and tetranuclear complexes, respectively, with ambiguous reaction stoichiometry. The reduced species is trapped with carbon dioxide to afford a carbonato-bridged, dianionic–dinuclear complex []. On the other hand, the oxidation of 35 with a quinone gives rise to the formation of a bis(semiquinone) Cr(III)2 complex [].
2.1.2. Modified 1H-Pyrazol-3-yl Pincer Complexes
Partial replacement of the components in the protic pincer ligand RLH2, the central pyridine and flanking pyrazoles, should tune the properties of their metal complexes as in other pincer-type complexes. This section provides an overview of the reactivities of protic pyrazole complexes obtained by ligand modification of RLH2.
Modification at Pincer Center
Introduction of a strong σ-donor as the central ligating atom in the protic pincer framework should affect the reactivities of the trans ligand in particular. We reported ligand substitution of the NCN pincer-type ruthenium complexes 41, which are obtained by cyclometalation of the corresponding 1,3-bis(1H-pyrazol-3-yl)benzenes (Scheme 13) []. Owing to the trans effect of the central aryl group in the pincer ligand, the substitution takes place even at room temperature to give 42, in contrast to the reaction of the NNN pincer-type analogue 4b at 100 °C with the aid of a Lewis acid []. An iridium complex, bearing this NCN pincer ligand, is also synthesized in a similar manner [].
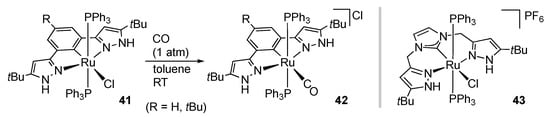
Scheme 13.
Ligand substitution of an NCN pincer-type protic pyrazole ruthenium complex.
We also installed an N-heterocyclic carbene (NHC) unit in the center of the pincer framework furnished with two protic pyrazoles []. The increase in the electron density of the resulting ruthenium complexes, such as 43, is indicated by the CV measurements. Meanwhile, the CO stretching frequency of the carbonyl derivative is higher than that of the pyridine-centered counterpart [Ru(CO)(PPh3)2(tBuL)] (1989 vs. 1964 cm−1). The DFT calculations suggest that the twist of the pincer ligand due to the increase in the chelate size in the NHC-centered pincer may lead to delocalization of the metal d-orbital to the pyrazole π-orbitals, which reduces π-back donation to the carbonyl ligand.
In addition to the C-centered pincer ligands, a novel triprotic NNN pincer ligand bearing a donating central nitrogen atom has been developed recently. The reaction of iron(II) chloride with 1,3-bis(1H-pyrazol-3-ylimino)isoindoline results in tautomerization of the ligand to give the 3-amino-1-imino-1H-isoindole complex 44 (Scheme 14). This dichloridoiron(II) complex exhibits reactivities similar to those of the tBuLH2 complex 19 shown in Scheme 7 []. The CO stretching frequency of the carbonyl complex 46 (1963 cm−1) is much lower than that of the tBuLH2 analogue 24a (2005 cm−1) [], indicating the donating nature of the central isoindole unit. Importantly, 46 undergoes deprotonation of the chelate backbone rather than the pyrazole groups to afford the isoindolin-2-yl complex 47 bearing a monoanionic bis(pyrazole) pincer ligand.
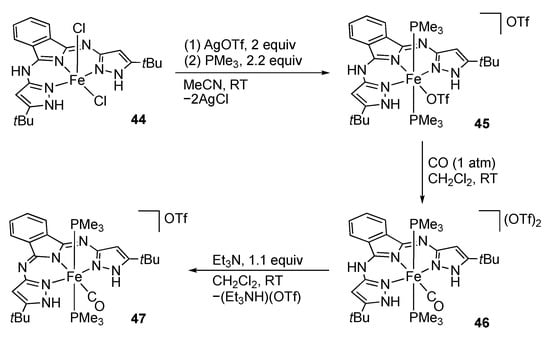
Scheme 14.
Reactions of isoindoline-based pincer-type protic pyrazole iron complexes.
Unsymmetrical Pincer-Type Complexes
Replacement of one of the two pyrazole arms in pincer-type RLH2 ligands by other donor groups has also been investigated. Such desymmetrization modifies the numbers and acidity of the protic sites as well as the electronic and steric properties of the metal center. We revealed that tautomerization of imidazole assisted by chelation with a pyrazolylpyridine unit results in the formation of the protic pincer-type ruthenium(II) complexes 48, having protic pyrazole and N-heterocyclic carbene (pNHC []) arms (Scheme 15) [,]. These complexes undergo reversible deprotonation to afford the corresponding pyrazolato complexes 49, showing that protic pyrazole is more acidic than pNHC. Exhaustive deprotonation of 48a under carbon monoxide gives the pyrazolato–imidazolyl carbonyl complex 50 []. Similar treatment of 48b under dihydrogen results in heterolytic cleavage of H2 to yield the hydrido complex 51, in which the proton derived from H2 goes to the more Brønsted basic imidazolyl arm [].
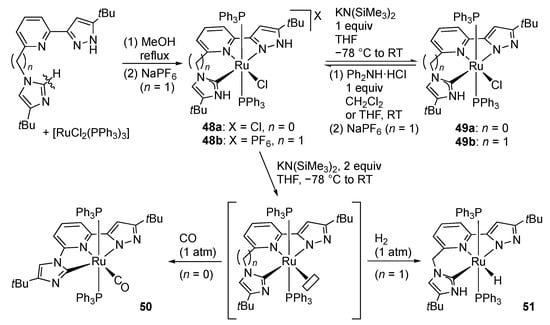
Scheme 15.
Synthesis and reactions of CNN pincer-type pyrazole ruthenium complexes bearing a pNHC arm.
A tertiary amino group can also be incorporated into the protic pincer framework to afford unsymmetrical pincer-type complexes with a hemilabile arm. After our isolation of the ruthenium(II) and iron(II) complexes, such as 52 [], Goldberg and co-workers reported the reactivities of the platinum(II) complexes bearing this unsymmetrical protic pincer ligand (Scheme 16) []. The methyl–pyrazolato complex 53, obtained from a dimethylplatinum(II) complex and the free ligand, undergoes protonation at the pyrazolato arm rather than the methyl ligand, as observed in the symmetrical bis(pyrazolato) complex 16 (vide supra). The reaction of 54 with hydrogen chloride leads to methane evolution to afford the chlorido complex 55. In contrast, intramolecular proton migration from the NH group to the methyl ligand appears more difficult. The methyl complex 54, with a deuterium-labeled NH group, releases only unlabeled CH4 after being subjected to the temperature higher than 100 °C. The absence of CDH3 generation indicates that the methane is derived from an external proton source rather than the NH group. Interestingly, heating of the methyl–pyrazolato complex 53 in benzene results in the formation of the phenyl complex 56. The reaction in C6D6 revealed concurrent site-specific deuteration of the methylene hydrogens in the ethyl groups of the pincer ligand, which implies the hemilabile nature of the dialkylaminomethyl arm.
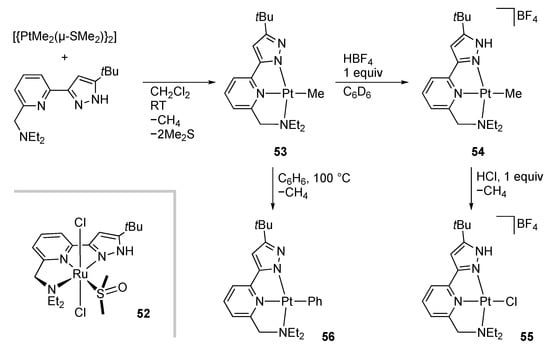
Scheme 16.
Synthesis and reactions of NNN pincer-type pyrazole platinum complexes bearing an aminomethyl arm.
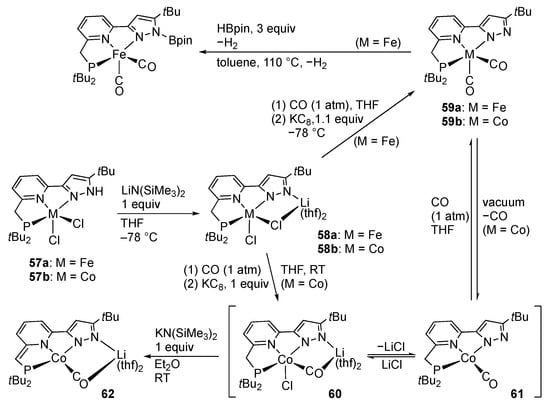
Caulton and co-workers reported the iron [] and cobalt [] complexes bearing a PNN pincer-type protic pyrazole ligand (Scheme 17). The dichlorido iron(II) and cobalt(II) complexes 57 with a distorted square-pyramidal geometry are deprotonated with lithium silylamide to afford the N-lithiated complexes 58. Subsequent treatment with KC8 under carbon monoxide yields the iron(I) and cobalt(I) complexes 59–61. Further reduction of the iron complex 59a with KC8 results in the formation of a monoanionic dicarbonyl iron(0) complex. The iron complex 59a also undergoes N-borylation of the pyrazolato arm with concurrent hydrogen evolution []. On the other hand, treatment of the mixture of the cobalt complexes 60 and 61 with an additional base leads to the second deprotonation at the methylene group of the pincer ligand to afford 62 with a dearomatized pyridine moiety [], as in the related lutidine-based pincer-type complexes []. The result substantiates the diprotic nature of the PNN pincer-type ligand bearing a protic pyrazole arm.
Scheme 17.
Reactions of iron and cobalt complexes bearing a protic PNN pincer-type ligand.
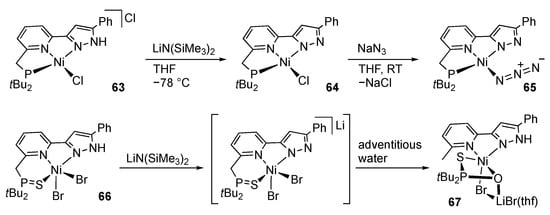
The square-planar nickel(II) complex 63, bearing a PNN pincer-type pyrazole ligand is also reported (Scheme 18) []. As expected, deprotonation of 63 affords the corresponding pyrazolato complex 64, which is subsequently converted to the azido complex 65. Meanwhile, deprotonation of the SNN pincer-type complex 66 results in P–C bond cleavage of the pincer ligand to give the pyrazole complex 67 with a trigonal bipyramidal geometry. The methyl hydrogen atom as well as pyrazole proton is believed to be derived from adventitious water. The reaction in rigorously dried THF leads to redistribution of the nickel–pincer unit in 66 to give a bis(pincer ligand)-type nickel complex.
Scheme 18.
Deprotonation and subsequent reactions of PNN and SNN pincer-type nickel complexes.
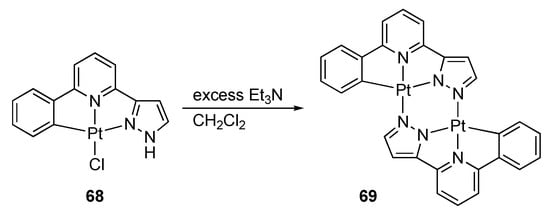
Previously, Lam and co-workers reported a CNN pincer-type pyrazole complex 68, wherein one of the two protic pyrazolyl groups in the HLH2 ligand is replaced by a phenyl group (Scheme 19) []. This complex undergoes dehydrochlorination to afford the pyrazolato-bridged dinuclear complex 69, as observed in bidentate 2-(1H-pyrazol-3-yl)pyridine complexes [,].
Scheme 19.
Dehydrochlorination and dimerization of a protic pincer complex of platinum.
2.2. Redox Reactions of Hydrazines and Azobenzene
Given the expanded π-conjugated structure, the pincer-type RLH2 ligands may be expected to function as an electron reservoir in addition to a proton source. The electron transfer coupled with proton transfer in the second coordination sphere appears crucial in various enzymatic transformations, including biological nitrogen fixation. In this context, reactions of protic pyrazole complexes with partially reduced dinitrogen species, such as hydrazine, have been investigated.
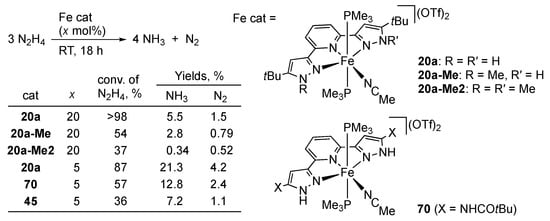
We revealed that the iron(II) tBuLH2 complex 20a catalyzes N–N bond cleavage of hydrazine as shown in Scheme 20 []. The pivotal role of the pyrazole NH groups is suggested by the reactions of the N-methylated derivatives, 20a-Me and 20a-Me2, which are sluggish and much more complicated. The catalytic activities of the amide-substituted complex 70 [] as well as the isoindoline-based pincer complex 45 [] are also lower than that of 20a. Control experiments and theoretical calculations [] led to the proposed mechanism summarized in Scheme 21, featuring multiple and bidirectional proton-coupled electron transfer (PCET) between the metal–ligand bifunctional platform and the hydrazine substrate. The pyrazole NH group promotes heterolytic N–N bond cleavage of the coordinated hydrazine in 71 through a hydrogen bond with the distal nitrogen atom. The second pyrazole NH group in the pincer ligand behaves as an acid–base catalyst for substitution of the amido ligand in 72 by the second molecule of hydrazine to afford the hydrazido(1−) complex 73. The calculations also suggest some radical character of the κN-nitrogen ligands in these high-valent iron species 72 and 73, and hence a mixed electronic structure of FeIV(NH2−) and FeIII(NH2•), for example []. Following PCET from the hydrazido(1−) ligand to the high-valent iron–bis(pyrazolato) fragment would yield the iron(II) diazene complexes 74. In fact, the phenyldiazene complex 74b is isolated in the reaction of phenylhydrazine. An X-ray analysis revealed that the phenyldiazene ligand in 74b benefits from stabilization by hydrogen-bonding interactions with the two pyrazole NH units and counteranion. Meanwhile, the reaction of 1,1-diphenylhydrazine results in reductive elimination of a hydrazinophosphonium salt from the hydrazido(1−) iron(IV) complex 73 bearing trimethylphosphine ligands (omitted in Scheme 21) due to the lack of the distal hydrogen atom. Finally, the diazene complex 74a releases free diazene, which disproportionates to dinitrogen and hydrazine. An alternative scenario that merits comments involves a direct reaction of 74a with hydrazine to give two moles of ammonia. The proposed 2H+/2e− shuttling may be applicable to other multielectron redox processes.
Scheme 20.
Catalytic disproportionation of hydrazine with protic pincer-type iron complexes.
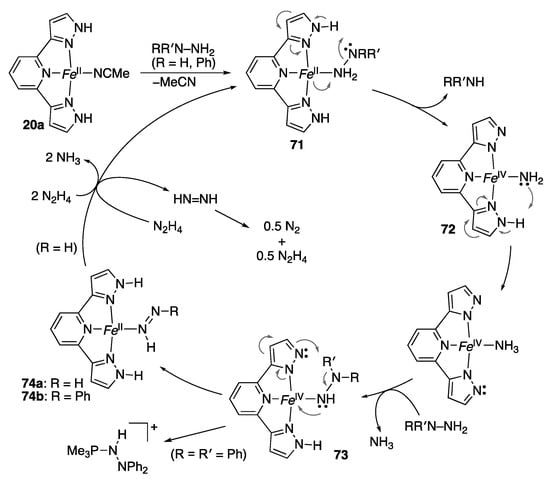
Scheme 21.
Proposed mechanism for catalytic disproportionation of hydrazine with a protic pincer-type iron complex. Fe = Fe(PMe3)22+. The total charge of the iron complexes (+2) and tert-butyl groups in the pincer ligand are omitted.
Similar disproportionation of a substituted hydrazine is reported for a triprotic, tripodal tris(1H-pyrazol-3-ylmethyl)amine complex []. Treatment of the chlorido-bridged diruthenium(II) complex 75 with 1,2-diphenylhydrazine results in N–N bond cleavage to afford the aniline complex 76 (Scheme 22) []. Concurrent formation of azobenzene along with free aniline indicates disproportionation of two moles of 1,2-diphenylhydrazine to azobenzene and two moles of aniline in this transformation. The aniline ligand in 76 is engaged in hydrogen bonding network with the protic pyrazole units along with the chloride counteranion. The disproportionation proceeds catalytically when an excess of 1,2-diphenylhydrazine is added to 75. An analogous complex of a tetradentate ligand having non-protic pyridylmethyl arms displays no catalytic activity, even in the presence of external protic pyrazole as a proton source, indicating that the proton-responsive unit in the second coordination sphere is responsible for the N–N bond cleavage.
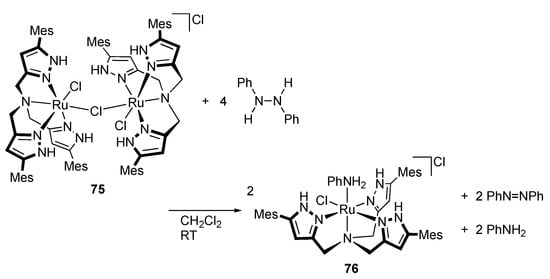
Scheme 22.
N–N bond cleavage of 1,2-diphenylhydrazine by a tripodal protic pyrazole ruthenium complex. Mes = 2,4,6-Me3C6H2.
Caulton’s group demonstrated that the reaction of dichromium(II) complex 35 (vide supra) [] with azobenzene yields the paramagnetic chromium(III) complex 77 having an unsymmetrically bridged PhNNPh unit (Scheme 23) []. The N–N distance of 1.471(9) Å indicates that the N=N bond is reduced by the chromium(II) centers in 35 to give the hydrazido(2−) ligand. A benzo[c]cinnoline derivative, featuring η2:η2-coordination of the azo group and lack of the THF ligand, is also characterized. Reduction of these hydrazido(2−) complexes with KC8 is further examined. Only the benzo[c]cinnoline derivative undergoes N–N bond cleavage upon treatment with an excess of the reductant.
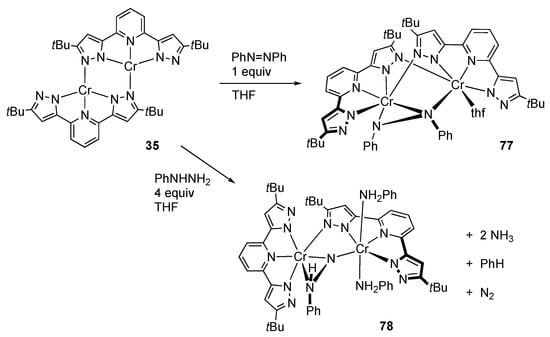
Scheme 23.
Reaction of the pyrazolato-bridged dichromium complex with azobenzene and phenylhydrazine.
Electron transfer from the dichloromium(II) complex 35 to phenylhydrazine gives rise to the formation of the phenylhydrazido(2−)-bridged di(aniline)dichromium(III) complex 78 (Scheme 23) []. The two aniline ligands, derived from N–N bond cleavage of phenylhydrazine, bind to one of the two chromium atoms at trans positions and form intramolecular hydrogen bonding with the protic pyrazoles on the other chromium center. While the reaction stoichiometry should be rather complicated, ammonia and benzene were detected as the fission products in the reaction mixture.
Even dinitrogen is coordinated to proton-responsive pyrazole-based complexes, as in the mononuclear tBuL complex 8 []. The NCN pincer-type dinuclear complex 79, supported by two linking diphosphine ligands, reacts with dinitrogen to afford the dinitrogen-bridged diruthenium(II) complex 80 (Scheme 24) []. Unfortunately, no further transformation of the dinitrogen ligand in this multiproton-responsive cavity has not been reported.
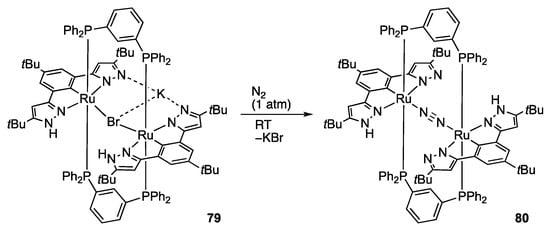
Scheme 24.
Synthesis of a dinitrogen-bridged diruthenium complex bearing protic pyrazole ligands.
2.3. Nitrate Reduction
Reduction of nitrogen oxides constitutes the inorganic nitrogen cycle in nature. Better understanding of the reaction may be important to address the issue of the coastal eutrophication caused by excessive use of nitrogen fertilizer. We found that the reaction of the ruthenium(II) complex 81 with silver nitrite leads to N–O bond cleavage of the nitro anion, giving the nitrosylruthenium(II) complex 82 (Scheme 25) []. The dehydrative conversion is most likely assisted by proton transfer from the pyrazole ligand, and the overall reaction is redox neutral.
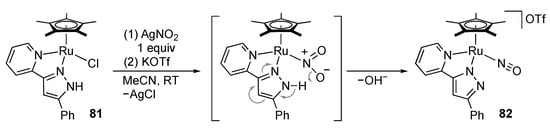
Scheme 25.
Dehydrative conversion of a nitrite ion to a nitrosyl ligand on a protic pyrazole complex of ruthenium.
Caulton’s group demonstrated that N,N′-disilyldihydropyrazines, whose usefulness in salt-free reduction was uncovered by Mashima and co-workers [], are effective for deoxygenation of NOx ligands on protic pyrazole complexes. Thus, the tris(nitrate) chromium(III) complex 83, bearing a protic bis(pyrazole)-type pincer ligand [], reacted with three equiv of an N,N′-disilyldihydropyrazine to afford the nitrosyl–nitrate chromium(I) complex 84 (Scheme 26) []. The reaction byproducts, hexamethyldisiloxane and pyrazine, were detected in the reaction mixture via 1H NMR spectroscopy. It is to be noted that the reaction takes place without any damage on the pyrazole NH protons.
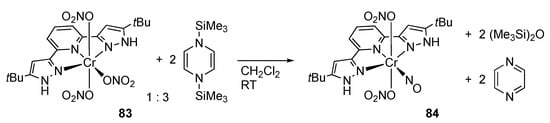
Scheme 26.
Deoxygenation of a nitrato ligand with N,N′-disilyldihydropyrazine on a protic pyrazole complex of chromium.
Similarly, treatment of the bis(1H-pyrazol-3-ylpyridine)nickel(II) nitrato complex 85 with an equimolar amount of the disilyldihydropyrazine results in deoxygenation of the nitrate and formation of mono-deprotonated product 86 (Scheme 27) []. The NH proton remaining on a pyrazole ring in 86 is identified by the larger NαΝβC angle. The deoxygenated product in this transformation, nitrite ion, was not detected; however, further deoxygenation of 86 affords the diamagnetic nickel(0) complex 87, having linear nitrosyl ligands derived from deoxygenation of the nitrate ligand. Thermodynamics of deoxygenation of the NOx ligands with a disilyldihydropyrazine was also investigated theoretically for manganese complexes bearing a PNN pincer-type protic pyrazole ligand [].
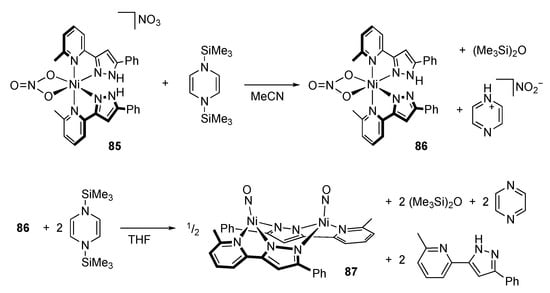
Scheme 27.
Deoxygenation of a nitrate ion with N,N′-disilyldihydropyrazine on a protic pyrazole complex of nickel.
The PNN pincer-type cobalt(II) dichlorido complex 88 bearing a protic pyrazole arm reacts with sodium nitrite to afford the tris(nitrito-N)cobalt(III) complex 89 and bent nitrosyl cobalt(III) complex 90 []. Although the yields and ratio of 89 and 90 are not described, the reaction stoichiometry shown in Scheme 28 has been proposed along with proton transfer from the pyrazole moiety to the bridging nitrite in a dinuclear intermediate. The compounds 89 and 90 are independently obtained by the reaction of the pincer ligand with a cobalt(III) complex Na3[Co(NO2)6] followed by deoxygenation and deprotonation with a disilyldihydropyrazine [].
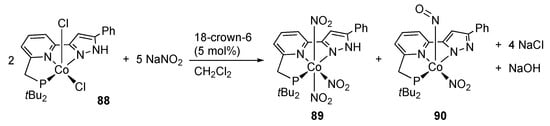
Scheme 28.
Reaction of a protic, PNN pincer-type cobalt complex with sodium nitrite.
2.4. CO2 Reduction
Liaw, Lu, and co-workers demonstrated that the pyrazolato-bridged {Fe(NO)2}9 complex 91 undergoes two-electron reduction to give the dianionic complex 92 (Scheme 29) []. Subsequent reaction with carbon dioxide results in nucleophilic attack of the pyrazolato ligand, resulting in the formation of the mononuclear CO2 adduct 93. Interestingly, addition of an equimolar amount of calcium triflate yields calcium oxalate and regenerates the dinuclear complex 91. A calcium-assisted one-electron transfer from the iron center to the CO2 unit followed by bimolecular coupling with 93 is proposed for the last step of this synthetic cycle.
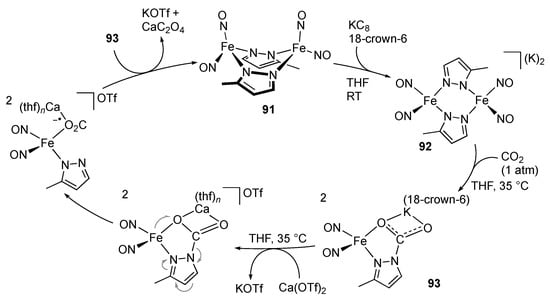
Scheme 29.
Synthetic cycle for reduction of carbon dioxide to oxalate promoted by a pyrazolato iron complex.
3. Catalysis of Protic Pyrazole Complexes
In addition to the aerobic oxidation of phosphines (Section 2.1.1) and catalytic disproportionation of hydrazines (Section 2.2), the catalytic application of protic pyrazole complexes to various chemical transformations has been investigated. This section focuses on recent work along with studies in which the role of the protic pyrazole ligand is evident or discussed. A previous review on this topic is available [].
3.1. Hydrogenation and Transfer Hydrogenation
In 2008, Yu’s group demonstrated that the protic pincer-type ruthenium(II) complex 94 catalyzes transfer hydrogenation of acetophenones with 2-propanol in the presence of an excess of a base (Scheme 30) []. Introduction of a non-protic pyrazole arm instead of the NHC in 94 significantly accelerates the reaction with 95 [,]. It is to be noted that the imidazole complex 96a with an NH group at a remoter position γ to the metal displays catalytic activity comparable with 95, whereas the non-protic analogue 96b is much less effective [,]. These results may suggest an inner-sphere mechanism involving β-hydrogen elimination of an alkoxide intermediate instead of a pyrazole-aided outer-sphere hydrogen transfer. The NH group in the pincer ligand may still operate to facilitate the dissociation of the halido ligand and to increase the nucleophilic character of the hydrido intermediate through deprotonation of the NH unit. Even asymmetric transfer hydrogenation of aryl ketones has been achieved with protic pincer-type complexes, such as 97, bearing an optically active oxazolinyl group in the chelate framework (Scheme 31) []. We [] and Halcrow [] also reported that RLH2 and related NCN pincer-type ruthenium(II) complexes 4 and 41 promote catalytic transfer hydrogenation of acetophenone with 2-propanol in the presence of alkoxide bases. The reactivities parallel with stoichiometric hydrogenation transfer in other protic pyrazole complexes [,].
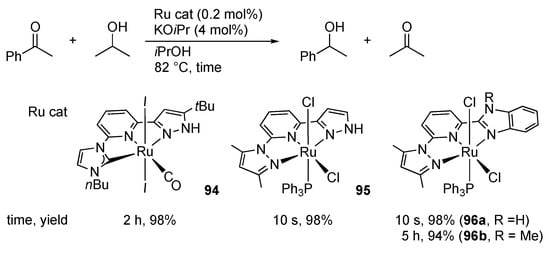
Scheme 30.
Transfer hydrogenation of acetophenone catalyzed by pincer-type pyrazole ruthenium complexes.
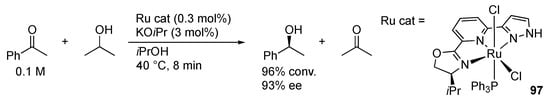
Scheme 31.
Asymmetric transfer hydrogenation of acetophenone catalyzed by a protic, pincer-type pyrazole ruthenium complex.
Thiel and co-workers demonstrated that the diprotic ruthenium(II) complexes 4a and 98 catalyze not only transfer hydrogenation but also hydrogenation of acetophenone (Scheme 32) []. Theoretical calculations suggest that outer sphere hydrogen transfer from the anticipated hydrido–pyrazole intermediate to the carbonyl substrate as well as the heterolytic cleavage of dihydrogen at the coordinatively unsaturated pyrazolato complex is facile. On the other hand, in the transfer hydrogenation reaction, the efficiency of the non-protic analogues, such as 99, is comparable or even higher in some cases [,,,], implying that such metal–pyrazole cooperating mechanism is less probable. Catalytic hydrogenation with the tBuLH2 analogue 4b is also reported [].
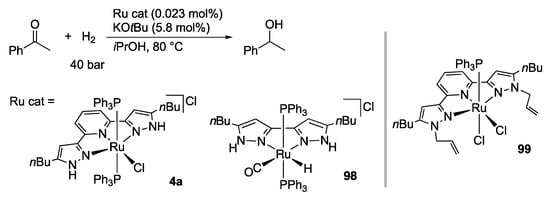
Scheme 32.
Hydrogenation of acetophenone catalyzed by diprotic pyrazole ruthenium complexes.
Nikonov’s group uncovered that the ruthenium(II) complexes, typified by 100, bearing a P–N chelate protic pyrazole ligand, promote transfer hydrogenation of not only acetophenone [] but also nitriles, heteroaromatics, alkynes, alkenes, and esters []. In the reaction of aromatic and aliphatic nitriles with 2-propanol, the initially formed amine product further reacts with acetone, a byproduct of the transfer hydrogenation in 2-propanol, to afford the ketimines as the final product (Scheme 33). No further reduction of the ketimines to secondary amines is observed. In the transfer hydrogenation of inner alkynes, semi-hydrogenation products, alkenes, are obtained with E-selectivity. The E-alkenes would be formed by the isomerization of Z-alkenes initially generated. Actually, cis-stilbene isomerizes to trans-stilbene under the catalytic conditions. When the substituents on alkynes are less bulky, further reduction takes place to give the corresponding alkanes. Meanwhile, the conversion of a terminal alkyne is very low. Scheme 33 also illustrates catalytic transfer hydrogenation of ethyl trifluoroacetate to 1,1,1-trifluoroethanol. In this reaction, ethanol is oxidized to ethyl acetate through Tishchenko reaction, and whole process can be viewed as ester metathesis [].
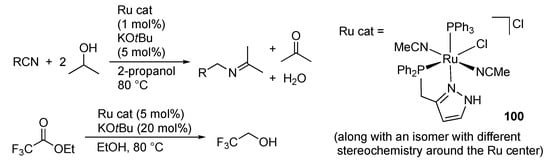
Scheme 33.
Transfer hydrogenation of nitriles and ethyl trifluoroacetate catalyzed by a protic pyrazole ruthenium complex.
Gong, Meggers, and co-workers revealed that the chiral-at-metal iridium(III) complex 101 catalyzes asymmetric transfer hydrogenation of ketones in the presence of protic pyrazoles (Scheme 34) []. Other additives, such as PnBu3, 2,6-diaminopyridine, and imidazole, leads to much lower catalytic activity and enantioselectivity as the reaction without the protic pyrazoles. An X-ray analysis of a related chlorido–pyrazole complex suggests that the NH unit would be placed in the optimum orientation for efficient bifunctional hydrogen transfer to the ketone substrate (inset). Further, attractive π–π interaction between the C–N chelate ligand and the arene ring in the substrate is proposed to realize the high enantioselectivity. This binary catalyst system is also effective for asymmetric hydrogenation of acetophenone []. In addition, the catalyst serves as a photoredox mediator, which allows asymmetric hydrogenation and photoredox transformation sequences without isolation of the chiral alcohol intermediate. On the other hand, half-sandwich C–N chelate pyrazole complexes of iridium(III) are known to catalyze transfer hydrogenation of acetophenone with 2-propanol [].
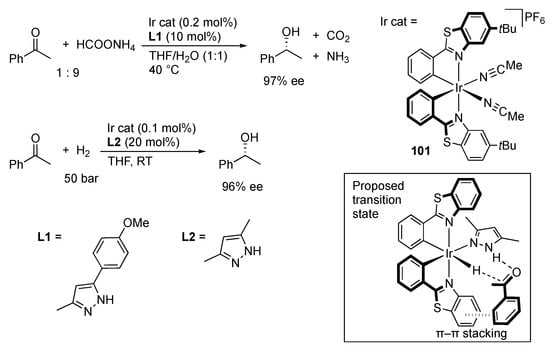
Scheme 34.
Asymmetric transfer hydrogenation and hydrogenation of acetonitrile mediated by a chiral iridium complex and protic pyrazoles binary catalysts. In inset, tert-butyl groups are omitted.
Niedner-Schatteburg, van Wüllen, and Thiel’s team demonstrated that the pyrazolatoruthenium(II) complex 102 catalyzes hydrogenation of carbon dioxide under supercritical conditions (Scheme 35), in addition to transfer hydrogenation of acetophenone with 2-propanol []. The CO2 hydrogenation activity of 102 is almost comparable with those of the conventional dichloridoruthenium(II) phosphine complexes. A mechanism without any proton response of the pyrazolato group is proposed on the basis of theoretical calculations (Scheme 36). A major role of the ionizable pyrazole in the chelate appears to be to make the chelate ligand anionic and more electron-donating []. The stronger trans influence of the pyrazolato moiety leads to generation of a vacant site, where carbon dioxide is coordinated. Subsequent insertion into the Ru–H bond gives a formato ligand, which mediates heterolytic cleavage of dihydrogen.
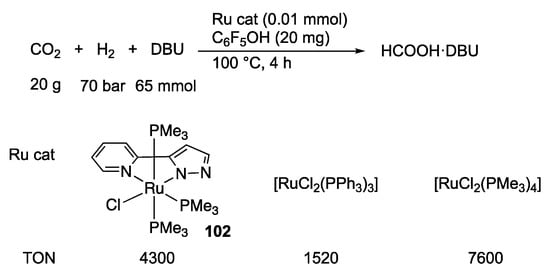
Scheme 35.
Hydrogenation of carbon dioxide catalyzed by ruthenium complexes. DBU = 1,8-diazabicyclo [5.4.0]undec-7-ene.
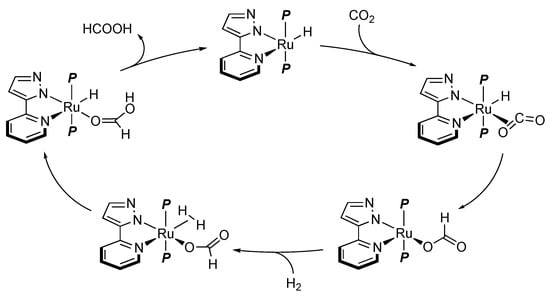
Scheme 36.
Proposed mechanism for hydrogenation of carbon dioxide with a pyrazolato ruthenium catalyst. P = PMe3.
Himeda, Ertem, and co-workers described the half-sandwich iridium(III) complexes bearing proton-responsive ligands as efficient CO2 hydrogenation catalysts in basic aqueous solutions (Scheme 37) [,]. The protic pyrazole group brings about better catalytic activity than the N-methylpyrazole group (103a vs. 103b and 104a vs. 104b). The performance of the pyrazole complexes 103 is, however, not so prominent when compared with that of the imidazole complexes 105. Meanwhile, introduction of an OH group at the 6-position of the pyridine ring significantly accelerates the reaction (104 and 106). These observations as well as DFT calculations led to the mechanistic proposal that the proton-responsive site on the diazole rings offers strong electron donation to the iridium center through its deprotonation. Still, the diazolato unit may also provide a proton acceptor site for the H2 heterolysis even in the absence of an OH group.
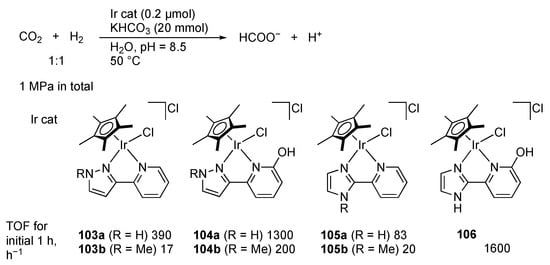
Scheme 37.
Hydrogenation of carbon dioxide catalyzed by iridium complexes.
3.2. Hydrogen Evolution
Dehydrogenation of formic acid, a reverse reaction of CO2 hydrogenation, is also catalyzed with protic pyrazole complexes. Wang, Himeda, and co-workers revealed that the 2-(1H-pyrazol-3-yl)pyridine iridium(III) complex 107 promotes hydrogen evolution from formic acid under acidic conditions (Scheme 38) []. The catalytic activity of 107 is less than that of the imidazole analogue 108 with a remoter γ-NH group, suggesting that the azole units mainly have a role in increasing the electron donation to the metal center through their deprotonation. Interestingly, introduction of a pendant pyridyl group on the pyrazole unit improves the catalytic activity []. The proposed mechanism involves a two-point hydrogen bonding between protic pyrazole–pyridinium unit and an external formic acid in the second coordination sphere (Scheme 39). The proton relay would facilitate the protonation to the hydrido ligand.
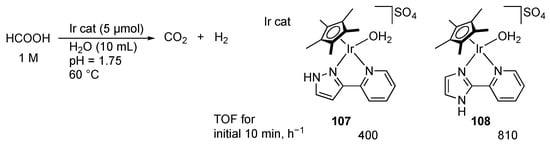
Scheme 38.
Dehydrogenation of formic acid catalyzed by iridium complexes.
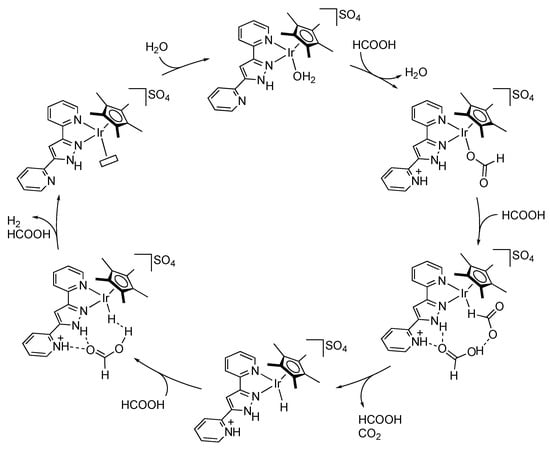
Scheme 39.
Proposed mechanism for dehydrogenation of formic acid catalyzed by an iridium complex bearing a pyridine-appended protic pyrazolylpyridine chelate ligand.
We reported hydrogen evolution from formic acid catalyzed by the CF3LH ruthenium(II) complex 109 (Scheme 40) []. The catalytic activity of the less acidic tBuLH analogue 6 is poor, indicating the importance of proton transfer from the protic pyrazole arm in the catalysis. The reaction with the NCN pincer-type complex 110 is also much slower.
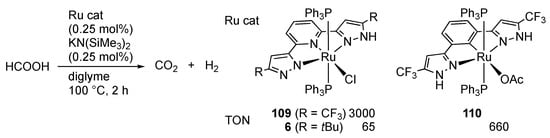
Scheme 40.
Dehydrogenation of formic acid catalyzed by protic pincer-type ruthenium complexes.
In addition to formic acid, amine–boranes have attracted much attention in terms of chemical H2-storage. Pal and Nozaki reported hydrogen evolution from dimethylamine–borane promoted by the pyrazole–pyrazolato rhodium(III) complex 111 []. On the basis of theoretical calculations as well as the fact that [{Cp*RhCl2}2] and the free pyrazole are catalytically inactive in separate runs, a metal–pyrazole cooperative mechanism is proposed, as shown in Scheme 41. Dehydrochlorination of the catalyst precursor 111 generates the coordinatively unsaturated bis(pyrazole) complex 112. The Lewis acidic rhodium center and Brønsted basic pyrazolato ligand in 112 dehydrogenates dimethylamine–borane substrate in a cooperative manner. The resulting hydrido complex 113 releases dihydrogen gas guided by an intramolecular hydrogen bond to regenerate 112.
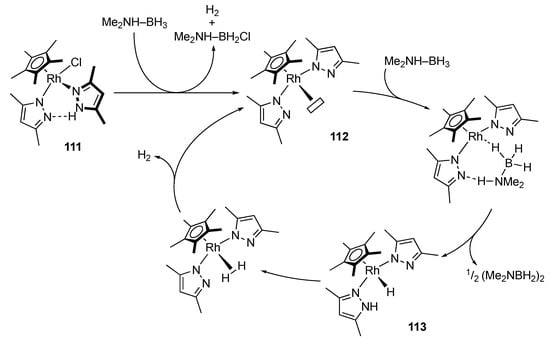
Scheme 41.
Proposed mechanism for dehydrogenation of an amine–borane catalyzed by a protic pyrazole rhodium complex.
3.3. Borrowing Hydrogen Catalysis
Owing to their hydrogen transfer ability, protic pyrazole complexes also exhibit borrowing hydrogen catalysis, wherein the catalyst first borrows hydrogen atoms from the alcohol substrate and then returns them after the bond formation of the resulting carbonyl intermediate []. Ryu and co-workers applied a pincer-type ligand in the Yu’s transfer hydrogenation catalyst (Section 3.1) to α-alkylation of amides with primary alcohols (Scheme 42) []. As in typical borrowing hydrogen transformations, initial dehydrogenative oxidation of the primary alcohol followed by dehydrative condensation of the resulting aldehyde and amide is proposed. Subsequent transfer hydrogenation from the catalyst would yield the α-alkylation product.
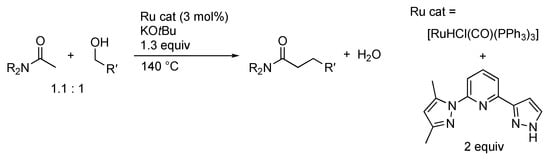
Scheme 42.
α-Alkylation of amides with primary alcohols catalyzed by a protic pyrazole ruthenium complex.
Bagh’s group demonstrated that the pyrazolato-bridged diiridium(III) complex 114 promotes α-alkylation of arylacetonitriles with secondary alcohols (Scheme 43) []. When the catalyst precursor 114 is dissolved in DMSO, the DMSO complex 115 is obtained, whereas reaction of 114 with cyclohexanol affords the hydrido–pyrazole complex 116 with liberation of cyclohexanone (Scheme 43b). Formation of these mononuclear N–O chelate complexes suggests that the catalysis involves initial split of the pyrazolato dimer complex 114 into a coordinatively unsaturated pyrazolato complex 117, which then undergoes transfer hydrogenation from the secondary alcohol to give the hydrido–pyrazole complex 116 (Scheme 43c). Following steps in borrowing hydrogen cycle were also supported by the stoichiometric reactions.
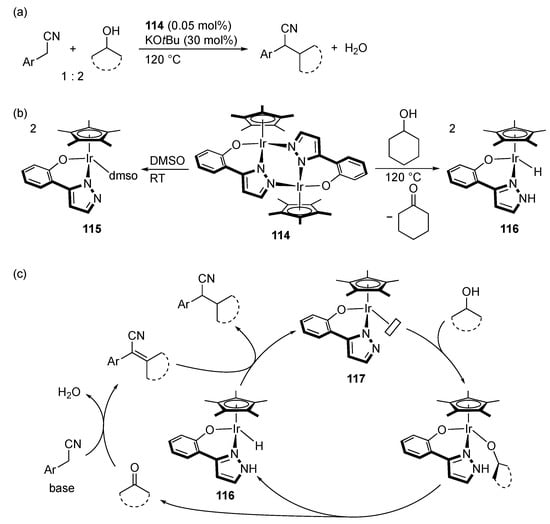
Scheme 43.
(a) α-Alkylation of arylacetonitriles with secondary alcohols catalyzed by a pyrazolato-bridged diiridium complex. (b) Stoichiometric reactivities of the dinuclear catalyst precursor. (c) Proposed mechanism.
3.4. Dehydrogenative Oxidation
If the hydride intermediate in the borrowing hydrogen catalysis somehow releases dihydrogen gas instead of returning the hydrogen atoms to give the redox-neutral product, the net reaction would turn to be dehydrogenative oxidation. Hölscher, Bera, and co-workers described acceptorless double dehydrogenation of primary amines catalyzed by the ruthenium(II) complex 118a bearing an N–N chelate protic pyrazole ligand (Scheme 44) []. Both aromatic and aliphatic nitriles are obtained in this manner. Secondary amines are also converted to the corresponding imines under the reaction conditions. The poor activity of the N-methylated analogue 118b indicates the crucial role of the pyrazole NH group in the catalysis. Meanwhile, the uncharged pyrazolato complex 119 with a Lewis acid displays catalytic performance comparable with that of 118a even in the absence of base. A coordinatively unsaturated pyrazolato complex is thus suggested to be a catalytically active species. Computational study proposed that the pyrazole unit is not involved directly in the abstraction of hydride from the amine substrate, but dehydrogenation of an imine intermediate occurs in a concerted, metal–ligand cooperative manner with the aid of an external substrate molecule in the second coordination sphere (inset). The resultant hydrido complex would undergo protonation with ammonium cation to evolve dihydrogen gas.
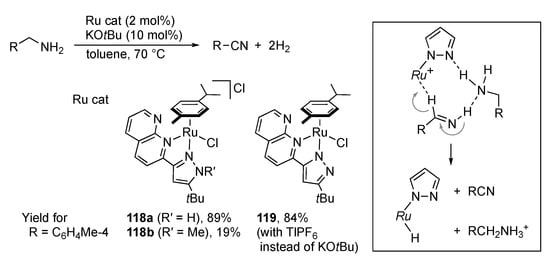
Scheme 44.
Acceptorless dehydrogenation of primary amines catalyzed by protic pyrazole ruthenium complexes.
Chai and co-workers reported synthesis of imines catalyzed by the protic pyrazole manganese(I) complex 120a (Scheme 45) []. The non-protic analogue 120b exhibits lower conversion and brings about increased formation of the amine byproduct through the borrowing hydrogen pathway, although the detailed reason is not mentioned. The reaction is proposed to proceed via dehydrative condensation of aniline and benzaldehyde, which is formed by dehydrogenative oxidation of benzyl alcohol with a coordinatively unsaturated pyrazolato complex.
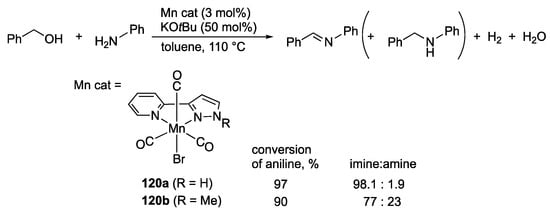
Scheme 45.
Acceptorless dehydrogenative coupling of benzyl alcohol and aniline catalyzed by pyrazole manganese complexes.
3.5. Transformation of Allylic and Propargylic Compounds
Satake and co-workers demonstrated that the N–N chelate pyridylpyrazole palladium(II) complex 121 catalyzes the reaction of allylic acetates and ketene silyl acetals, as shown in Scheme 46 []. The significance of the NH group therein is evident by comparison with the catalytic performance of the non-protic complex 122. On the other hand, the position of the NH group appears unimportant since the protic imidazole complex 123 bearing a γ-NH group displays similar catalytic activity and even better selectivity for cyclopropanation over allylation []. The proposed mechanism is illustrated in Scheme 47. Deprotonation of the catalyst precursor 121 gives the uncharged pyrazolato complex 124, which undergoes nucleophilic attack of the ketene silyl acetal to afford the palladacyclobutane complex 125. The major role of the NH group would be to make the chelate ligand a better σ-donor, which directs the attack of nucleophile at the central carbon rather than the terminal carbon atoms []. Use of chiral oxazolidines as the chelate tether realizes asymmetric cyclopropanation of a ketene silyl acetal with moderate stereoselectivity [].
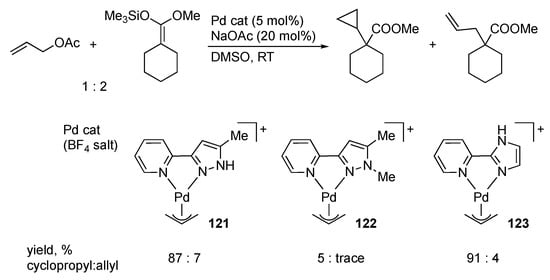
Scheme 46.
Cyclopropanation of ketene silyl acetal with allyl acetate catalyzed by pyrazole palladium complexes.
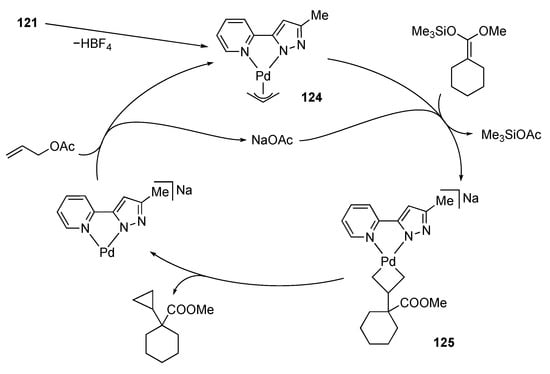
Scheme 47.
Proposed mechanism for catalytic cyclopropanation.
Gimeno, Lledós, and co-workers described isomerization of allylic alcohols mediated by protic azole ruthenium(IV) catalyst precursors in water (Scheme 48) [,]. Because the N-methylated pyrazole and γ-protic imidazole complexes exhibit similar and much higher catalytic performance, respectively, the aqua ligand rather than the protic azoles is proposed as an acid–base catalytic site to promote the metal–ligand cooperative hydrido migration from the allylic carbon to the vinyl carbon atom.
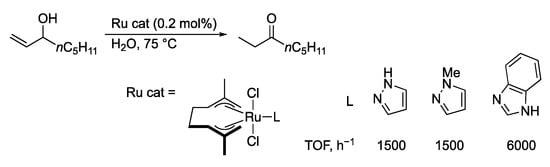
Scheme 48.
Isomerization of an allylic alcohol to a ketone catalyzed by azole ruthenium complexes.
We uncovered that the protic pyrazole ruthenium(II) complexes 126 catalyze isomerization of 1,1-dimethyl-3-phenylprop-2-yn-1-ol to 3-methyl-1-phenylbut-2-en-1-one in methanol (Scheme 49) []. The reaction, known as Meyer–Schuster rearrangement [,], does not occur with non-protic analogue 127 as well as a substitution-inert isocyanide complex, whereas the complex 126b with a less electron-withdrawing phenyl substituent requires a more elevated temperature (reflux, 87%). These observations indicate that both Lewis acidic metal center and Brønsted acidic NH group in 126 are necessary for this catalysis. A proposed mechanism is shown in Scheme 50. The π-bound propargylic alcohol in 128 undergoes an SN2′-type propargylic substitution by the solvent methanol with the aid of the intramolecular NH···O hydrogen bonding in the second coordination sphere to afford the allene complex 129. Dissociation and solvolysis of the allene would yield an acetal, which is finally converted to the enone product via hydrolysis.
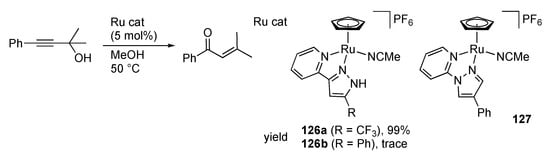
Scheme 49.
Isomerization of a propargylic alcohol to an enone catalyzed by pyrazole ruthenium complexes.
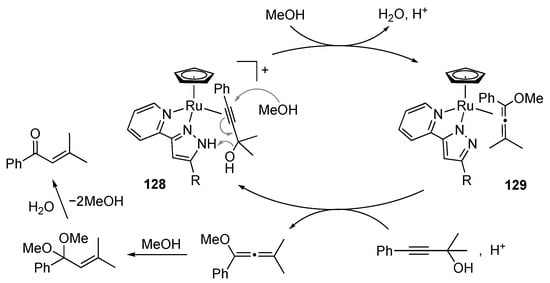
Scheme 50.
Proposed mechanism for isomerization of a propargylic alcohol catalyzed by protic pyrazole ruthenium complexes.
Use of less nucleophilic solvents, such as 1,2-dichloroethane and 1,4-dioxane, completely switches the reaction outcome. In the absence of the external nucleophile, a two-point interaction between the protic pyrazole complex and the substrate would lead to facile C–O bond cleavage, giving the η3-propargyl complex 130 (Scheme 51). Involvement of 130 in the Meyer–Schuster rearrangement in methanol (Scheme 50) is less likely because the nucleophilic addition to η3-propargyl ligands generally takes place at the central carbon atom [] and would fail to provide the observed product. The pyrazolato unit in 130 mediates proton migration in the η3-propargyl ligand to afford the η3-butadienyl complex 131, which is isolable in the case of R = CF3. When the pyrazolato ligand in 131 is more nucleophilic (R = Ph), intramolecular addition to the terminal carbon in the η3-butadienyl ligand occurs to give the N-allenylmethylpyrazole complex 132.
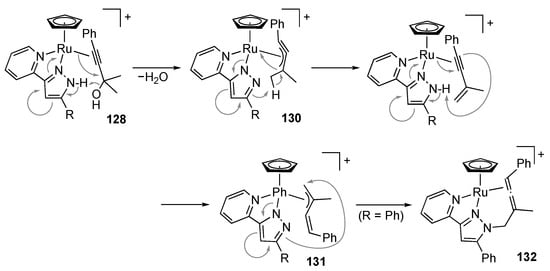
Scheme 51.
Transformation of a propargylic alcohol on protic pyrazole ruthenium complexes in less nucleophilic solvents.
3.6. Hydroamination of Alkenes
The metal–ligand bifunctional nature of protic pyrazole complexes is further applied to catalytic hydroamination of alkenes, in which both amine and olefin functional groups are activated simultaneously. We disclosed that the C–N chelate protic pyrazole iridium(III) complex 133 promotes cyclization of aminoalkenes with the aid of an equimolar amount of an alkoxide base (Scheme 52). The catalyst is compatible with various functional groups such as ester, bromo, cyano, and hydroxy groups. The pyrazolato-bridged dimer 134, obtained by dehydrochlorination of 133, exhibits catalytic activity similar to 133 even in the absence of the base. Meanwhile, the catalytic performance of the related complexes, 135 and 136, having a proton-responsive site at the positions γ and α to the iridium center, respectively, is poor. Additionally, catalytically inactive are the N-methylated derivative 137 and the six-membered chelate analogue 138 []. These results indicate that an exquisitely positioned proton-responsive site with appropriate direction and acidity is crucial for the catalysis. Scheme 53 illustrates a proposed mechanism featuring the metal–ligand cooperation. The olefin part in the aminoalkene substrate binds to the coordinatively unsaturated mononuclear pyrazolato complex 139, derived from dehydrochlorination of the chlorido complex 133 or split of the pyrazolato dimer 134. The activated olefin is attacked by the amino group with increased nucleophilic character owing to intramolecular hydrogen bond with the pyrazolato unit. The tight-fitting assembly in transition state 140 is supported by the large negative activation entropy provided by kinetic experiments. Subsequent proton transfer from the pyrazole unit to the Ir–C bond yields the product and regenerates the unsaturated pyrazolato complex 139.
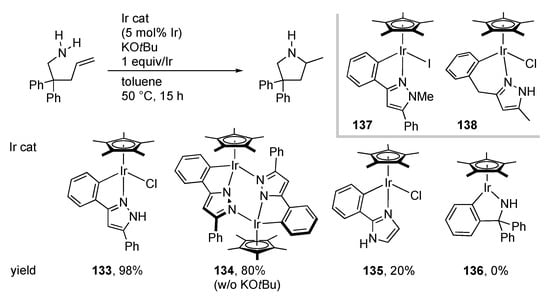
Scheme 52.
Cyclization of an aminoalkene catalyzed by iridium complexes.
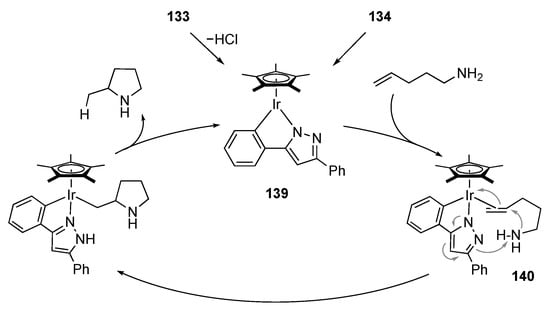
Scheme 53.
Proposed mechanism for cyclization of aminoalkenes catalyzed by a protic pyrazole iridium complex. Substituents of aminoalkenes are omitted.
3.7. Hydration of Nitriles
Metal-mediated coupling of nitrile and pyrazole to give a stable chelate pyrazolylamidino ligand, illustrated in Scheme 54, has been known for a long time. Nevertheless, a certain type of protic pyrazole complex catalyzes hydration of nitriles. Rodríguez, Romero, and co-workers reported hydration of benzonitriles and acrylonitrile catalyzed by the pyrazole complexes, such as 141 and 142 (Scheme 55) [,]. The reaction is proposed to proceed via nucleophilic attack of water or hydroxide anion to the coordinated nitrile. Although the role of the pyrazole ligand in this catalytic reaction is not mentioned, the proton-responsive pyrazole ligand may increase the nucleophilic character of water through hydrogen bonding in the second coordination sphere.
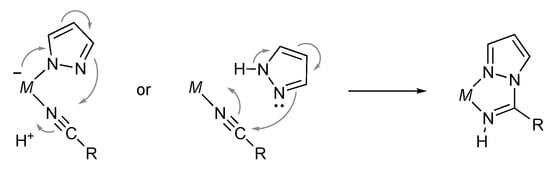
Scheme 54.
Coupling of pyrazole and nitrile on a metal center.
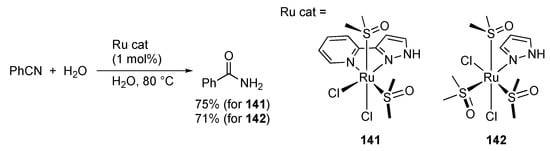
Scheme 55.
Catalytic hydration of benzonitrile with protic pyrazole ruthenium complexes.
3.8. Catalysis with Coordinatively Saturated Complexes
In their seminal work on chiral-at-metal complexes [,], Gong and Meggers described ligand-centered catalysis of protic pyrazole complexes. For example, the iridium complex 143 bearing an amido-substituted protic pyrazole ligand catalyzes asymmetric 1,4-addition of indoles to β-nitroacrylates (Scheme 56) []. As the well-known thiourea organocatalysis [], the amidopyrazole unit serves as a hydrogen bond donor to the nitroalkene. The chiral metal center in the catalyst 143 is coordinatively saturated and inert; however, the carboxamide substituent in the C–N chelate acts as a hydrogen bond acceptor for the indole, making the complex bifunctional.
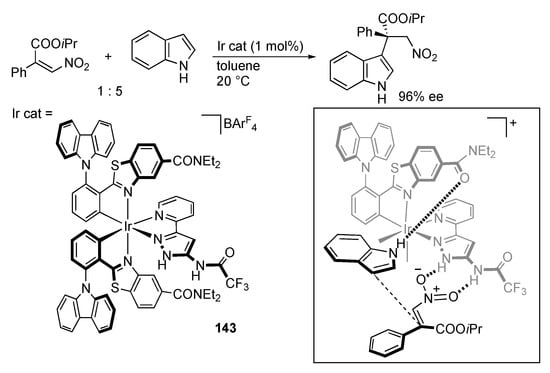
Scheme 56.
Asymmetric 1,4-addition of indole to a β-nitroacrylate catalyzed by a protic pyrazole iridium complex. ArF = C6H3(CF3)2-3,5.
3.9. Miscellaneous
Suzuki–Miyaura coupling reactions with bis(pyrazole) palladium(II) complexes [] as well as a binary system of PdCl2 and PhLH2 [] are reported. Garralda and co-workers described hydrolysis of ammonia–borane and amine–boranes catalyzed by the rhodium(III) bis(pyrazole) complexes, such as 144 (Scheme 57) []. Rodríguez and Romero reported the ruthenium(II) complex 145 bearing a protic pyrazole ligand catalyzes photochemical oxidation of alcohols in water in the presence of a sacrificial oxidant along with [Ru(bpy)3]2+ as a photosensitizer (Scheme 58) []. Chemical oxidation of alkenes to epoxides is also promoted. Unfortunately, the mechanisms for these reactions and the role of the protic pyrazole ligand therein are not discussed.
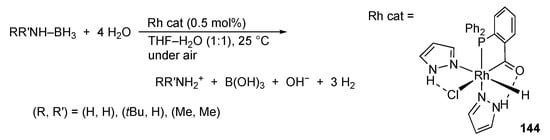
Scheme 57.
Hydrolysis of amine–boranes catalyzed by a protic pyrazole rhodium complex.
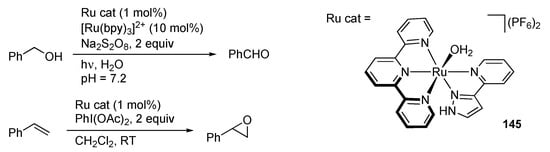
Scheme 58.
Oxidation of an alcohol and alkene catalyzed by a protic pyrazole ruthenium complex.
4. Conclusions
It is now apparent that the protic pyrazoles are versatile non-innocent ligands, owing to their ability to place a proton-responsive site in the second coordination sphere. The deprotonation of the β-NH unit turns the pyrazole ligand to a stronger σ-donor. In both protonated and deprotonated forms, the ligand-based hydrogen bonding and electrostatic interactions at the β-position allows efficient substrate recognition and activation as well as additional tuning of the electronic properties of the metal center. Importantly, these events in the outer coordination sphere are linked with the metal-centered reactions in some cases. Such metal–ligand cooperation can be compared with those of related proton-responsive ligand systems (Scheme 1); however, the stiff five-membered structure that fixes the proton with an azole acidity in the metal–pyrazole plane makes the reactivity of protic pyrazole complexes distinctive. Additionally, facile redox of the metal–pyrazole framework coupled with deprotonation is implied for π-delocalized, bis(pyrazole)-type tBuLH2 complexes (Scheme 21 and compound 40 in Scheme 12). The metal–ligand cooperative catalysis thus has been explored for various types of reactions, typified by transfer hydrogenation and functionalization of unsaturated carbon–carbon bonds. The role of the NH groups therein, however, appears to depend on the catalyst systems. In some reactions, control experiments with γ-protic imidazole complexes indicate that the presence of the ionizable NH group is prerequisite for the catalysis but the position is unimportant. The protic pyrazole ligand in this case would remain deprotonated during the catalytic turnover and serve only to increase the electron density of the metal center. Further studies are needed to elucidate the conditions under which the metal–ligand cooperation is exerted. These efforts will lead to improvement and deeper mechanistic understanding of the catalysis of protic pyrazole complexes. The metal–ligand cooperation has also been applied to activation of small inorganic molecules. A future target in this direction will undoubtedly be multiproton-coupled multielectron reduction of inert molecules, such as carbon dioxide and dinitrogen, with polyprotic pyrazole complexes. In these studies, novel design of protic pyrazole ligands will continue to be a major subject to create more sophisticated metal–ligand cooperation platforms for both stoichiometric and catalytic transformations unique to protic pyrazole complexes. Facile construction of the pyrazole rings will be advantageous in the synthesis of the pyrazole ligands whose Brønsted acidity and deployment are controlled by the substituents on the ring and chelate framework.
Author Contributions
Writing—original draft preparation, W.-S.L.; writing—review and editing, S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI Grant Number JP19H02732.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Trofimenko, S. Recent Advances in Poly(pyrazolyl)borate (Scorpionate) Chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Halcrow, M.A. Pyrazoles and Pyrazolides—Flexible Synthons in Self-Assembly. Dalton Trans. 2009, 38, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.S.S.; Zeni, G. Alkynes and Nitrogen Compounds: Useful Substrates for the Synthesis of Pyrazoles. Chem. Eur. J. 2020, 26, 8175–8189. [Google Scholar] [CrossRef] [PubMed]
- Sloop, J.C.; Holder, C.; Henary, M. Selective Incorporation of Fluorine in Pyrazoles. Eur. J. Org. Chem. 2015, 2015, 3405–3422. [Google Scholar] [CrossRef]
- Bhaumik, P.K.; Ghosh, K.; Chattopadhyay, S. Synthetic Strategies, Crystal Structures and Biological Activities of Metal Complexes with the Members of Azole Family: A Review. Polyhedron 2021, 200, 115093. [Google Scholar] [CrossRef]
- Ionel, H. Inverse Coordination. Organic Nitrogen Heterocycles as Coordination Centers. A Survey of Molecular Topologies and Systematization. Part Five-Membered and Smaller Rings. J. Coord. Chem. 2019, 72, 2127–2159. [Google Scholar] [CrossRef]
- Sethi, S.; Jena, S.; Das, P.K.; Behera, N. Synthetic Approach and Structural Diversities of Pyridylpyrazole Derived Late Transition Metal Complexes. J. Mol. Struct. 2019, 1193, 495–521. [Google Scholar] [CrossRef]
- Doidge, E.D.; Roebuck, J.W.; Healy, M.R.; Tasker, P.A. Phenolic Pyrazoles: Versatile Polynucleating Ligands. Coord. Chem. Rev. 2015, 288, 98–117. [Google Scholar] [CrossRef]
- Lu, C.-W.; Wang, Y.; Chi, Y. Metal Complexes with Azolate-Functionalized Multidentate Ligands: Tactical Designs and Optoelectronic Applications. Chem. Eur. J. 2016, 22, 17892–17908. [Google Scholar] [CrossRef]
- Kuwata, S.; Ikariya, T. β-Protic Pyrazole and N-Heterocyclic Carbene Complexes: Synthesis, Properties, and Metal–Ligand Cooperative Bifunctional Catalysis. Chem. Eur. J. 2011, 17, 3542–3556. [Google Scholar] [CrossRef]
- Kuwata, S.; Ikariya, T. Metal–Ligand Bifunctional Reactivity and Catalysis of Protic N-Heterocyclic Carbene and Pyrazole Complexes Featuring β-NH Units. Chem. Commun. 2014, 50, 14290–14300. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, S. Design of Multiproton-Responsive Metal Complexes as Molecular Technology for Transformation of Small Molecules. In Molecular Technology: Energy Innovation; Yamamoto, H., Kato, T., Eds.; Wiley-VCH: Weinheim, Germany, 2018; pp. 81–103. ISBN 978-3-527-34163-4. [Google Scholar]
- Pérez, J.; Riera, L. Pyrazole Complexes and Supramolecular Chemistry. Eur. J. Inorg. Chem. 2009, 2009, 4913–4925. [Google Scholar] [CrossRef]
- Perez, J.; Riera, L. Organometallic Complexes as Anion Hosts. Chem. Commun. 2008, 44, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Riera, L. Stable Metal–Organic Complexes as Anion Hosts. Chem. Soc. Rev. 2008, 37, 2658–2667. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Shamsuzzaman Review: Biologically Active Pyrazole Derivatives. New J. Chem. 2016, 41, 16–41. [Google Scholar] [CrossRef]
- Khusnutdinova, J.R.; Milstein, D. Metal–Ligand Cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef]
- Kar, S.; Milstein, D. Sustainable Catalysis with Fluxional Acridine-Based PNP Pincer Complexes. Chem. Commun. 2022, 58, 3731–3746. [Google Scholar] [CrossRef]
- Elsby, M.R.; Baker, R.T. Strategies and Mechanisms of Metal–Ligand Cooperativity in First-Row Transition Metal Complex Catalysts. Chem. Soc. Rev. 2020, 49, 8933–8987. [Google Scholar] [CrossRef]
- Cotman, A.E. Escaping from Flatland: Stereoconvergent Synthesis of Three-Dimensional Scaffolds via Ruthenium(II)-Catalyzed Noyori–Ikariya Transfer Hydrogenation. Chem. Eur. J. 2021, 27, 39–53. [Google Scholar] [CrossRef]
- Akter, M.; Anbarasan, P. (Cyclopentadienone)iron Complexes: Synthesis, Mechanism and Applications in Organic Synthesis. Chem. Asian J. 2021, 16, 1703–1724. [Google Scholar] [CrossRef]
- Gonçalves, T.P.; Dutta, I.; Huang, K.-W. Aromaticity in Catalysis: Metal Ligand Cooperation via Ligand Dearomatization and Rearomatization. Chem. Commun. 2021, 57, 3070–3082. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K. Development and Application of New Iridium Catalysts for Efficient Dehydrogenative Reactions of Organic Molecules. Bull. Chem. Soc. Jpn. 2019, 92, 344–351. [Google Scholar] [CrossRef]
- Matsunami, A.; Kayaki, Y. Upgrading and Expanding the Scope of Homogeneous Transfer Hydrogenation. Tetrahedron Lett. 2018, 59, 504–513. [Google Scholar] [CrossRef]
- Hale, L.V.A.; Szymczak, N.K. Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere. ACS Catal. 2018, 8, 6446–6461. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The Role of the Metal-Bound N–H Functionality in Noyori-Type Molecular Catalysts. Nat. Rev. Chem. 2018, 2, 396–408. [Google Scholar] [CrossRef]
- Theuergarten, E.; Schlüns, D.; Grunenberg, J.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. Intramolecular Heterolytic Dihydrogen Cleavage by a Bifunctional Frustrated Pyrazolylborane Lewis Pair. Chem. Commun. 2010, 46, 8561–8563. [Google Scholar] [CrossRef]
- Theuergarten, E.; Schlösser, J.; Schlüns, D.; Freytag, M.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. Fixation of Carbon Dioxide and Related Small Molecules by a Bifunctional Frustrated Pyrazolylborane Lewis Pair. Dalton Trans. 2012, 41, 9101–9110. [Google Scholar] [CrossRef]
- Cook, L.J.K.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin State Behavior of Iron(II)/Dipyrazolylpyridine Complexes. New Insights from Crystallographic and Solution Measurements. Coord. Chem. Rev. 2015, 289–290, 2–12. [Google Scholar] [CrossRef]
- Halcrow, M.A. Recent Advances in the Synthesis and Applications of 2,6-Dipyrazolylpyridine Derivatives and Their Complexes. New J. Chem. 2014, 38, 1868–1882. [Google Scholar] [CrossRef]
- Craig, G.A.; Roubeau, O.; Aromí, G. Spin State Switching in 2,6-Bis(pyrazol-3-yl)pyridine (3-Bpp) Based Fe(II) Complexes. Coord. Chem. Rev. 2014, 269, 13–31. [Google Scholar] [CrossRef]
- Olguín, J.; Brooker, S. Spin Crossover Active Iron(II) Complexes of Selected Pyrazole-Pyridine/Pyrazine Ligands. Coord. Chem. Rev. 2011, 255, 203–240. [Google Scholar] [CrossRef]
- Halcrow, M.A. The Synthesis and Coordination Chemistry of 2,6-Bis(pyrazolyl)pyridines and Related Ligands—Versatile Terpyridine Analogues. Coord. Chem. Rev. 2005, 249, 2880–2908. [Google Scholar] [CrossRef]
- Li, T.-Y.; Wu, J.; Wu, Z.-G.; Zheng, Y.-X.; Zuo, J.-L.; Pan, Y. Rational Design of Phosphorescent Iridium(III) Complexes for Emission Color Tunability and Their Applications in OLEDs. Coord. Chem. Rev. 2018, 374, 55–92. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Van, S.; Combs, D.; Lev, D.A.; Schneider, C.; Incarvito, C.D.; Lam, K.-C.; Rossi, G.; Rheingold, A.L.; Rideout, M.; et al. Substituent Control of Hydrogen Bonding in Palladium(II)-Pyrazole Complexes. Inorg. Chem. 2003, 42, 3347–3355. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Van, S.; Combs, D.; Lev, D.A.; Schneider, C.; Rideout, M.; Meyer, C.; Hernandez, G.; Mejorado, L. New Flexible Synthesis of Pyrazoles with Different, Functionalized Substituents at C3 and C5. J. Org. Chem. 2002, 67, 9200–9209. [Google Scholar] [CrossRef]
- Grotjahn, D.B. Bifunctional Catalysts and Related Complexes: Structures and Properties. Dalton Trans. 2008, 37, 6497–6508. [Google Scholar] [CrossRef]
- Labrum, N.S.; Chen, C.-H.; Caulton, K.G. A New Face for Bis(pyrazol-3-yl)pyridine: Incompatible Geometric Preferences Dictates Unprecedented Pincer Ligand Connectivity. Inorg. Chim. Acta 2019, 485, 54–57. [Google Scholar] [CrossRef]
- Jozak, T.; Zabel, D.; Schubert, A.; Thiel, W.R. Ruthenium Complexes Bearing N–H Acidic Pyrazole Ligands. Eur. J. Inorg. Chem. 2010, 2010, 5135–5145. [Google Scholar] [CrossRef]
- Yoshinari, A.; Tazawa, A.; Kuwata, S.; Ikariya, T. Synthesis, Structures, and Reactivities of Pincer-Type Ruthenium Complexes Bearing Two Proton-Responsive Pyrazole Arms. Chem. Asian J. 2012, 7, 1417–1425. [Google Scholar] [CrossRef]
- Kashiwame, Y.; Watanabe, M.; Araki, K.; Kuwata, S.; Ikariya, T. Synthesis, Structure, and Proton-Transfer Reactions of Brønsted Acidic Pyridylpyrazole Complexes of Ruthenium. Bull. Chem. Soc. Jpn. 2011, 84, 251–258. [Google Scholar] [CrossRef]
- Liao, J.-L.; Chi, Y.; Su, Y.-D.; Huang, H.-X.; Chang, C.-H.; Liu, S.-H.; Lee, G.-H.; Chou, P.-T. Os(II) Metal Phosphors Bearing Tridentate 2,6-Di(pyrazol-3-yl)pyridine Chelate: Synthetic Design, Characterization and Application in OLED Fabrication. J. Mater. Chem. C 2014, 2, 6269–6282. [Google Scholar] [CrossRef]
- Petrović, A.Z.; Ćoćić, D.C.; Bockfeld, D.; Živanović, M.; Milivojević, N.; Virijević, K.; Janković, N.; Scheurer, A.; Vraneš, M.; Bogojeski, J.V. Biological Activity of Bis(pyrazolylpyridine) and Terpiridine Os(II) Complexes in the Presence of Biocompatible Ionic Liquids. Inorg. Chem. Front. 2021, 8, 2749–2770. [Google Scholar] [CrossRef]
- Kuo, J.L.; Goldberg, K.I. Metal/Ligand Proton Tautomerism Facilitates Dinuclear H2 Reductive Elimination. J. Am. Chem. Soc. 2020, 142, 21439–21449. [Google Scholar] [CrossRef] [PubMed]
- Milutinović, M.M.; Bogojeski, J.V.; Klisurić, O.; Scheurer, A.; Elmroth, S.K.C.; Bugarčić, Z.D. Synthesis and Structures of a Pincer-Type Rhodium(III) Complex: Reactivity toward Biomolecules. Dalton Trans. 2016, 45, 15481–15491. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Saitoh, K.; Yoshinari, A.; Ikariya, T.; Kuwata, S. Synthesis and Structures of NCN Pincer-Type Ruthenium and Iridium Complexes Bearing Protic Pyrazole Arms. Organometallics 2017, 36, 1188–1195. [Google Scholar] [CrossRef]
- Zahora, B.A.; Gau, M.R.; Goldberg, K.I. Synthesis and Reactivity of PtII Methyl Complexes Supported by Pyrazolate Pincer Ligands. Organometallics 2020, 39, 1230–1237. [Google Scholar] [CrossRef]
- Galstyan, A.; Naziruddin, A.R.; Cebrián, C.; Iordache, A.; Daniliuc, C.G.; Cola, L.D.; Strassert, C.A. Correlating the Structural and Photophysical Features of Pincer Luminophores and Monodentate Ancillary Ligands in PtII Phosphors. Eur. J. Inorg. Chem. 2015, 2015, 5822–5831. [Google Scholar] [CrossRef]
- Umehara, K.; Kuwata, S.; Ikariya, T. Synthesis, Structures, and Reactivities of Iron, Cobalt, and Manganese Complexes Bearing a Pincer Ligand with Two Protic Pyrazole Arms. Inorg. Chim. Acta 2014, 413, 136–142. [Google Scholar] [CrossRef]
- Umehara, K.; Kuwata, S.; Ikariya, T. N–N Bond Cleavage of Hydrazines with a Multiproton-Responsive Pincer-Type Iron Complex. J. Am. Chem. Soc. 2013, 135, 6754–6757. [Google Scholar] [CrossRef]
- Nikovsky, I.A.; Polezhaev, A.V.; Melnikova, E.K.; Nelyubina, Y.V. New Iron(III) Oxo Complex with Substituted 2,6-Bis(pyrazol-3-yl)pyridine. Russ. J. Inorg. Chem. 2020, 65, 864–869. [Google Scholar] [CrossRef]
- Cook, B.J.; Polezhaev, A.V.; Chen, C.-H.; Pink, M.; Caulton, K.G. Deprotonation, Chloride Abstraction, and Dehydrohalogenation as Synthetic Routes to Bis-Pyrazolate Pyridyl Iron(II) Complexes. Eur. J. Inorg. Chem. 2017, 2017, 3999–4012. [Google Scholar] [CrossRef]
- Cook, B.J.; Chen, C.-H.; Pink, M.; Caulton, K.G. Dehydrohalogenation of Proton Responsive Complexes: Versatile Aggregation Viapyrazolate Pincer Ligand Arms. Dalton Trans. 2018, 47, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.J.; Pink, M.; Chen, C.-H.; Caulton, K.G. Electrophile Recruitment as a Structural Element in Bis-Pyrazolate Pyridine Complex Aggregation. Eur. J. Inorg. Chem. 2018, 2018, 5160–5166. [Google Scholar] [CrossRef]
- Cook, B.J.; Chen, C.-H.; Caulton, K.G. A Multifunctional Pincer Ligand for Cobalt-Promoted Oxidation by N2O. Chem. Eur. J. 2018, 24, 5962–5966. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.J.; Pink, M.; Pal, K.; Caulton, K.G. Electron and Oxygen Atom Transfer Chemistry of Co(II) in a Proton Responsive, Redox Active Ligand Environment. Inorg. Chem. 2018, 57, 6176–6185. [Google Scholar] [CrossRef]
- Labrum, N.S.; Pink, M.; Chen, C.-H.; Caulton, K.G. Reactivity of an Unusual Divalent Chromium Aggregate Supported by a Multifunctional Bis(pyrazolate) Pincer Ligand. Eur. J. Inorg. Chem. 2019, 2019, 1932–1940. [Google Scholar] [CrossRef]
- Labrum, N.S.; Curtin, G.M.; Jakubikova, E.; Caulton, K.G. The Influence of Nucleophilic and Redox Pincer Character as Well as Alkali Metals on the Capture of Oxygen Substrates: The Case of Chromium(II). Chem. Eur. J. 2020, 26, 9547–9555. [Google Scholar] [CrossRef]
- Labrum, N.S.; Chen, C.-H.; Caulton, K.G. A Bis-Pyrazolate Pincer on Reduced Cr Deoxygenates CO2: Selective Capture of the Derived Oxide by CrII. Chem. Eur. J. 2019, 25, 7935–7940. [Google Scholar] [CrossRef]
- Toda, T.; Yoshinari, A.; Ikariya, T.; Kuwata, S. Protic N-Heterocyclic Carbene Versus Pyrazole: Rigorous Comparison of Proton- and Electron-Donating Abilities in a Pincer-Type Framework. Chem. Eur. J. 2016, 22, 16675–16683. [Google Scholar] [CrossRef]
- Toda, T.; Kuwata, S. Central N-Heterocyclic Carbene Moieties in Protic Pincer-Type Bis(pyrazole) Ligands: Perturbation on Steric and Electronic Properties of Ruthenium Center. J. Organomet. Chem. 2020, 917, 121270. [Google Scholar] [CrossRef]
- Toda, T.; Kuwata, S. Synthesis, Structures, and Reactivities of Iron Complexes Bearing an Isoindoline-Based, Polyprotic Pincer-Type Pyrazole Ligand. Z. Anorg. Allg. Chem. 2021, 647, 1471–1477. [Google Scholar] [CrossRef]
- Kuwata, S.; Hahn, F.E. Complexes Bearing Protic N-Heterocyclic Carbene Ligands. Chem. Rev. 2018, 118, 9642–9677. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Kuwata, S.; Ikariya, T. Unsymmetrical Pincer-Type Ruthenium Complex Containing β-Protic Pyrazole and N-Heterocyclic Carbene Arms: Comparison of Brønsted Acidity of NH Groups in Second Coordination Sphere. Chem. Eur. J. 2014, 20, 9539–9542. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Kuwata, S.; Ikariya, T. Synthesis and Structures of Ruthenium and Iron Complexes Bearing an Unsymmetrical Pincer-Type Ligand with Protic Pyrazole and Tertiary Aminoalkyl Arms. Z. Anorg. Allg. Chem. 2015, 641, 2135–2139. [Google Scholar] [CrossRef]
- Polezhaev, A.V.; Liss, C.J.; Telser, J.; Chen, C.-H.; Caulton, K.G. A PNNH Pincer Ligand Allows Access to Monovalent Iron. Chem. Eur. J. 2018, 24, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Polezhaev, A.V.; Chen, C.; Losovyj, Y.; Caulton, K.G. A Multifunctional Pincer Ligand Supports Unsaturated Cobalt: Five Functionalities in One Pincer. Chem. Eur. J. 2017, 23, 8039–8050. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, A.C.; Carta, V.; Chen, C.-H.; Pink, M.; Caulton, K.G. Pincers with Diverse Donors and Their Interconversion: Application to Ni(II). Z. Anorg. Allg. Chem. 2021, 647, 1524–1529. [Google Scholar] [CrossRef]
- Koo, C.-K.; Ho, Y.-M.; Chow, C.-F.; Lam, M.H.-W.; Lau, T.-C.; Wong, W.-Y. Synthesis and Spectroscopic Studies of Cyclometalated Pt(II) Complexes Containing a Functionalized Cyclometalating Ligand, 2-Phenyl-6-(1H-pyrazol-3-yl)-pyridine. Inorg. Chem. 2007, 46, 3603–3612. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.; Gómez-Iglesias, P.; Anton, N.; García-Rodríguez, R.; Alegria, E.C.B.A.; Pombeiro, A.J.L.; Miguel, D.; Villafañe, F. Homo- and Heteropolymetallic 3-(2-Pyridyl)pyrazolate Manganese and Rhenium Complexes. Dalton Trans. 2014, 43, 4009–4020. [Google Scholar] [CrossRef]
- Cabelof, A.C.; Carta, V.; Caulton, K.G. A Proton-Responsive Ligand Becomes a Dimetal Linker for Multisubstrate Assembly via Nitrate Deoxygenation. Chem. Commun. 2021, 57, 2780–2783. [Google Scholar] [CrossRef]
- Nakahara, Y.; Toda, T.; Kuwata, S. Iron and Ruthenium Complexes Having a Pincer-Type Ligand with Two Protic Amidepyrazole Arms: Structures and Catalytic Application. Polyhedron 2018, 143, 105–110. [Google Scholar] [CrossRef]
- Tanaka, H.; Hitaoka, S.; Umehara, K.; Yoshizawa, K.; Kuwata, S. Mechanistic Study on Catalytic Disproportionation of Hydrazine by a Protic Pincer-Type Iron Complex through Proton-Coupled Electron Transfer. Eur. J. Inorg. Chem. 2020, 2020, 1472–1482. [Google Scholar] [CrossRef]
- Yamagishi, H.; Konuma, H.; Kuwata, S. Stereoselective Synthesis of Chlorido–Phosphine Ruthenium Complexes Bearing a Pyrazole-Based Protic Tripodal Amine Ligand. Polyhedron 2017, 125, 173–178. [Google Scholar] [CrossRef]
- Yamagishi, H.; Nabeya, S.; Ikariya, T.; Kuwata, S. Protic Ruthenium Tris(pyrazol-3-ylmethyl)amine Complexes Featuring a Hydrogen-Bonding Network in the Second Coordination Sphere. Inorg. Chem. 2015, 54, 11584–11586. [Google Scholar] [CrossRef]
- Labrum, N.S.; Cabelof, A.C.; Caulton, K.G. A Dimeric Chromium(II) Pincer as an Electron Shuttle for N=N Bond Scission. Chem. Eur. J. 2020, 26, 13915–13926. [Google Scholar] [CrossRef]
- Labrum, N.S.; Caulton, K.G. [Cr(Pincer2−)]2 as an Electron Shuttle for Reductively Promoted Hydrazine Disproportionation. Dalton Trans. 2019, 48, 11642–11646. [Google Scholar] [CrossRef]
- Toda, T.; Suzuki, S.; Kuwata, S. Metallo-Supramolecular Assembly of Protic Pincer-Type Complexes: Encapsulation of Dinitrogen and Carbon Disulfide into a Multiproton-Responsive Diruthenium Cage. Chem. Commun. 2019, 55, 1028–1031. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, H.; Tanahashi, H.; Kawakita, K.; Tsurugi, H.; Mashima, K. 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes as Strong Salt-Free Reductants for Generating Low-Valent Early Transition Metals with Electron-Donating Ligands. J. Am. Chem. Soc. 2014, 136, 5161–5170. [Google Scholar] [CrossRef]
- Labrum, N.S.; Seo, J.; Chen, C.-H.; Pink, M.; Beagan, D.M.; Caulton, K.G. Di- and Trivalent Chromium Bis(pyrazol-3-yl)pyridine Pincer Complexes with Good Leaving Groups. Inorg. Chim. Acta 2019, 486, 483–491. [Google Scholar] [CrossRef]
- Seo, J.; Cabelof, A.C.; Chen, C.-H.; Caulton, K.G. Selective Deoxygenation of Nitrate to Nitrosyl Using Trivalent Chromium and the Mashima Reagent: Reductive Silylation. Chem. Sci. 2018, 10, 475–479. [Google Scholar] [CrossRef]
- Cabelof, A.C.; Erny, A.M.; Beagan, D.M.; Caulton, K.G. A Redox Cascade of NOx− Complexes: Structures and Nitrogen Deoxygenation Thermodynamics. Polyhedron 2021, 200, 115119. [Google Scholar] [CrossRef]
- Cabelof, A.C.; Carta, V.; Chen, C.-H.; Caulton, K.G. Nitrogen Oxyanion Reduction by Co(II) Augmented by a Proton Responsive Ligand: Recruiting Multiple Metals. Dalton Trans. 2020, 49, 7891–7896. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-T.; Ching, W.-M.; Liaw, W.-F.; Lu, T.-T. Dinitrosyl Iron Complex [K-18-crown-6-ether][(NO)2Fe(MePyrCO2)]: Intermediate for Capture and Reduction of Carbon Dioxide. Angew. Chem. Int. Ed. 2020, 59, 11819–11823. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Yu, Z. Ruthenium(II) Complexes Bearing a Pyridyl-Supported Pyrazolyl-N-Heterocyclic Carbene (NNC) Ligand and Their Catalytic Activity in the Transfer Hydrogenation of Ketones. Organometallics 2008, 27, 6025–6028. [Google Scholar] [CrossRef]
- Jin, W.; Wang, L.; Yu, Z. A Highly Active Ruthenium(II) Pyrazolyl–Pyridyl–Pyrazole Complex Catalyst for Transfer Hydrogenation of Ketones. Organometallics 2012, 31, 5664–5667. [Google Scholar] [CrossRef]
- Liu, T.; Wang, L.; Wu, K.; Wang, Q.; Yu, Z. Mono- and Multinuclear Pincer-Type Ru(II) Complex Catalysts and Their Catalytic Applications. Inorg. Chim. Acta 2023, 551, 121458. [Google Scholar] [CrossRef]
- Zeng, F.; Yu, Z. Exceptionally Efficient Unsymmetrical Ruthenium(II) NNN Complex Catalysts Bearing a Pyridyl-Based Pyrazolyl-Imidazolyl Ligand for Transfer Hydrogenation of Ketones. Organometallics 2008, 27, 2898–2901. [Google Scholar] [CrossRef]
- Zeng, F.; Yu, Z. Construction of Highly Active Ruthenium(II) NNN Complex Catalysts Bearing a Pyridyl-Supported Pyrazolyl-Imidazolyl Ligand for Transfer Hydrogenation of Ketones. Organometallics 2009, 28, 1855–1862. [Google Scholar] [CrossRef]
- Ye, W.; Zhao, M.; Yu, Z. Ruthenium(II) Pyrazolyl–Pyridyl–Oxazolinyl Complex Catalysts for the Asymmetric Transfer Hydrogenation of Ketones. Chem. Eur. J. 2012, 18, 10843–10846. [Google Scholar] [CrossRef]
- Roberts, T.D.; Halcrow, M.A. Supramolecular Assembly and Transfer Hydrogenation Catalysis with Ruthenium(II) Complexes of 2,6-Di(1H-pyrazol-3-yl)pyridine Derivatives. Polyhedron 2016, 103, 79–86. [Google Scholar] [CrossRef]
- Kashiwame, Y.; Kuwata, S.; Ikariya, T. Metal–Pyrazole Bifunction in Half-Sandwich C–N Chelate Iridium Complexes: Pyrazole–Pyrazolato Interconversion and Application to Catalytic Intramolecular Hydroamination of Aminoalkene. Chem. Eur. J. 2010, 16, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Kashiwame, Y.; Ikariya, T.; Kuwata, S. Synthesis, Structures, and Reactivities of Six-Membered C–N Chelate Protic Pyrazole Complexes of Iridium. Polyhedron 2021, 197, 115036. [Google Scholar] [CrossRef]
- Ghoochany, L.T.; Farsadpour, S.; Sun, Y.; Thiel, W.R. New N,N,N-Donors Resulting in Highly Active Ruthenium Catalysts for Transfer Hydrogenation at Room Temperature. Eur. J. Inorg. Chem. 2011, 2011, 3431–3437. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, J.; Fu, H.; Yuan, M.; Zheng, X.; Chen, H.; Li, R. Construction of Pincer-Type Symmetrical Ruthenium(II) Complexes Bearing Pyridyl-2,6-Pyrazolyl Arms: Catalytic Behavior in Transfer Hydrogenation of Ketones. RSC Adv. 2014, 4, 52734–52739. [Google Scholar] [CrossRef]
- Dayan, O.; Dayan, S.; Kani, İ.; Cetinkaya, B. Ruthenium(II) Complexes Bearing Pyridine-Based Tridentate and Bidentate Ligands: Catalytic Activity for Transfer Hydrogenation of Aryl Ketones. Adv. Organomet. Chem. 2012, 26, 663–670. [Google Scholar] [CrossRef]
- Gunnaz, S.; Ozdemir, N.; Dayan, S.; Dayan, O.; Cetinkaya, B. Synthesis of Ruthenium(II) Complexes Containing Tridentate Triamine (‘NNN’) and Bidentate Diamine Ligands (NN′): As Catalysts for Transfer Hydrogenation of Ketones. Organometallics 2011, 30, 4165–4173. [Google Scholar] [CrossRef]
- Alshakova, I.D.; Korobkov, I.; Kuzmina, L.G.; Nikonov, G.I. Ruthenium Complexes with a Pyrazole-Phosphine Ligand. J. Organomet. Chem. 2017, 853, 68–73. [Google Scholar] [CrossRef]
- Alshakova, I.D.; Gabidullin, B.; Nikonov, G.I. Ru-Catalyzed Transfer Hydrogenation of Nitriles, Aromatics, Olefins, Alkynes and Esters. ChemCatChem 2018, 10, 4860–4869. [Google Scholar] [CrossRef]
- Dubey, A.; Khaskin, E. Catalytic Ester Metathesis Reaction and Its Application to Transfer Hydrogenation of Esters. ACS Catal. 2016, 6, 3998–4002. [Google Scholar] [CrossRef]
- Tian, C.; Gong, L.; Meggers, E. Chiral-at-Metal Iridium Complex for Efficient Enantioselective Transfer Hydrogenation of Ketones. Chem. Commun. 2016, 52, 4207–4210. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, J.; Huang, X.; Meggers, E. Sequential Asymmetric Hydrogenation and Photoredox Chemistry with a Single Catalyst. Org. Chem. Front. 2018, 5, 166–170. [Google Scholar] [CrossRef]
- Muller, K.; Sun, Y.; Heimermann, A.; Menges, F.; Niedner-Schatteburg, G.; van Wüllen, C.; Thiel, W.R. Structure–Reactivity Relationships in the Hydrogenation of Carbon Dioxide with Ruthenium Complexes Bearing Pyridinylazolato Ligands. Chem. Eur. J. 2013, 19, 7825–7834. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, B.G.; Young, K.J.H.; Yousufuddin, M.; Goddard, W.A.; Periana, R.A. Acceleration of Nucleophilic CH Activation by Strongly Basic Solvents. J. Am. Chem. Soc. 2010, 132, 12542–12545. [Google Scholar] [CrossRef] [PubMed]
- Suna, Y.; Himeda, Y.; Fujita, E.; Muckerman, J.T.; Ertem, M.Z. Iridium Complexes with Proton-Responsive Azole-Type Ligands as Effective Catalysts for CO2 Hydrogenation. ChemSusChem 2017, 10, 4535–4543. [Google Scholar] [CrossRef] [PubMed]
- Onishi, N.; Xu, S.; Manaka, Y.; Suna, Y.; Wang, W.-H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. CO2 Hydrogenation Catalyzed by Iridium Complexes with a Proton-Responsive Ligand. Inorg. Chem. 2015, 54, 5114–5123. [Google Scholar] [CrossRef]
- Manaka, Y.; Wang, W.-H.; Suna, Y.; Kambayashi, H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Efficient H2 Generation from Formic Acid Using Azole Complexes in Water. Catal. Sci. Technol. 2014, 4, 34–37. [Google Scholar] [CrossRef]
- Wang, W.-H.; Wang, H.; Yang, Y.; Lai, X.; Li, Y.; Wang, J.; Himeda, Y.; Bao, M. Synergistic Effect of Pendant N Moieties for Proton Shuttling in the Dehydrogenation of Formic Acid Catalyzed by Biomimetic IrIII Complexes. ChemSusChem 2020, 13, 5015–5022. [Google Scholar] [CrossRef]
- Nakahara, Y.; Toda, T.; Matsunami, A.; Kayaki, Y.; Kuwata, S. Protic NNN and NCN Pincer-Type Ruthenium Complexes Featuring (Trifluoromethyl)pyrazole Arms: Synthesis and Application to Catalytic Hydrogen Evolution from Formic Acid. Chem. Asian J. 2018, 13, 73–80. [Google Scholar] [CrossRef]
- Pal, S.; Iwasaki, T.; Nozaki, K. Metal–Ligand Cooperative κ1-N-Pyrazolate Cp*RhIII-Catalysts for Dehydrogenation of Dimethylamine-Borane at Room Temperature. Dalton Trans. 2021, 50, 7938–7943. [Google Scholar] [CrossRef]
- Reed-Berendt, B.G.; Latham, D.E.; Dambatta, M.B.; Morrill, L.C. Borrowing Hydrogen for Organic Synthesis. ACS Cent. Sci. 2021, 7, 570–585. [Google Scholar] [CrossRef]
- Kuwahara, T.; Fukuyama, T.; Ryu, I. Ruthenium Hydride/Nitrogen Tridentate Ligand-Catalyzed α-Alkylation of Acetamides with Primary Alcohols. RSC Adv. 2013, 3, 13702–13704. [Google Scholar] [CrossRef]
- Panda, S.; Saha, R.; Sethi, S.; Ghosh, R.; Bagh, B. Efficient α-Alkylation of Arylacetonitriles with Secondary Alcohols Catalyzed by a Phosphine-Free Air-Stable Iridium(III) Complex. J. Org. Chem. 2020, 85, 15610–15621. [Google Scholar] [CrossRef] [PubMed]
- Dutta, I.; Yadav, S.; Sarbajna, A.; De, S.; Hölscher, M.; Leitner, W.; Bera, J.K. Double Dehydrogenation of Primary Amines to Nitriles by a Ruthenium Complex Featuring Pyrazole Functionality. J. Am. Chem. Soc. 2018, 140, 8662–8666. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Yu, K.; Liu, B.; Tan, W.; Zhang, G. A Highly Selective Manganese-Catalyzed Synthesis of Imines under Phosphine-Free Conditions. Organometallics 2020, 39, 217–226. [Google Scholar] [CrossRef]
- Satake, A.; Nakata, T. Novel η3-Allylpalladium–Pyridinylpyrazole Complex: Synthesis, Reactivity, and Catalytic Activity for Cyclopropanation of Ketene Silyl Acetal with Allylic Acetates. J. Am. Chem. Soc. 1998, 120, 10391–10396. [Google Scholar] [CrossRef]
- Satake, A.; Koshino, H.; Nakata, T. Cyclopropanation of Ketene Silyl Acetals with Allylic Acetates Using η3-Allylpalladium-Pyridinylimidazole Catalysts. Chem. Lett. 1999, 28, 49–50. [Google Scholar] [CrossRef]
- Aranyos, A.; Szabó, K.J.; Castaño, A.M.; Bäckvall, J.-E. Central versus Terminal Attack in Nucleophilic Addition to (π-Allyl)palladium Complexes. Ligand Effects and Mechanism. Organometallics 1997, 16, 1058–1064. [Google Scholar] [CrossRef]
- Satake, A.; Kadohama, H.; Koshino, H.; Nakata, T. Asymmetric Cyclopropanation of Ketene Silyl Acetal with Allylic Acetate Catalyzed by a Palladium Complex. Tetrahedron Lett. 1999, 40, 3597–3600. [Google Scholar] [CrossRef]
- Diez, J.; Gimeno, J.; Lledós, A.; Suárez, F.J.; Vicent, C. Imidazole Based Ruthenium(IV) Complexes as Highly Efficient Bifunctional Catalysts for the Redox Isomerization of Allylic Alcohols in Aqueous Medium: Water as Cooperating Ligand. ACS Catal. 2012, 2, 2087–2099. [Google Scholar] [CrossRef]
- Bellarosa, L.; Diez, J.; Gimeno, J.; Lledós, A.; Suárez, F.J.; Ujaque, G.; Vicent, C. Highly Efficient Redox Isomerisation of Allylic Alcohols Catalysed by Pyrazole-Based Ruthenium(IV) Complexes in Water: Mechanisms of Bifunctional Catalysis in Water. Chem. Eur. J. 2012, 18, 7749–7765. [Google Scholar] [CrossRef]
- Tashima, N.; Ohta, S.; Kuwata, S. Metal–Ligand Cooperative C–O Bond Cleavage of Propargylic Alcohol with Protic Pyrazole Complexes of Ruthenium. Faraday Discuss. 2019, 220, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Justaud, F.; Hachem, A.; Grée, R. Recent Developments in the Meyer-Schuster Rearrangement. Eur. J. Org. Chem. 2021, 2021, 514–542. [Google Scholar] [CrossRef]
- Cadierno, V.; Crochet, P.; García-Garrido, S.E.; Gimeno, J. Metal-Catalyzed Transformations of Propargylic Alcohols into α,β-Unsaturated Carbonyl Compounds: From the Meyer–Schuster and Rupe Rearrangements to Redox Isomerizations. Dalton Trans. 2010, 39, 4015–4031. [Google Scholar] [CrossRef] [PubMed]
- Wojcicki, A. Allenyls and Propargyls: Versatile Ligands in Transition-Metal Chemistry. Inorg. Chem. Commun. 2002, 5, 82–97. [Google Scholar] [CrossRef]
- Ferrer, Í.; Rich, J.; Fontrodona, X.; Rodríguez, M.; Romero, I. Ru(II) Complexes Containing DMSO and Pyrazolyl Ligands as Catalysts for Nitrile Hydration in Environmentally Friendly Media. Dalton Trans. 2013, 42, 13461–13469. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, Í.; Fontrodona, X.; Rodríguez, M.; Romero, I. Ru(II)-DMSO Complexes Containing Azole-Based Ligands: Synthesis, Linkage Isomerism and Catalytic Behaviour. Dalton Trans. 2016, 45, 3163–3174. [Google Scholar] [CrossRef]
- Zhang, L.; Meggers, E. Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res. 2017, 50, 320–330. [Google Scholar] [CrossRef]
- Zhang, L.; Meggers, E. Stereogenic-Only-at-Metal Asymmetric Catalysts. Chem. Asian J. 2017, 12, 2335–2342. [Google Scholar] [CrossRef]
- Chen, L.-A.; Tang, X.; Xi, J.; Xu, W.; Gong, L.; Meggers, E. Chiral-at-Metal Octahedral Iridium Catalyst for the Asymmetric Construction of an All-Carbon Quaternary Stereocenter. Angew. Chem. Int. Ed. 2013, 52, 14021–14025. [Google Scholar] [CrossRef]
- Vera, S.; García-Urricelqui, A.; Mielgo, A.; Oiarbide, M. Progress in (Thio)urea- and Squaramide-Based Brønsted Base Catalysts with Multiple H-Bond Donors. Eur. J. Org. Chem. 2023, 26, e202201254. [Google Scholar] [CrossRef]
- Agarwal, P.; Thirupathi, N.; Nethaji, M. Syntheses, Structural Aspects, Solution Behavior, and Catalytic Utility of Cyclopalladated N,N′,N″-Triarylguanidines [κ2(C,N)Pd(Pyrazole)2X] (X = Br, OC(O)CF3, and PF6) in Suzuki–Miyaura Coupling Reactions of Aryl Bromides. Organometallics 2016, 35, 3112–3123. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, L.; Lei, L.; Zheng, X.-L.; Fu, H.; Yuan, M.; Chen, H.; Li, R.-X. PdCl2-2,6-Bis(1,5-diphenyl-1H-pyrazol-3-yl)pyridine Catalyzed Suzuki–Miyaura Cross-Coupling. Catal. Commun. 2012, 29, 194–197. [Google Scholar] [CrossRef]
- Nacianceno, V.S.; Azpeitia, S.; Ibarlucea, L.; Mendicute-Fierro, C.; Rodriguez-Dieguez, A.; Seco, J.M.; Sebastian, E.S.; Garralda, M.A. Stereoselective Formation and Catalytic Activity of Hydrido(acylphosphane)(chlorido)(pyrazole)rhodium(III) Complexes. Experimental and DFT Studies. Dalton Trans. 2015, 44, 13141–13155. [Google Scholar] [CrossRef] [PubMed]
- Manrique, E.; Fontrodona, X.; Rodríguez, M.; Romero, I. A Ruthenium(II) Aqua Complex as Efficient Chemical and Photochemical Catalyst for Alkene and Alcohol Oxidation. Eur. J. Inorg. Chem. 2019, 2019, 2124–2133. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).