
Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
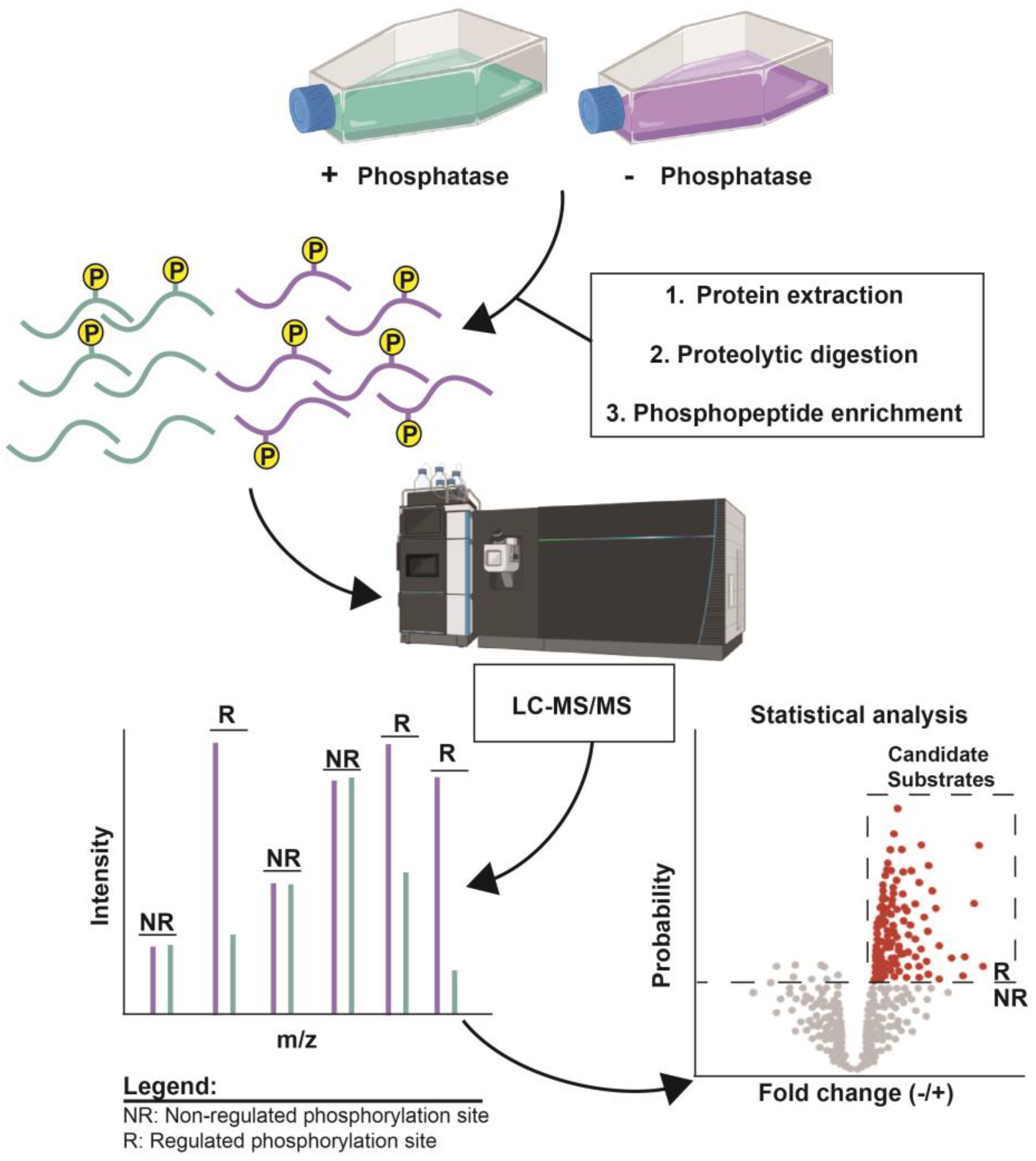
2. Methods Involving In Vivo Enzyme Perturbation
- Genetic disruption of target expression.
- Chemical inhibition of target enzyme activity.
- Advantages and disadvantages of target perturbation methods.
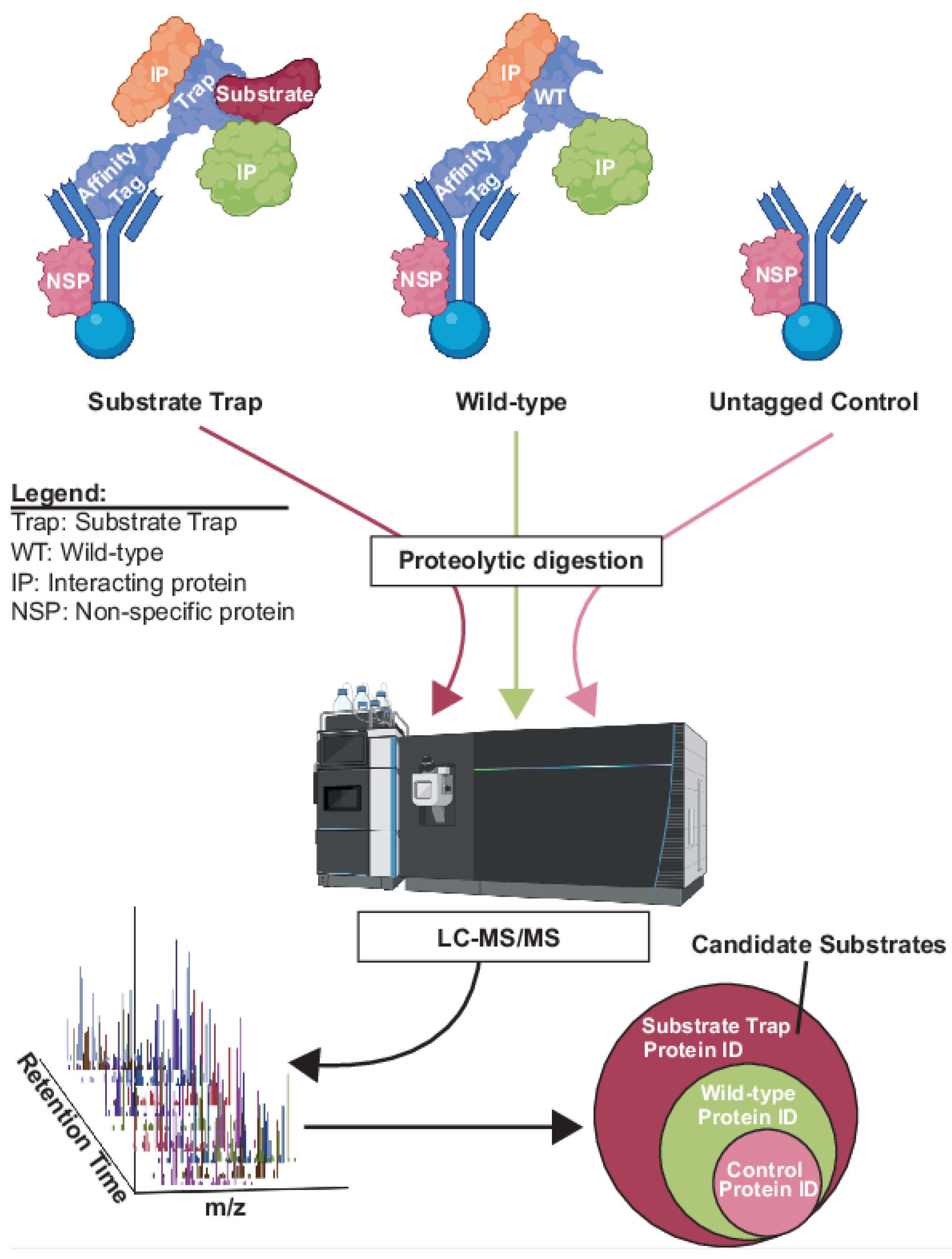
3. Methods Involving Affinity Capture of Candidate Substrates
- Co-purification of enzyme-substrate complexes.
- Proximity labeling.
- Advantages and disadvantages of affinity capture methods.
4. Methods Involving In Vitro Enzyme Reactions
- In vitro experiments using cell extracts.
- In vitro experiments using peptide substrate pools.
- Advantages and disadvantages of in vitro phosphoproteomic approaches.
5. Combining Multiple Methods Can Improve Substrate Identification Confidence
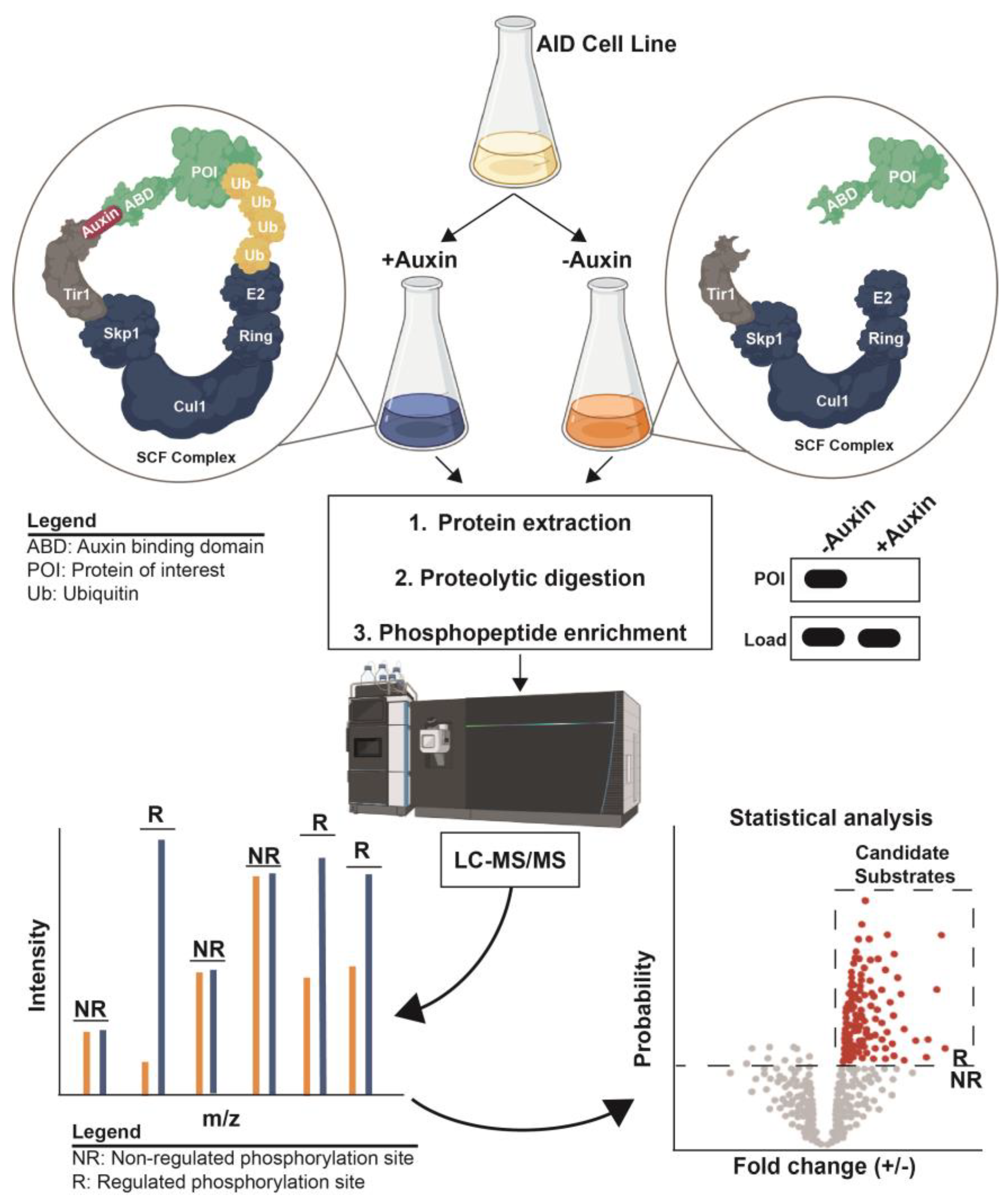
6. Coupling Inducible Degradation with Phosphoproteomics to Define Kinase and Phosphatase Substrates
7. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- Palomba, A.; Abbondio, M.; Fiorito, G.; Uzzau, S.; Pagnozzi, D.; Tanca, A. Comparative Evaluation of MaxQuant and Proteome Discoverer MS1-Based Protein Quantification Tools. J. Proteome Res. 2021, 20, 3497–3507. [Google Scholar] [CrossRef]
- Zhao, L.; Cong, X.; Zhai, L.; Hu, H.; Xu, J.-Y.; Zhao, W.; Zhu, M.; Tan, M.; Ye, B.-C. Comparative evaluation of label-free quantification strategies. J. Proteom. 2020, 215, 103669. [Google Scholar] [CrossRef]
- Brandi, J.; Noberini, R.; Bonaldi, T.; Cecconi, D. Advances in enrichment methods for mass spectrometry-based proteomics analysis of post-translational modifications. J. Chromatogr. A 2022, 1678, 463352. [Google Scholar] [CrossRef]
- Li, J.; Zhan, X. Mass spectrometry analysis of phosphotyrosine-containing proteins. Mass Spectrom. Rev. 2023. [Google Scholar] [CrossRef]
- Winter, D.L.; Wilkins, M.R.; Donald, W.A. Differential Ion Mobility–Mass Spectrometry for Detailed Analysis of the Proteome. Trends Biotechnol. 2019, 37, 198–213. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics 2020, 20, 1900276. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd. Recent technical advances in proteomics. F1000Res 2019, 8, F1000 Faculty Rev-351. [Google Scholar] [CrossRef]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Sing, J.C.; Gingras, A.-C.; Röst, H.L. Improving Phosphoproteomics Profiling Using Data-Independent Mass Spectrometry. J. Proteome Res. 2022, 21, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Low, T.Y.; Mohtar, M.A.; Lee, P.Y.; Omar, N.; Zhou, H.; Ye, M. Widening the Bottleneck of Phosphoproteomics: Evolving Strategies for Phosphopeptide Enrichment. Mass Spectrom. Rev. 2021, 40, 309–333. [Google Scholar] [CrossRef]
- Potel, C.M.; Lemeer, S.; Heck, A.J.R. Phosphopeptide Fragmentation and Site Localization by Mass Spectrometry: An Update. Anal. Chem. 2019, 91, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Ulintz, P.J.; Yocum, A.K.; Bodenmiller, B.; Aebersold, R.; Andrews, P.C.; Nesvizhskii, A.I. Comparison of MS2-Only, MSA, and MS2/MS3 Methodologies for Phosphopeptide Identification. J. Proteome Res. 2009, 8, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Refsgaard, J.C.; Munk, S.; Jensen, L.J. Search Databases and Statistics: Pitfalls and Best Practices in Phosphoproteomics. Methods Mol. Biol. 2016, 1355, 323–339. [Google Scholar] [CrossRef]
- Lee, D.C.; Jones, A.R.; Hubbard, S.J. Computational phosphoproteomics: From identification to localization. Proteomics 2015, 15, 950–963. [Google Scholar] [CrossRef]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef]
- Macek, B.; Mann, M.; Olsen, J.V. Global and site-specific quantitative phosphoproteomics: Principles and applications. Annu. Rev. Pharm. Toxicol. 2009, 49, 199–221. [Google Scholar] [CrossRef]
- Paulo, J.A.; Schweppe, D.K. Advances in quantitative high-throughput phosphoproteomics with sample multiplexing. Proteomics 2021, 21, 2000140. [Google Scholar] [CrossRef]
- Engholm-Keller, K.; Larsen, M.R. Technologies and challenges in large-scale phosphoproteomics. Proteomics 2013, 13, 910–931. [Google Scholar] [CrossRef]
- Kanshin, E.; Michnick, S.; Thibault, P. Sample preparation and analytical strategies for large-scale phosphoproteomics experiments. Semin. Cell Dev. Biol. 2012, 23, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Jünger, M.A.; Aebersold, R. Mass spectrometry-driven phosphoproteomics: Patterning the systems biology mosaic. WIREs Dev. Biol. 2014, 3, 83–112. [Google Scholar] [CrossRef]
- Bodenmiller, B.; Wanka, S.; Kraft, C.; Urban, J.; Campbell, D.; Pedrioli, P.G.; Gerrits, B.; Picotti, P.; Lam, H.; Vitek, O.; et al. Phosphoproteomic Analysis Reveals Interconnected System-Wide Responses to Perturbations of Kinases and Phosphatases in Yeast. Sci. Signal. 2010, 3, rs4. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Paulo, J.A.; Nusinow, D.P.; Huttlin, E.L.; Gygi, S.P. Investigation of Proteomic and Phosphoproteomic Responses to Signaling Network Perturbations Reveals Functional Pathway Organizations in Yeast. Cell Rep. 2019, 29, 2092–2104.e4. [Google Scholar] [CrossRef] [PubMed]
- Owusu, M.; Bannauer, P.; Ferreira da Silva, J.; Mourikis, T.P.; Jones, A.; Májek, P.; Caldera, M.; Wiedner, M.; Lardeau, C.-H.; Mueller, A.C.; et al. Mapping the Human Kinome in Response to DNA Damage. Cell Rep. 2019, 26, 555–563.e6. [Google Scholar] [CrossRef]
- Rusin, S.F.; Schlosser, K.A.; Adamo, M.E.; Kettenbach, A.N. Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Sci. Signal. 2015, 8, rs12. [Google Scholar] [CrossRef]
- Kauko, O.; Imanishi, S.Y.; Kulesskiy, E.; Yetukuri, L.; Laajala, T.D.; Sharma, M.; Pavic, K.; Aakula, A.; Rupp, C.; Jumppanen, M.; et al. Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J. Biol. Chem. 2020, 295, 4194–4211. [Google Scholar] [CrossRef]
- Iswahyudi; Mukamolova, G.V.; Straatman-Iwanowska, A.A.; Allcock, N.; Ajuh, P.; Turapov, O.; O’Hare, H.M. Mycobacterial phosphatase PstP regulates global serine threonine phosphorylation and cell division. Sci. Rep. 2019, 9, 8337. [Google Scholar] [CrossRef]
- Velázquez, D.; Albacar, M.; Zhang, C.; Calafí, C.; López-Malo, M.; Torres-Torronteras, J.; Martí, R.; Kovalchuk, S.I.; Pinson, B.; Jensen, O.N.; et al. Yeast Ppz1 protein phosphatase toxicity involves the alteration of multiple cellular targets. Sci. Rep. 2020, 10, 15613. [Google Scholar] [CrossRef] [PubMed]
- Bansal, P.; Antil, N.; Kumar, M.; Yamaryo-Botté, Y.; Rawat, R.S.; Pinto, S.; Datta, K.K.; Katris, N.J.; Botté, C.Y.; Prasad, T.S.K.; et al. Protein kinase TgCDPK7 regulates vesicular trafficking and phospholipid synthesis in Toxoplasma gondii. PLoS Pathog. 2021, 17, e1009325. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. Analysing kinase inhibitor selectivity. Nat. Rev. Drug Discov. 2012, 11, 21. [Google Scholar] [CrossRef]
- Wang, B.; Wu, H.; Hu, C.; Wang, H.; Liu, J.; Wang, W.; Liu, Q. An overview of kinase downregulators and recent advances in discovery approaches. Signal Transduct. Target. Ther. 2021, 6, 423. [Google Scholar] [CrossRef]
- Müller, S.; Chaikuad, A.; Gray, N.S.; Knapp, S. The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol. 2015, 11, 818–821. [Google Scholar] [CrossRef]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef]
- Rimel, J.K.; Poss, Z.C.; Erickson, B.; Maas, Z.L.; Ebmeier, C.C.; Johnson, J.L.; Decker, T.-M.; Yaron, T.M.; Bradley, M.J.; Hamman, K.B.; et al. Selective inhibition of CDK7 reveals high-confidence targets and new models for TFIIH function in transcription. Genes Dev. 2020, 34, 1452–1473. [Google Scholar] [CrossRef]
- Byrne, D.P.; Clarke, C.J.; Brownridge, P.J.; Kalyuzhnyy, A.; Perkins, S.; Campbell, A.; Mason, D.; Jones, A.R.; Eyers, P.A.; Eyers, C.E. Use of the Polo-like kinase 4 (PLK4) inhibitor centrinone to investigate intracellular signalling networks using SILAC-based phosphoproteomics. Biochem. J. 2020, 477, 2451–2475. [Google Scholar] [CrossRef]
- Waldrip, Z.J.; Burdine, L.; Harrison, D.K.; Azevedo-Pouly, A.C.; Storey, A.J.; Moffett, O.G.; Mackintosh, S.G.; Burdine, M.S. DNA-PKcs kinase activity stabilizes the transcription factor Egr1 in activated immune cells. J. Biol. Chem. 2021, 297, 101209. [Google Scholar] [CrossRef]
- Pan, C.; Olsen, J.V.; Daub, H.; Mann, M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell Proteom. 2009, 8, 2796–2808. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Nakayama, M.; Amano, M.; Kaibuchi, K. Proteomic Screening for Rho-kinase Substrates by Combining Kinase and Phosphatase Inhibitors with 14-3-3ζ; Affinity Chromatography. Cell Struct. Funct. 2012, 37, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Grosstessner-Hain, K.; Hegemann, B.; Novatchkova, M.; Rameseder, J.; Joughin, B.A.; Hudecz, O.; Roitinger, E.; Pichler, P.; Kraut, N.; Yaffe, M.B.; et al. Quantitative Phospho-proteomics to Investigate the Polo-like Kinase 1-Dependent Phospho-proteome. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.C.; Shah, K.; Liu, Y.; Witucki, L.; Kung, C.; Shokat, K.M. Design of allele-specific inhibitors to probe protein kinase signaling. Curr. Biol. 1998, 8, 257–266. [Google Scholar] [CrossRef]
- Bishop, A.C.; Ubersax, J.A.; Petsch, D.T.; Matheos, D.P.; Gray, N.S.; Blethrow, J.; Shimizu, E.; Tsien, J.Z.; Schultz, P.G.; Rose, M.D.; et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Liu, Y.; Deirmengian, C.; Shokat, K.M. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc. Natl. Acad. Sci. USA 1997, 94, 3565–3570. [Google Scholar] [CrossRef]
- Bishop, A.C.; Kung, C.-y.; Shah, K.; Witucki, L.; Shokat, K.M.; Liu, Y. Generation of Monospecific Nanomolar Tyrosine Kinase Inhibitors via a Chemical Genetic Approach. J. Am. Chem. Soc. 1999, 121, 627–631. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villén, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 2016, 167, 1750–1761.e16. [Google Scholar] [CrossRef]
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008, 9, R149. [Google Scholar] [CrossRef]
- Decker, T.-M.; Forné, I.; Straub, T.; Elsaman, H.; Ma, G.; Shah, N.; Imhof, A.; Eick, D. Analog-sensitive cell line identifies cellular substrates of CDK9. Oncotarget 2019, 10, 6934–6943. [Google Scholar] [CrossRef] [PubMed]
- Umaña, A.C.; Iwahori, S.; Kalejta, R.F. Direct Substrate Identification with an Analog Sensitive (AS) Viral Cyclin-Dependent Kinase (v-Cdk). ACS Chem. Biol. 2018, 13, 189–199. [Google Scholar] [CrossRef]
- Plank, M.; Perepelkina, M.; Müller, M.; Vaga, S.; Zou, X.; Bourgoint, C.; Berti, M.; Saarbach, J.; Haesendonckx, S.; Winssinger, N.; et al. Chemical Genetics of AGC-kinases Reveals Shared Targets of Ypk1, Protein Kinase A and Sch9. Mol. Cell. Proteom. 2020, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Kubiniok, P.; Finicle, B.T.; Piffaretti, F.; McCracken, A.N.; Perryman, M.; Hanessian, S.; Edinger, A.L.; Thibault, P. Dynamic Phosphoproteomics Uncovers Signaling Pathways Modulated by Anti-oncogenic Sphingolipid Analogs. Mol. Cell Proteom. 2019, 18, 408–422. [Google Scholar] [CrossRef]
- He, R.; Zeng, L.-F.; He, Y.; Zhang, S.; Zhang, Z.-Y. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013, 280, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Vemulapalli, V.; Chylek, L.A.; Erickson, A.; Pfeiffer, A.; Gabriel, K.-H.; LaRochelle, J.; Subramanian, K.; Cao, R.; Stegmaier, K.; Mohseni, M.; et al. Time-resolved phosphoproteomics reveals scaffolding and catalysis-responsive patterns of SHP2-dependent signaling. eLife 2021, 10, e64251. [Google Scholar] [CrossRef] [PubMed]
- Batth, T.S.; Papetti, M.; Pfeiffer, A.; Tollenaere, M.A.X.; Francavilla, C.; Olsen, J.V. Large-Scale Phosphoproteomics Reveals Shp-2 Phosphatase-Dependent Regulators of Pdgf Receptor Signaling. Cell Rep. 2018, 22, 2784–2796. [Google Scholar] [CrossRef]
- Shah, K.; Kim, H. The significant others: Global search for direct kinase substrates using chemical approaches. IUBMB Life 2019, 71, 721–737. [Google Scholar] [CrossRef]
- Lowery, D.M.; Clauser, K.R.; Hjerrild, M.; Lim, D.; Alexander, J.; Kishi, K.; Ong, S.E.; Gammeltoft, S.; Carr, S.A.; Yaffe, M.B. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. 2007, 26, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Schlosser, K.A.; Lyons, S.P.; Nasa, I.; Gui, J.; Adamo, M.E.; Gerber, S.A. Global assessment of its network dynamics reveals that the kinase Plk1 inhibits the phosphatase PP6 to promote Aurora A activity. Sci. Signal 2018, 11, eaaq1441. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.-F. Mass spectrometry-based proteomic analysis of the epitope-tag affinity purified protein complexes in eukaryotes. Proteomics 2006, 6, 6158–6166. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. SAINT: Probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.J.; Tiganis, T.; Barford, D.; Tonks, N.K. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 1997, 94, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, Y.-L.; Zhang, Z.-Y. Design and Characterization of an Improved Protein Tyrosine Phosphatase Substrate-Trapping Mutant. Biochemistry 2002, 41, 4032–4039. [Google Scholar] [CrossRef]
- Kaneva, I.N.; Sudbery, I.M.; Dickman, M.J.; Sudbery, P.E. Proteins that physically interact with the phosphatase Cdc14 in Candida albicans have diverse roles in the cell cycle. Sci. Rep. 2019, 9, 6258. [Google Scholar] [CrossRef]
- Bloom, J.; Cristea, I.M.; Procko, A.L.; Lubkov, V.; Chait, B.T.; Snyder, M.; Cross, F.R. Global Analysis of Cdc14 Phosphatase Reveals Diverse Roles in Mitotic Processes. J. Biol. Chem. 2011, 286, 5434–5445. [Google Scholar] [CrossRef]
- Fearnley, G.W.; Young, K.A.; Edgar, J.R.; Antrobus, R.; Hay, I.M.; Liang, W.-C.; Martinez-Martin, N.; Lin, W.; Deane, J.E.; Sharpe, H.J. The homophilic receptor PTPRK selectively dephosphorylates multiple junctional regulators to promote cell–cell adhesion. eLife 2019, 8, e44597. [Google Scholar] [CrossRef]
- Wu, J.; Katrekar, A.; Honigberg, L.A.; Smith, A.M.; Conn, M.T.; Tang, J.; Jeffery, D.; Mortara, K.; Sampang, J.; Williams, S.R.; et al. Identification of Substrates of Human Protein-tyrosine Phosphatase PTPN22. J. Biol. Chem. 2006, 281, 11002–11010. [Google Scholar] [CrossRef]
- Bonham, C.; Mandati, V.; Singh, R.; Pappin, D.; Tonks, N. Coupling substrate-trapping with proximity-labeling to identify protein tyrosine phosphatase PTP1B signaling networks. J. Biol. Chem. 2023, 299, 104582. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-S.; Broadus, M.R.; McLean, J.R.; Feoktistova, A.; Ren, L.; Gould, K.L. Comprehensive Proteomics Analysis Reveals New Substrates and Regulators of the Fission Yeast Clp1/Cdc14 Phosphatase. Mol. Cell. Proteom. 2013, 12, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Guo, A.; Possemato, A.; Wang, C.; Beard, L.; Carlin, C.; Markowitz, S.D.; Polakiewicz, R.D.; Wang, Z. Identification and functional characterization of p130Cas as a substrate of protein tyrosine phosphatase nonreceptor 14. Oncogene 2013, 32, 2087–2095. [Google Scholar] [CrossRef]
- Wu, D.; De Wever, V.; Derua, R.; Winkler, C.; Beullens, M.; Van Eynde, A.; Bollen, M. A substrate-trapping strategy for protein phosphatase PP1 holoenzymes using hypoactive subunit fusions. J. Biol. Chem. 2018, 293, 15152–15162. [Google Scholar] [CrossRef]
- Garre, S.; Gamage, A.K.; Faner, T.R.; Dedigama-Arachchige, P.; Pflum, M.K.H. Identification of Kinases and Interactors of p53 Using Kinase-Catalyzed Cross-Linking and Immunoprecipitation. J. Am. Chem. Soc. 2018, 140, 16299–16310. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Maly, D.J.; Seeliger, M.A.; Fabian, M.A.; Biggs, W.H.; Lockhart, D.J.; Zarrinkar, P.P.; Kuriyan, J.; Shokat, K.M. Tuning a Three-Component Reaction For Trapping Kinase Substrate Complexes. J. Am. Chem. Soc. 2008, 130, 17568–17574. [Google Scholar] [CrossRef] [PubMed]
- Beltman, R.J.; Pflum, M.K.H. Kinase-Catalyzed Crosslinking and Immunoprecipitation (K-CLIP) to Explore Kinase-Substrate Pairs. Curr. Protoc. 2022, 2, e539. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 2021, 18, 133–143. [Google Scholar] [CrossRef]
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined proximity labeling and affinity purification−mass spectrometry workflow for mapping and visualizing protein interaction networks. Nat. Protoc. 2020, 15, 3182–3211. [Google Scholar] [CrossRef]
- Nguyen, T.M.T.; Kim, J.; Doan, T.T.; Lee, M.-W.; Lee, M. APEX Proximity Labeling as a Versatile Tool for Biological Research. Biochemistry 2020, 59, 260–269. [Google Scholar] [CrossRef]
- Cho, K.F.; Branon, T.C.; Udeshi, N.D.; Myers, S.A.; Carr, S.A.; Ting, A.Y. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 2020, 15, 3971–3999. [Google Scholar] [CrossRef]
- Sears, R.M.; May, D.G.; Roux, K.J. BioID as a Tool for Protein-Proximity Labeling in Living Cells. Methods Mol. Biol. 2019, 2012, 299–313. [Google Scholar] [CrossRef]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Dumont, A.A.; Dumont, L.; Berthiaume, J.; Auger-Messier, M. p38α MAPK proximity assay reveals a regulatory mechanism of alternative splicing in cardiomyocytes. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Fassl, A.; Vaites, L.P.; Fu, S.; Sicinski, P.; Paulo, J.A.; Gygi, S.P. Interrogating Kinase–Substrate Relationships with Proximity Labeling and Phosphorylation Enrichment. J. Proteome Res. 2022, 21, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Uçkun, E.; Wolfstetter, G.; Anthonydhason, V.; Sukumar, S.K.; Umapathy, G.; Molander, L.; Fuchs, J.; Palmer, R.H. In vivo Profiling of the Alk Proximitome in the Developing Drosophila Brain. J. Mol. Biol. 2021, 433, 167282. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Park, C.H.; Hsu, C.C.; Kim, Y.W.; Ko, Y.W.; Zhang, Z.; Zhu, J.Y.; Hsiao, Y.C.; Branon, T.; Kaasik, K.; et al. Mapping the signaling network of BIN2 kinase using TurboID-mediated biotin labeling and phosphoproteomics. Plant Cell 2023, 35, 975–993. [Google Scholar] [CrossRef]
- Knight, J.D.; Tian, R.; Lee, R.E.; Wang, F.; Beauvais, A.; Zou, H.; Megeney, L.A.; Gingras, A.C.; Pawson, T.; Figeys, D.; et al. A novel whole-cell lysate kinase assay identifies substrates of the p38 MAPK in differentiating myoblasts. Skelet Muscle 2012, 2, 5. [Google Scholar] [CrossRef]
- Xue, L.; Geahlen, R.L.; Tao, W.A. Identification of direct tyrosine kinase substrates based on protein kinase assay-linked phosphoproteomics. Mol. Cell Proteom. 2013, 12, 2969–2980. [Google Scholar] [CrossRef]
- Müller, A.C.; Giambruno, R.; Weißer, J.; Májek, P.; Hofer, A.; Bigenzahn, J.W.; Superti-Furga, G.; Jessen, H.J.; Bennett, K.L. Identifying Kinase Substrates via a Heavy ATP Kinase Assay and Quantitative Mass Spectrometry. Sci. Rep. 2016, 6, 28107. [Google Scholar] [CrossRef]
- Embogama, D.M.; Pflum, M.K.H. K-BILDS: A Kinase Substrate Discovery Tool. Chembiochem 2017, 18, 136–141. [Google Scholar] [CrossRef]
- Li, Y.; Cross, F.R.; Chait, B.T. Method for identifying phosphorylated substrates of specific cyclin/cyclin-dependent kinase complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 11323. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Hamaguchi, T.; Shohag, M.H.; Kozawa, K.; Kato, K.; Zhang, X.; Yura, Y.; Matsuura, Y.; Kataoka, C.; Nishioka, T.; et al. Kinase-interacting substrate screening is a novel method to identify kinase substrates. J. Cell Biol. 2015, 209, 895–912. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Clurman, B.E. Mass Spectrometry-Based Identification of Protein Kinase Substrates Utilizing Engineered Kinases and Thiophosphate Labeling. Curr. Protoc. Chem. Biol. 2010, 2, 219–234. [Google Scholar] [CrossRef]
- Allen, J.J.; Li, M.; Brinkworth, C.S.; Paulson, J.L.; Wang, D.; Hübner, A.; Chou, W.-H.; Davis, R.J.; Burlingame, A.L.; Messing, R.O.; et al. A semisynthetic epitope for kinase substrates. Nat. Methods 2007, 4, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Blethrow, J.D.; Glavy, J.S.; Morgan, D.O.; Shokat, K.M. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. USA 2008, 105, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Banko, M.R.; Allen, J.J.; Schaffer, B.E.; Wilker, E.W.; Tsou, P.; White, J.L.; Villén, J.; Wang, B.; Kim, S.R.; Sakamoto, K.; et al. Chemical Genetic Screen for AMPKα2 Substrates Uncovers a Network of Proteins Involved in Mitosis. Mol. Cell 2011, 44, 878–892. [Google Scholar] [CrossRef]
- Schaffer, B.E.; Levin, R.S.; Hertz, N.T.; Maures, T.J.; Schoof, M.L.; Hollstein, P.E.; Benayoun, B.A.; Banko, M.R.; Shaw, R.J.; Shokat, K.M.; et al. Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab. 2015, 22, 907–921. [Google Scholar] [CrossRef]
- Kruse, T.; Gnosa, S.P.; Nasa, I.; Garvanska, D.H.; Hein, J.B.; Nguyen, H.; Samsøe-Petersen, J.; Lopez-Mendez, B.; Hertz, E.P.T.; Schwarz, J.; et al. Mechanisms of site-specific dephosphorylation and kinase opposition imposed by PP2A regulatory subunits. EMBO J. 2020, 39, e103695. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.B.; Garvanska, D.H.; Nasa, I.; Kettenbach, A.N.; Nilsson, J. Coupling of Cdc20 inhibition and activation by BubR1. J. Cell Biol. 2021, 220, e202012081. [Google Scholar] [CrossRef]
- Hoermann, B.; Kokot, T.; Helm, D.; Heinzlmeir, S.; Chojnacki, J.E.; Schubert, T.; Ludwig, C.; Berteotti, A.; Kurzawa, N.; Kuster, B.; et al. Dissecting the sequence determinants for dephosphorylation by the catalytic subunits of phosphatases PP1 and PP2A. Nat. Commun. 2020, 11, 3583. [Google Scholar] [CrossRef]
- Xue, L.; Wang, W.-H.; Iliuk, A.; Hu, L.; Galan, J.A.; Yu, S.; Hans, M.; Geahlen, R.L.; Tao, W.A. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc. Natl. Acad. Sci. USA 2012, 109, 5615. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Song, C.; Torres Robles, J.; Salichos, L.; Lou, H.J.; Lam, T.T.; Gerstein, M.; Turk, B.E. Proteome-wide screening for mitogen-activated protein kinase docking motifs and interactors. Sci. Signal. 2023, 16, eabm5518. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.-Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, L.C.; Kuo, H.Y.; Mrksich, M. Peptide Arrays: Development and Application. Anal. Chem. 2018, 90, 266–282. [Google Scholar] [CrossRef]
- DeMarco, A.G.; Pascuzzi, P.E.; Tao, W.A.; Hall, M.C. Phosphatase and Kinase Substrate SpecificitySubstrate specificities Profiling with Pooled Synthetic PeptidesSynthetic peptides and Mass SpectrometryMass Spectrometry. In Cell Cycle Oscillators: Methods and Protocols; Coutts, A.S., Weston, L., Eds.; Springer: New York, NY, USA, 2021; pp. 51–70. [Google Scholar]
- Meyer, N.O.; O’Donoghue, A.J.; Schulze-Gahmen, U.; Ravalin, M.; Moss, S.M.; Winter, M.B.; Knudsen, G.M.; Craik, C.S. Multiplex Substrate Profiling by Mass Spectrometry for Kinases as a Method for Revealing Quantitative Substrate Motifs. Anal. Chem. 2017, 89, 4550–4558. [Google Scholar] [CrossRef]
- Deng, Z.; Mao, J.; Wang, Y.; Zou, H.; Ye, M. Enzyme Kinetics for Complex System Enables Accurate Determination of Specificity Constants of Numerous Substrates in a Mixture by Proteomics Platform. Mol. Cell. Proteom. 2017, 16, 135. [Google Scholar] [CrossRef]
- Eissler, C.L.; Mazón, G.; Powers, B.L.; Savinov, S.N.; Symington, L.S.; Hall, M.C. The Cdk/cDc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol. Cell 2014, 54, 80–93. [Google Scholar] [CrossRef]
- Huang, Y.; Thelen, J.J. KiC Assay: A Quantitative Mass Spectrometry-Based Approach; Humana Press: Totowa, NJ, USA, 2012; pp. 359–370. [Google Scholar]
- Kokot, T.; Hoermann, B.; Helm, D.; Chojnacki, J.E.; Savitski, M.M.; Köhn, M. PLDMS: Phosphopeptide Library Dephosphorylation Followed by Mass Spectrometry Analysis to Determine the Specificity of Phosphatases for Dephosphorylation Site Sequences. In Computational Methods for Predicting Post-Translational Modification Sites; Kc, D.B., Ed.; Springer: New York, NY, USA, 2022; pp. 43–64. [Google Scholar]
- Wang, P.; Fu, H.; Snavley, D.F.; Freitas, M.A.; Pei, D. Screening Combinatorial Libraries by Mass Spectrometry. 2. Identification of Optimal Substrates of Protein Tyrosine Phosphatase SHP-1. Biochemistry 2002, 41, 6202–6210. [Google Scholar] [CrossRef]
- Ren, L.; Chen, X.; Luechapanichkul, R.; Selner, N.G.; Meyer, T.M.; Wavreille, A.-S.; Chan, R.; Iorio, C.; Zhou, X.; Neel, B.G.; et al. Substrate Specificity of Protein Tyrosine Phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry 2011, 50, 2339–2356. [Google Scholar] [CrossRef]
- Zhu, P.; Wu, X.; Zhang, R.-Y.; Hsu, C.-C.; Zhang, Z.-Y.; Tao, W.A. An Integrated Proteomic Strategy to Identify SHP2 Substrates. J. Proteome Res. 2022, 21, 2515–2525. [Google Scholar] [CrossRef]
- Malik, N.; Nirujogi, R.S.; Peltier, J.; Macartney, T.; Wightman, M.; Prescott, A.R.; Gourlay, R.; Trost, M.; Alessi, D.R.; Karapetsas, A. Phosphoproteomics reveals that the hVPS34 regulated SGK3 kinase specifically phosphorylates endosomal proteins including Syntaxin-7, Syntaxin-12, RFIP4 and WDR44. Biochem. J. 2019, 476, 3081–3107. [Google Scholar] [CrossRef] [PubMed]
- Prozzillo, Y.; Fattorini, G.; Santopietro, M.V.; Suglia, L.; Ruggiero, A.; Ferreri, D.; Messina, G. Targeted Protein Degradation Tools: Overview and Future Perspectives. Biology 2020, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Natsume, T.; Kanemaki, M.T. Conditional Degrons for Controlling Protein Expression at the Protein Level. Annu. Rev. Genet. 2017, 51, 83–102. [Google Scholar] [CrossRef]
- Kanemaki, M.T. Ligand-induced degrons for studying nuclear functions. Curr. Opin. Cell Biol. 2022, 74, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Yesbolatova, A.; Tominari, Y.; Kanemaki, M.T. Ligand-induced genetic degradation as a tool for target validation. Drug Discov. Today Technol. 2019, 31, 91–98. [Google Scholar] [CrossRef]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 2009, 6, 917–922. [Google Scholar] [CrossRef]
- Nicastro, R.; Raucci, S.; Michel, A.H.; Stumpe, M.; Garcia Osuna, G.M.; Jaquenoud, M.; Kornmann, B.; De Virgilio, C. Indole-3-acetic acid is a physiological inhibitor of TORC1 in yeast. PLoS Genet. 2021, 17, e1009414. [Google Scholar] [CrossRef]
- Snyder, N.A.; Kim, A.; Kester, L.; Gale, A.N.; Studer, C.; Hoepfner, D.; Roggo, S.; Helliwell, S.B.; Cunningham, K.W. Auxin-Inducible Depletion of the Essentialome Suggests Inhibition of TORC1 by Auxins and Inhibition of Vrg4 by SDZ 90-215, a Natural Antifungal Cyclopeptide. G3 Genes Genomes Genet. 2019, 9, 829–840. [Google Scholar] [CrossRef]
- Niwa, T.; Ise, M.; Miyazaki, T. Progression of glomerular sclerosis in experimental uremic rats by administration of indole, a precursor of indoxyl sulfate. Am. J. Nephrol. 1994, 14, 207–212. [Google Scholar] [CrossRef]
- Yesbolatova, A.; Saito, Y.; Kitamoto, N.; Makino-Itou, H.; Ajima, R.; Nakano, R.; Nakaoka, H.; Fukui, K.; Gamo, K.; Tominari, Y.; et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 2020, 11, 5701. [Google Scholar] [CrossRef]
- Nishimura, K.; Yamada, R.; Hagihara, S.; Iwasaki, R.; Uchida, N.; Kamura, T.; Takahashi, K.; Torii, K.U.; Fukagawa, T. A super-sensitive auxin-inducible degron system with an engineered auxin-TIR1 pair. Nucleic Acids Res. 2020, 48, e108. [Google Scholar] [CrossRef]
- Kreidenweiss, A.; Hopkins, A.V.; Mordmüller, B. 2A and the auxin-based degron system facilitate control of protein levels in Plasmodium falciparum. PLoS ONE 2013, 8, e78661. [Google Scholar] [CrossRef] [PubMed]
- Trost, M.; Blattner, A.C.; Lehner, C.F. Regulated protein depletion by the auxin-inducible degradation system in Drosophila melanogaster. Fly 2016, 10, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ward, J.D.; Cheng, Z.; Dernburg, A.F. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 2015, 142, 4374–4384. [Google Scholar] [CrossRef] [PubMed]
- Camlin, N.J.; Evans, J.P. Auxin-inducible protein degradation as a novel approach for protein depletion and reverse genetic discoveries in mammalian oocytes†. Biol. Reprod. 2019, 101, 704–718. [Google Scholar] [CrossRef]
- Zhang, X.-R.; Zhao, L.; Suo, F.; Gao, Y.; Wu, Q.; Qi, X.; Du, L.-L. An improved auxin-inducible degron system for fission yeast. G3 Genes|Genomes|Genet. 2022, 12, jkab393. [Google Scholar] [CrossRef]
- Hollenstein, D.M.; Gérecová, G.; Romanov, N.; Ferrari, J.; Veis, J.; Janschitz, M.; Beyer, R.; Schüller, C.; Ogris, E.; Hartl, M.; et al. A phosphatase-centric mechanism drives stress signaling response. EMBO Rep. 2021, 22, e52476. [Google Scholar] [CrossRef]
- Plank, M.; Berti, M.; Loewith, R. Phosphoproteomic Effects of Acute Depletion of PP2A Regulatory Subunit Cdc55. Proteomics 2021, 21, 2000166. [Google Scholar] [CrossRef]
- Hards, R.; Howarth, C.L.; Wiredu, K.; LaCroix, I.; Valle, J.M.d.; Adamo, M.; Kettenbach, A.N.; Holland, A.J.; Gerber, S.A. Development and validation of inducible protein degradation and quantitative phosphoproteomics to identify kinase-substrate relationships. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mariano, N.C.; Rusin, S.F.; Nasa, I.; Kettenbach, A.N. Inducible protein degradation as a strategy to identify Phosphoprotein Phosphatase 6 substrates in RAS-mutant colorectal cancer cells. bioRxiv 2023. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeMarco, A.G.; Hall, M.C. Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates. Molecules 2023, 28, 3675. https://doi.org/10.3390/molecules28093675
DeMarco AG, Hall MC. Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates. Molecules. 2023; 28(9):3675. https://doi.org/10.3390/molecules28093675
Chicago/Turabian StyleDeMarco, Andrew G., and Mark C. Hall. 2023. "Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates" Molecules 28, no. 9: 3675. https://doi.org/10.3390/molecules28093675
APA StyleDeMarco, A. G., & Hall, M. C. (2023). Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates. Molecules, 28(9), 3675. https://doi.org/10.3390/molecules28093675