
PROTACs in the Management of Prostate Cancer


Abstract
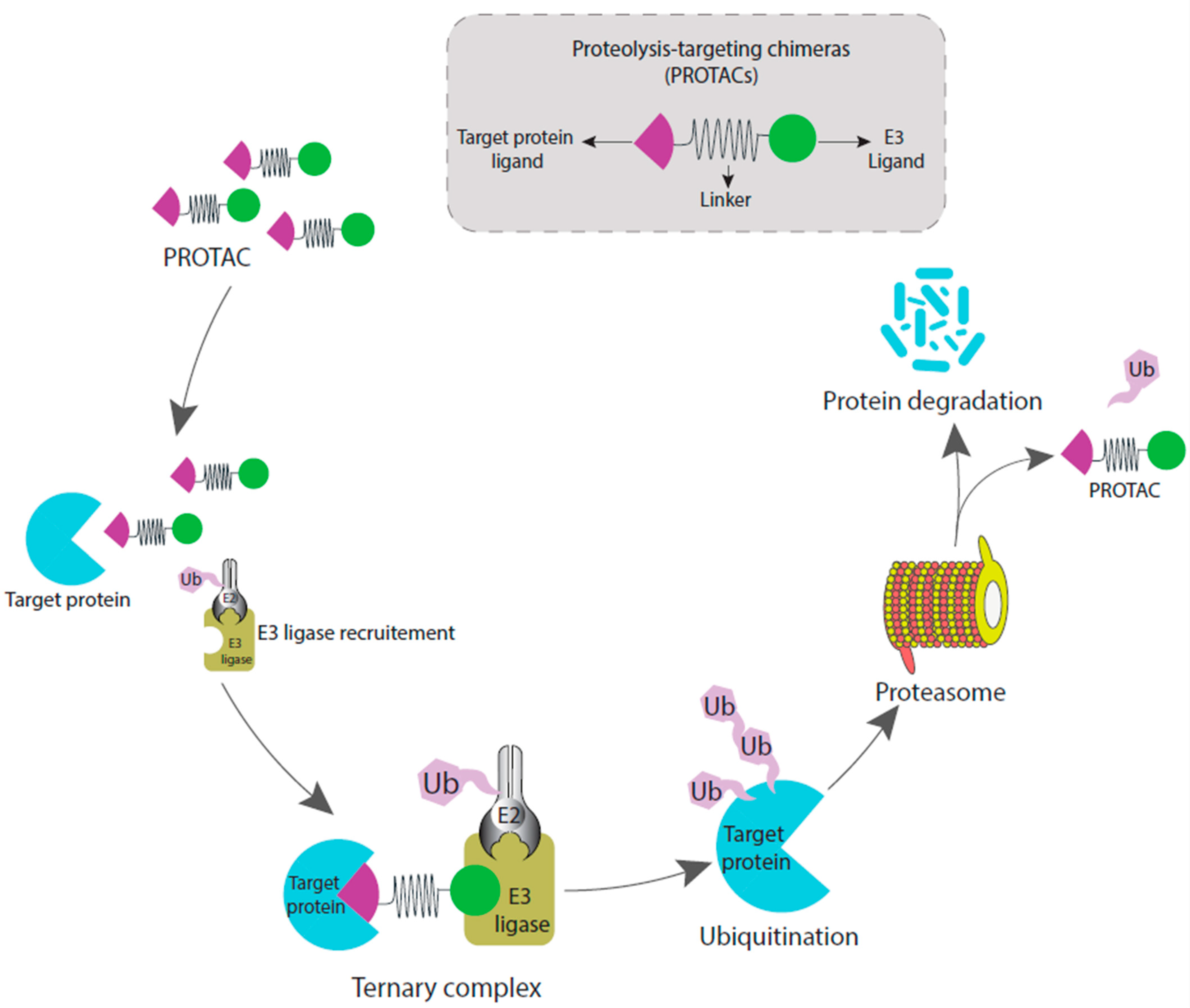
:1. Introduction
2. Ubiquitination and Proteolysis-Targeting Mechanism
2.1. Types of Targeted Protein Degraders
2.2. Mechanism of Action of ARV110 in Treating Prostate Cancer
3. Advantages and Disadvantages of PROTACs over SMIs
4. Genetic Alterations Affecting Prostate Cancer
4.1. Non-AR-Related Pathways
4.2. AR Pathway
5. Prominent PROTACs Targeting Prostate Cancer
5.1. Androgen Receptor Targeting PROTACs
5.1.1. CRBN-Based PROTACs
5.1.2. VHL-Based PROTACs
5.1.3. Miscellaneous
5.2. Non-AR-Targeting PROTACs
6. Design and Medicinal Chemistry of PROTACs
6.1. Design of E3 Ligands
6.2. Design of Linker
6.3. Design of a Target Protein-Binding Ligand
7. Conclusions and Future Recommendations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Baudino, T. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-Based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Xiang, Y.; Kang, X. Small-Molecule PROTACs for Cancer Immunotherapy. Molecules 2022, 27, 5439. [Google Scholar] [CrossRef] [PubMed]
- Prozzillo, Y.; Fattorini, G.; Santopietro, M.V.; Suglia, L.; Ruggiero, A.; Ferreri, D.; Messina, G. Targeted Protein Degradation Tools: Overview and Future Perspectives. Biology 2020, 9, 421. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. A History of Prostate Cancer Treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, S.; Zhu, C.; Shen, J. The Role of Ferroptosis in Esophageal Cancer. Cancer Cell Int. 2022, 22, 2–6. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- NIH. Available online: https://seer.cancer.gov/statfacts/html/prost.html (accessed on 7 September 2022).
- Gandhi, J.; Afridi, A.; Vatsia, S.; Joshi, G.; Joshi, G.; Kaplan, S.A.; Smith, N.L.; Khan, S.A. The Molecular Biology of Prostate Cancer: Current Understanding and Clinical Implications. Prostate Cancer Prostatic. Dis. 2018, 21, 22–36. [Google Scholar] [CrossRef]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; De Bono, J.S. Drug Discovery in Advanced Prostate Cancer: Translating Biology into Therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef]
- Mullard, A. Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The Ubiquitin-Proteasome System. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The Life Cycle of the 26S Proteasome: From Birth, through Regulation and Function, and onto Its Death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Kumar, D.; Hassan, M.I. Targeted Protein Degraders March towards the Clinic for Neurodegenerative Diseases. Ageing Res. Rev. 2022, 78, 101616. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, Present and Future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef]
- Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606–10620. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, Y.; Ni, Z.; Yang, C.; Tong, Y.; Liu, Y.; Li, H.; Rao, Y. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. 2021, 31, 1315–1318. [Google Scholar] [CrossRef]
- Bradbury, R.H.; Acton, D.G.; Broadbent, N.L.; Brooks, A.N.; Carr, G.R.; Hatter, G.; Hayter, B.R.; Hill, K.J.; Howe, N.J.; Jones, R.D.; et al. Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorg. Med. Chem. Lett. 2013, 23, 1945–1948. [Google Scholar] [CrossRef]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Kaniskan, H.U.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef]
- Zheng, M.; Huo, J.; Gu, X.; Wang, Y.; Wu, C.; Zhang, Q.; Wang, W.; Liu, Y.; Liu, Y.; Zhou, X.; et al. Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 2021, 64, 7839–7852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Li, X.; Wu, J.; Zhao, L.; Li, W.; Liu, J. Dynamics-Based Discovery of Novel, Potent Benzoic Acid Derivatives as Orally Bioavailable Selective Estrogen Receptor Degraders for ERalpha+ Breast Cancer. J. Med. Chem. 2021, 64, 7575–7595. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Miao, Y.; Gao, Q.; Mao, M.; Zhang, C.; Yang, L.; Yang, Y.; Han, D. Bispecific aptamer chimeras enable targeted protein degradation on cell membranes. Angew. Chem. 2021, 133, 11367–11371. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Han, Y.; Yang, Y.; Lin, F.; Li, K.; Kong, L.; Liu, H.; Dang, Y.; Lin, J.; Chen, P.R. Covalently Engineered Nanobody Chimeras for Targeted Membrane Protein Degradation. J. Am. Chem. Soc. 2021, 143, 16377–16382. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187. [Google Scholar] [CrossRef]
- Ji, C.H.; Kim, H.Y.; Lee, M.J.; Heo, A.J.; Park, D.Y.; Lim, S.; Shin, S.; Ganipisetti, S.; Yang, W.S.; Jung, C.A.; et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 2022, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted Protein Degradation: Mechanisms, Strategies and Application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, Present and Future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e3. [Google Scholar] [CrossRef]
- Han, X.; Sun, Y. Review Strategies for the Discovery of Oral PROTAC Degraders Aimed at Cancer Therapy. Cell Rep. Phys. Sci. 2022, 3, 101062. [Google Scholar] [CrossRef]
- Clague, M.J.; Heride, C.; Urbé, S. The Demographics of the Ubiquitin System. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Zaidman, D.; Prilusky, J.; London, N. ProsetTac: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J. Chem. Inf. Model. 2020, 60, 4894–4903. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, D.; Spring, D.J.; Depinho, R.A. Genetics and Biology of Prostate Cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef]
- Dong, J.T. Prevalent Mutations in Prostate Cancer. J. Cell Biochem. 2006, 97, 433–447. [Google Scholar] [CrossRef]
- Robinson, D.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Eeles, R.; Goh, C.; Castro, E.; Bancroft, E.; Guy, M.; Olama, A.A.; Easton, D.; Kote-Jarai, Z. The Genetic Epidemiology of Prostate Cancer and Its Clinical Implications. Nat. Rev. Urol. 2014, 11, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Castro, E.; Aragón, I.M.; Cendón, Y.; Cattrini, C.; López-Casas, P.P.; Olmos, D. Genetic Aberrations in DNA Repair Pathways: A Cornerstone of Precision Oncology in Prostate Cancer. Br. J. Cancer 2021, 124, 552–563. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS Family of Oncogenic Transcription Factors in Solid Tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Montagna, C.; Leighton, X.; Glasman, M.; Naga, S.; Eidelman, O.; Ried, T.; Pollard, H.B. Haploinsufficiency of Anx7 Tumor Suppressor Gene and Consequent Genomic Instability Promotes Tumorigenesis in the Anx7(+/−) Mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 14287–14292. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Frierson, H.F.; Chen, C.; Li, C.; Ran, Q.; Otto, K.B.; Cantarel, B.M.; Vessella, R.L.; Gao, A.C.; Petros, J.; et al. Frequent Somatic Mutations of the Transcription Factor ATBF1 in Human Prostate Cancer. Nat. Genet. 2005, 37, 407–412. [Google Scholar] [CrossRef]
- Kibel, A.S.; Suarez, B.K.; Belani, J.; Oh, J.; Webster, R.; Brophy-Ebbers, M.; Guo, C.; Catalona, W.J.; Picus, J.; Goodfellow, P.J. CDKN1A and CDKN1B Polymorphisms and Risk of Advanced Prostate Carcinoma. Cancer Res. 2003, 63, 2033–2036. [Google Scholar]
- Dong, X.; Wang, L.; Taniguchi, K.; Wang, X.; Cunningham, J.M.; McDonnell, S.K.; Qian, C.; Marks, A.F.; Slager, S.L.; Peterson, B.J.; et al. Mutations in CHEK2 Associated with Prostate Cancer Risk. Am. J. Hum. Genet. 2003, 72, 270–280. [Google Scholar] [CrossRef]
- Ntais, C.; Polycarpou, A.; Ioannidis, J.P.A. Association of GSTM1, GSTT1, and GSTP1 Gene Polymorphisms with the Risk of Prostate Cancer: A Meta-Analysis. Cancer Epidemiol. Biomark Prev. 2005, 14, 176–181. [Google Scholar] [CrossRef]
- Chen, C.; Bhalala, H.v.; Vessella, R.L.; Dong, J.T. KLF5 Is Frequently Deleted and Down-Regulated but Rarely Mutated in Prostate Cancer. Prostate 2003, 55, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Narla, G.; Heath, K.E.; Reeves, H.L.; Li, D.; Giono, L.E.; Kimmelman, A.C.; Glucksman, M.J.; Narla, J.; Eng, F.J.; Chan, A.M.; et al. KLF6, a Candidate Tumor Suppressor Gene Mutated in Prostate Cancer. Science 2001, 294, 2563–2566. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lilly Zheng, S.; Komiya, A.; Mychaleckyj, J.C.; Isaacs, S.D.; Chang, B.; Turner, A.R.; Ewing, C.M.; Wiley, K.E.; Hawkins, G.A.; et al. Common Sequence Variants of the Macrophage Scavenger Receptor 1 Gene Are Associated with Prostate Cancer Risk. Am. J. Hum. Genet. 2003, 72, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, G.K.; Mutton, L.N.; Khalili, M.; McMullin, R.P.; Hicks, J.L.; Bianchi-Frias, D.; Horn, L.A.; Kulac, I.; Moubarek, M.S.; Nelson, P.S.; et al. Combined MYC Activation and Pten Loss Are Sufficient to Create Genomic Instability and Lethal Metastatic Prostate Cancer. Cancer Res. 2016, 76, 283–292. [Google Scholar] [CrossRef]
- Abdulkadir, S.A.; Magee, J.A.; Peters, T.J.; Kaleem, Z.; Naughton, C.K.; Humphrey, P.A.; Milbrandt, J. Conditional Loss of Nkx3.1 in Adult Mice Induces Prostatic Intraepithelial Neoplasia. Mol. Cell Biol. 2002, 22, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Marchesani, M.; Hakkarainen, A.; Tuomainen, T.P.; Kaikkonen, J.; Pukkala, E.; Uimari, P.; Seppälä, E.; Matikainen, M.; Kallioniemi, O.P.; Schleutker, J.; et al. New Paraoxonase 1 Polymorphism I102V and the Risk of Prostate Cancer in Finnish Men. J. Natl. Cancer Inst. 2003, 95, 812–818. [Google Scholar] [CrossRef]
- Carpten, J.; Nupponen, N.; Isaacs, S.; Sood, R.; Robbins, C.; Xu, J.; Faruque, M.; Moses, T.; Ewing, C.; Gillanders, E.; et al. Germline Mutations in the Ribonuclease L Gene in Families Showing Linkage with HPC1. Nat. Genet. 2002, 30, 181–184. [Google Scholar] [CrossRef]
- Cicek, M.S.; Conti, D.v.; Curran, A.; Neville, P.J.; Paris, P.L.; Casey, G.; Witte, J.S. Association of Prostate Cancer Risk and Aggressiveness to Androgen Pathway Genes: SRD5A2, CYP17, and the AR. Prostate 2004, 59, 69–76. [Google Scholar] [CrossRef]
- Medeiros, R.; Morais, A.; Vasconcelos, A.; Costa, S.; Pinto, D.; Oliveira, J.; Lopes, C. The Role of Vitamin D Receptor Gene Polymorphisms in the Susceptibility to Prostate Cancer of a Southern European Population. J. Hum. Genet. 2002, 47, 413–418. [Google Scholar] [CrossRef]
- Newmark, J.R.; Hardy, D.; Tonb, D.C.; Carter, B.S.; Epsteint, J.I.; Isaacs, W.B.; Browns, T.R.; Barrack, E.R. Androgen Receptor Gene Mutations in Human Prostate Cancer (Polymerase Chain Reaction/Denaturing Gradient Gel Electrophoresis/Somatic Mutation). Proc. Natl. Acad. Sci. USA 1992, 89, 6319–6323. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Stampfer, M.J.; Krithivas, K.; Brown, M.; Brufsky, A.; Talcott, J.; Hennekens, C.H.; Kantoff, P.W. The CAG Repeat within the Androgen Receptor Gene and Its Relationship to Prostate Cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 3320–3323. Available online: www.pnas.org (accessed on 1 April 1997). [CrossRef] [PubMed]
- Gockel, L.M.; Pfeifer, V.; Baltes, F.; Bachmaier, R.D.; Wagner, K.G.; Bendas, G.; Gütschow, M.; Sosič, I.; Steinebach, C. Design, Synthesis, and Characterization of PROTACs Targeting the Androgen Receptor in Prostate and Lung Cancer Models. Arch. Pharm. 2022, 355, 156. [Google Scholar] [CrossRef]
- Takwale, A.D.; Jo, S.H.; Jeon, Y.U.; Kim, H.S.; Shin, C.H.; Lee, H.K.; Ahn, S.; Lee, C.O.; du Ha, J.; Kim, J.H.; et al. Design and Characterization of Cereblon-Mediated Androgen Receptor Proteolysis-Targeting Chimeras. Eur. J. Med. Chem. 2020, 208, 112769. [Google Scholar] [CrossRef]
- Liang, J.J.; Xie, H.; Yang, R.H.; Wang, N.; Zheng, Z.J.; Zhou, C.; Wang, Y.L.; Wang, Z.J.; Liu, H.M.; Shan, L.H.; et al. Designed, Synthesized and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) as AR Degraders for Prostate Cancer Treatment. Bioorg. Med. Chem. 2021, 45, 16331. [Google Scholar] [CrossRef]
- Kim, G.Y.; Song, C.W.; Yang, Y.S.; Lee, N.R.; Yoo, H.S.; Son, S.H.; Lee, S.J.; Park, J.S.; Lee, J.K.; Inn, K.S. Chemical Degradation of Androgen Receptor (Ar) Using Bicalutamide Analog–Thalidomide Protacs. Molecules 2021, 26, 2525. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Wang, L.; Matvekas, A.; et al. Strategies toward Discovery of Potent and Orally Bioavailable Proteolysis Targeting Chimera Degraders of Androgen Receptor for the Treatment of Prostate Cancer. J. Med. Chem. 2021, 64, 12831–12854. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Zhao, L.; Han, X.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Kirchhoff, P.D.; Wang, L.; et al. Discovery of ARD-2585 as an Exceptionally Potent and Orally Active PROTAC Degrader of Androgen Receptor for the Treatment of Advanced Prostate Cancer. J. Med. Chem. 2021, 64, 13487–13509. [Google Scholar] [CrossRef]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; Xu, T.; Wang, M.; Yang, C.Y.; Chinnaswamy, K.; Stuckey, J.; et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J. Med. Chem. 2019, 62, 11218–11231. [Google Scholar] [CrossRef]
- Chen, L.; Han, L.; Mao, S.; Xu, P.; Xu, X.; Zhao, R.; Wu, Z.; Zhong, K.; Yu, G.; Wang, X. Discovery of A031 as Effective Proteolysis Targeting Chimera (PROTAC) Androgen Receptor (AR) Degrader for the Treatment of Prostate Cancer. Eur. J. Med. Chem. 2021, 216, 113307. [Google Scholar] [CrossRef] [PubMed]
- Munoz, E.; Chen, G.; Hossain, A.; Wu, S.; Oceguera Nava, E.; Hang, J.; Lee, T.; Zhang, Q.; Wang, G.; Chen, Q.-H. Synthesis and Biological Evaluation of Niclosamide PROTACs. Bioorg. Med. Chem. Lett. 2022, 72, 128870. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Mol. Cancer Ther. 2021, 20, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen Receptor Degradation by the Proteolysis-Targeting Chimera ARCC-4 Outperforms Enzalutamide in Cellular Models of Prostate Cancer Drug Resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen Receptor Degraders Overcome Common Resistance Mechanisms Developed during Prostate Cancer Treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Ma, B.; Fan, Y.; Zhang, D.; Wei, Y.; Jian, Y.; Liu, D.; Wang, Z.; Gao, Y.; Ma, J.; Chen, Y.; et al. De Novo Design of an Androgen Receptor DNA Binding Domain-Targeted Peptide PROTAC for Prostate Cancer Therapy. Adv. Sci. 2022, 9, 2201859. [Google Scholar] [CrossRef]
- Xie, H.; Liang, J.J.; Wang, Y.L.; Hu, T.X.; Wang, J.Y.; Yang, R.H.; Yan, J.K.; Zhang, Q.R.; Xu, X.; Liu, H.M.; et al. The Design, Synthesis and Anti-Tumor Mechanism Study of New Androgen Receptor Degrader. Eur. J. Med. Chem. 2020, 204, 112512. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef]
- Hu, R.; Wang, W.L.; Yang, Y.Y.; Hu, X.T.; Wang, Q.W.; Zuo, W.Q.; Xu, Y.; Feng, Q.; Wang, N.Y. Identification of a Selective BRD4 PROTAC with Potent Antiproliferative Effects in AR-Positive Prostate Cancer Based on a Dual BET/PLK1 Inhibitor. Eur. J. Med. Chem. 2022, 227, 113922. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, L.; Cao, C.; Yu, J.; Luo, X.; Zhou, P.; Zhao, L.; Du, W.; Cheng, J.; Xie, Y.; et al. Development of Selective Mono or Dual PROTAC Degrader Probe of CDK Isoforms. Eur. J. Med. Chem. 2020, 187, 111952. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A.P. Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 That Regulates the Organelle’s Dynamics and Signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef]
- Guenette, R.G.; Yang, S.W.; Min, J.; Pei, B.; Potts, P.R. Target and Tissue Selectivity of PROTAC Degraders. Chem. Soc. Rev. 2022, 51, 5740–5756. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; He, M.; Wang, L.; He, Y.; Rao, Y. Chemistries of Bifunctional PROTAC Degraders. Chem. Soc. Rev. 2022, 51, 7066–7114. [Google Scholar] [CrossRef]
- Ishida, T.; Ciulli, A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov. 2021, 26, 484–502. [Google Scholar] [CrossRef] [PubMed]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review. Explor. Target Antitumor. Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef]
- Hughes, S.J.; Testa, A.; Thompson, N.; Churcher, I. The rise and rise of protein degradation: Opportunities and challenges ahead. Drug Discov. Today 2021, 26, 2889–2897. [Google Scholar] [CrossRef]
- Dey, S.K.; Jaffrey, S.R. RIBOTACs: Small Molecules Target RNA for Degradation. Cell Chem. Biol. 2019, 26, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hu, Q.; Zhang, X.; Sun, R.; Liu, Z.; Wu, S.; Tian, S.; Ma, X.; Dai, Z.; Yang, X.; et al. DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs. Nat. Commun. 2022, 13, 7133. [Google Scholar] [CrossRef]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the role of linker design in proteolysis targeting chimeras: Miniperspective. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Yang, J.; Chen, Y.; Zhou, P.; Wang, Y.; Du, W.; Zhao, L.; Chen, Y. Discovery of SK-575 as a Highly Potent and Efficacious ProteolysisTargeting Chimera Degrader of PARP1 for Treating Cancers. J. Med. Chem. 2020, 63, 11012–11033. [Google Scholar] [CrossRef]
- Qu, X.; Liu, H.; Song, X.; Sun, N.; Zhong, H.; Qiu, X.; Yang, X.; Jiang, B. Effective degradation of EGFRL858R+T790M mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem. 2021, 218, 113328. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, M.; Saeedi, M.; Larijani, B.; Mahdavi, M. Recent advances in biological activities of rhodium complexes: Their applications in drug discovery research. Eur. J. Med. Chem. 2021, 216, 113308. [Google Scholar] [CrossRef]
- Jansa, J.; Jorda, R.; Skerlova, J.; Pachl, P.; Perina, M.; Reznickova, E.; Heger, T.; Gucky, T.; Rezacova, P.; Lycka, A.; et al. Imidazo[1,2c]pyrimidin-5(6H)-one inhibitors of CDK2: Synthesis, kinase inhibition and co-crystal structure. Eur. J. Med. Chem. 2021, 216, 113309. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Song, Q.; Cao, Z.; Deng, Y.; Tan, Z.; Zhang, L. Design, synthesis and evaluation of novel dimethylamino chalcone-O-alkylamines derivatives as potential multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 216, 113310. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Guo, Z.; Wang, J.Y.; Wang, H.M.; Da Qi, J.; Ma, J.; Piao, H.R.; Jin, C.H.; Jin, X. Synthesis and evaluation of the epithelial-to- mesenchymal inhibitory activity of indazole-derived imidazoles as dual ALK5/p38alpha MAP inhibitors. Eur. J. Med. Chem. 2021, 216, 113311. [Google Scholar] [CrossRef]
- Singh, S.S.; Mattheolabakis, G.; Gu, X.; Withers, S.; Dahal, A.; Jois, S. A grafted peptidomimetic for EGFR heterodimerization inhibition: Implications in NSCLC models. Eur. J. Med. Chem. 2021, 216, 113312. [Google Scholar] [CrossRef]
- Lu, R.; Wang, Y.; Liu, C.; Zhang, Z.; Li, B.; Meng, Z.; Jiang, C.; Hu, Q. Design, synthesis and evaluation of 3-amide-5-aryl benzoic acid derivatives as novel P2Y(14)R antagonists with potential high efficiency against acute gouty arthritis. Eur. J. Med. Chem. 2021, 216, 113313. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, X.; Guan, P.; Song, S.; You, G.; Xia, C.; Liu, T. Recent progress in agents targeting polo-like kinases: Promising therapeutic strategies. Eur. J. Med. Chem. 2021, 217, 113314. [Google Scholar] [CrossRef]
- Villard, A.L.; Aubertin, A.M.; Peyrottes, S.; Perigaud, C. An original pronucleotide strategy for the simultaneous delivery of two bioactive drugs. Eur. J. Med. Chem. 2021, 216, 113315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, W.; Shuai, W.; Liu, Y.; Yang, L.; Tan, Y.; Zheng, T.; Yao, H.; Xu, J.; Zhu, Z.; et al. Discovery of novel Nbenzylbenzamide derivatives as tubulin polymerization inhibitors with potent antitumor activities. Eur. J. Med. Chem. 2021, 216, 113316. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, M.; Liu, S.; Zou, Z.; Ren, R.; Zhang, C.; Xie, H.; Sun, J.; Qi, Y.; Qu, Q.; et al. Pyxinol bearing amino acid residues: Easily achievable and promising modulators of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2021, 216, 113317. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.; Jo, J.; Chang, J.W.; Sim, J.; Yun, H. Recent advances in development of hetero-bivalent kinase inhibitors. Eur. J. Med. Chem. 2021, 216, 113318. [Google Scholar] [CrossRef]
- Li, Z.; Xin, W.; Wang, Q.; Zhu, M.; Zhou, H. Design and synthesis of N-(3-sulfamoylphenyl)amides as Trypanosoma brucei leucyl-tRNA synthetase inhibitors. Eur. J. Med. Chem. 2021, 217, 113319. [Google Scholar] [CrossRef]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef]
- Aaghaz, S.; Sharma, K.; Jain, R.; Kamal, A. beta-Carbolines as potential anticancer agents. Eur. J. Med. Chem. 2021, 216, 113321. [Google Scholar] [CrossRef]
- Li, Y.S.; He, M.; Zhou, T.S.; Wang, Q.; He, L.; Wang, S.J.; Hu, B.; Wei, B.; Wang, H.; Cui, Z.N. 2,5-Disubstituted furan derivatives containing 1,3,4-thiadiazole moiety as potent alpha-glucosidase and E. coli beta-glucuronidase inhibitors. Eur. J. Med. Chem. 2021, 216, 113322. [Google Scholar] [CrossRef]
- Jo, J.; Lee, D.; Park, Y.H.; Choi, H.; Han, J.; Park, D.H.; Choi, Y.K.; Kwak, J.; Yang, M.K.; Yoo, J.W.; et al. Discovery and optimization of novel 3-benzyl-N-phenyl-1H-pyrazole-5-carboxamides as bifunctional antidiabetic agents stimulating both insulin secretion and glucose uptake. Eur. J. Med. Chem. 2021, 217, 113325. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, C.; Dong, G.; Qiao, H.; Yang, J.; Liu, H.; Ding, L.; Sun, K.; Zhao, W. Development of phenyltriazole thiol-based derivatives as highly potent inhibitors of DCN1-UBC12 interaction. Eur. J. Med. Chem. 2021, 217, 113326. [Google Scholar] [CrossRef]
- Wu, S.; Xu, C.; Xia, K.; Lin, Y.; Tian, S.; Ma, H.; Ji, Y.; Zhu, F.; He, S.; Zhang, X. Ring closure strategy leads to potent RIPK3 inhibitors. Eur. J. Med. Chem. 2021, 217, 113327. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, C.; Wang, D.; Zhang, J.; Zhang, T. Emerging small-molecule inhibitors of the Bruton’s tyrosine kinase (BTK): Current development. Eur. J. Med. Chem. 2021, 217, 113329. [Google Scholar] [CrossRef] [PubMed]
- Kayamba, F.; Malimabe, T.; Ademola, I.K.; Pooe, O.J.; Kushwaha, N.D.; Mahlalela, M.; van Zyl, R.L.; Gordon, M.; Mudau, P.T.; Zininga, T.; et al. Design and synthesis of quinoline-pyrimidine inspired hybrids as potential plasmodial inhibitors. Eur. J. Med. Chem. 2021, 217, 113330. [Google Scholar] [CrossRef] [PubMed]
- Grieco, I.; Bissaro, M.; Tiz, D.B.; Perez, D.I.; Perez, C.; Martinez, A.; Redenti, S.; Mariotto, E.; Bortolozzi, R.; Viola, G.; et al. Developing novel classes of protein kinase CK1delta inhibitors by fusing [1,2,4]triazole with different bicyclic heteroaromatic systems. Eur. J. Med. Chem. 2021, 216, 113331. [Google Scholar] [CrossRef]
- Gediya, P.; Parikh, P.K.; Vyas, V.K.; Ghate, M.D. Histone deacetylase 2: A potential therapeutic target for cancer and neurodegenerative disorders. Eur. J. Med. Chem. 2021, 216, 113332. [Google Scholar] [CrossRef]
- Gao, D.; Jin, N.; Fu, Y.; Zhu, Y.; Wang, Y.; Wang, T.; Chen, Y.; Zhang, M.; Xiao, Q.; Huang, M.; et al. Rational drug design of benzothiazole-based derivatives as potent signal transducer and activator of transcription 3 (STAT3) signaling pathway inhibitors. Eur. J. Med. Chem. 2021, 216, 113333. [Google Scholar] [CrossRef]
- Dhokne, P.; Sakla, A.P.; Shankaraiah, N. Structural insights of oxindole based kinase inhibitors as anticancer agents: Recent advances. Eur. J. Med. Chem. 2021, 216, 113334. [Google Scholar] [CrossRef]
- Ren, C.; Sun, N.; Kong, Y.; Qu, X.; Liu, H.; Zhong, H.; Song, X.; Yang, X.; Jiang, B. Structure-based discovery of SIAIS001 as an oral bioavailability ALK degrader constructed from Alectinib. Eur. J. Med. Chem. 2021, 217, 113335. [Google Scholar] [CrossRef]
- Shim, J.H. Inhibitory Effects of Cycloheterophyllin on Melanin Synthesis. Molecules 2021, 26, 2526. [Google Scholar] [CrossRef]
- Azzouz, Z.; Bettache, A.; Boucherba, N.; Prieto, A.; Martinez, M.J.; Benallaoua, S.; de Eugenio, L.I. Optimization of beta-1,4-Endoxylanase Production by an Aspergillus niger Strain Growing on Wheat Straw and Application in Xylooligosaccharides Production. Molecules 2021, 26, 2527. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.M. The Redox Potential of the beta-(93)-Cysteine Thiol Group in Human Hemoglobin Estimated from In Vitro Oxidant Challenge Experiments. Molecules 2021, 26, 2528. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yang, W.S.; Htwe, K.M.; Lee, M.N.; Kim, Y.D.; Yoon, K.D.; Lee, B.H.; Lee, S.; Cho, J.Y. Dipterocarpus tuberculatus Roxb. Ethanol Extract Has Anti-Inflammatory and Hepatoprotective Effects In Vitro and In Vivo by Targeting the IRAK1/AP-1 Pathway. Molecules 2021, 26, 2529. [Google Scholar] [CrossRef] [PubMed]
- Budeev, A.; Kantin, G.; Dar’in, D.; Krasavin, M. Diazocarbonyl and Related Compounds in the Synthesis of Azoles. Molecules 2021, 26, 2530. [Google Scholar] [CrossRef]
- Dao, T.B.; Nguyen, T.M.; Nguyen, V.Q.; Tran, T.M.; Tran, N.M.; Nguyen, C.H.; Nguyen, T.H.; Nguyen, H.H.; Sichaem, J.; Tran, C.L.; et al. Flavones from Combretum quadrangulare Growing in Vietnam and Their Alpha-Glucosidase Inhibitory Activity. Molecules 2021, 26, 2531. [Google Scholar] [CrossRef] [PubMed]
- Zhen, L.; Lange, H.; Crestini, C. An Analytical Toolbox for Fast and Straightforward Structural Characterisation of Commercially Available Tannins. Molecules 2021, 26, 2532. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Ukai, Y.; Kondo, N.; Nozu, K.; Kimura, C.; Hashimoto, K.; Mizusawa, E.; Maki, H.; Naito, A.; Kawai, M. Identification of Thiazoyl Guanidine Derivatives as Novel Antifungal Agents Inhibiting Ergosterol Biosynthesis for Treatment of Invasive Fungal Infections. J. Med. Chem. 2021, 64, 10482–10496. [Google Scholar] [CrossRef]
- Jimenez, T.; Botero, J.; Otaegui, D.; Calvo, J.; Hernandez, F.J.; San Sebastian, E. Rational Design and Experimental Analysis of Short-Oligonucleotide Substrate Specificity for Targeting Bacterial Nucleases. J. Med. Chem. 2021, 64, 12855–12864. [Google Scholar] [CrossRef]
- Rossino, G.; Rui, M.; Linciano, P.; Rossi, D.; Boiocchi, M.; Peviani, M.; Poggio, E.; Curti, D.; Schepmann, D.; Wunsch, B.; et al. Bitopic Sigma 1 Receptor Modulators to Shed Light on Molecular Mechanisms Underpinning Ligand Binding and Receptor Oligomerization. J. Med. Chem. 2021, 64, 14997–15016. [Google Scholar] [CrossRef]
- Plesselova, S.; Garcia-Cerezo, P.; Blanco, V.; Reche-Perez, F.J.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F.; Giron-Gonzalez, M.D.; Salto-Gonzalez, R. Polyethylenimine-Bisphosphonate-Cyclodextrin Ternary Conjugates: Supramolecular Systems for the Delivery of Antineoplastic Drugs. J. Med. Chem. 2021, 64, 12245–12260. [Google Scholar] [CrossRef]
- Ryan, M.D.; Parkes, A.L.; Corbett, D.; Dickie, A.P.; Southey, M.; Andersen, O.A.; Stein, D.B.; Barbeau, O.R.; Sanzone, A.; Thommes, P.; et al. Discovery of Novel UDP-N-Acetylglucosamine Acyltransferase (LpxA) Inhibitors with Activity against Pseudomonas aeruginosa. J. Med. Chem. 2021, 64, 14377–14425. [Google Scholar] [CrossRef]
- Mann, M.K.; Zepeda-Velazquez, C.A.; Gonzalez-Alvarez, H.; Dong, A.; Kiyota, T.; Aman, A.M.; Loppnau, P.; Li, Y.; Wilson, B.; Arrowsmith, C.H.; et al. Structure-Activity Relationship of USP5 Inhibitors. J. Med. Chem. 2021, 64, 1501715036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ghosh, A.; Connolly, P.J.; King, P.; Wilde, T.; Wang, J.; Dong, Y.; Li, X.; Liao, D.; Chen, H.; et al. GutRestricted Selective Cyclooxygenase-2 (COX-2) Inhibitors for Chemoprevention of Colorectal Cancer. J. Med. Chem. 2021, 64, 11570–11596. [Google Scholar] [CrossRef]
- Singh, H.; Thirupathi, A.; Das, B.; Janni, M.; Kumari, R.; Singh, S.; Rashid, M.; Wahajuddin, M.; Balamurali, M.M.; Jagavelu, K.; et al. 2,3-Difunctionalized Benzo[b]thiophene Scaffolds Possessing Potent Antiangiogenic Properties. J. Med. Chem. 2022, 65, 120134. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.A.; Minotti, A.; Luraghi, A.; Romerio, A.; Gotri, N.; Matamoros-Recio, A.; Iannucci, A.; Palmer, C.; Wang, G.; Ingram, R.; et al. Synthetic Glycolipids as Molecular Vaccine Adjuvants: Mechanism of Action in Human Cells and In Vivo Activity. J. Med. Chem. 2021, 64, 12261–12272. [Google Scholar] [CrossRef]
- Chen, H.; Liu, J.; Kaniskan, H.U.; Wei, W.; Jin, J. Folate-Guided Protein Degradation by Immunomodulatory Imide Drug-Based Molecular Glues and Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 12273–12285. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.D.; Stotz, S.; Kinzler, J.; Geibel, C.; Lammerhofer, M.; Pichler, B.J.; Maurer, A. DoE Optimization Empowers the Automated Preparation of Enantiomerically Pure [(18)F]Talazoparib and its In Vivo Evaluation as a PARP Radiotracer. J. Med. Chem. 2021, 64, 1569015701. [Google Scholar] [CrossRef]
- Bursavich, M.G.; Harrison, B.A.; Acharya, R.; Costa, D.E.; Freeman, E.A.; Hrdlicka, L.A.; Jin, H.; Kapadnis, S.; Moffit, J.S.; Murphy, D.; et al. Discovery of the Oxadiazine FRM-024: A Potent CNSPenetrant Gamma Secretase Modulator. J. Med. Chem. 2021, 64, 14426–14447. [Google Scholar] [CrossRef]
- Heightman, T.D.; Berdini, V.; Bevan, L.; Buck, I.M.; Carr, M.G.; Courtin, A.; Coyle, J.E.; Day, J.E.H.; East, C.; Fazal, L.; et al. Discovery of ASTX029, A Clinical Candidate Which Modulates the Phosphorylation and Catalytic Activity of ERK1/2. J. Med. Chem. 2021, 64, 12286–12303. [Google Scholar] [CrossRef]
- Xiong, B.; Wang, Y.; Chen, Y.; Xing, S.; Liao, Q.; Chen, Y.; Li, Q.; Li, W.; Sun, H. Strategies for Structural Modification of Small Molecules to Improve Blood-Brain Barrier Penetration: A Recent Perspective. J. Med. Chem. 2021, 64, 13152–13173. [Google Scholar] [CrossRef]
- Deng, X.; Salgado-Polo, F.; Shao, T.; Xiao, Z.; Van, R.; Chen, J.; Rong, J.; Haider, A.; Shao, Y.; Josephson, L.; et al. Imaging Autotaxin In Vivo with (18)F-Labeled Positron Emission Tomography Ligands. J. Med. Chem. 2021, 64, 15053–15068. [Google Scholar] [CrossRef]
- Fallica, A.N.; Barbaraci, C.; Amata, E.; Pasquinucci, L.; Turnaturi, R.; Dichiara, M.; Intagliata, S.; Gariboldi, M.B.; Marras, E.; Orlandi, V.T.; et al. Nitric Oxide Photo-Donor Hybrids of Ciprofloxacin and Norfloxacin: A Shift in Activity from Antimicrobial to Anticancer Agents. J. Med. Chem. 2021, 64, 11597–11613. [Google Scholar] [CrossRef]
- Monsarrat, C.; Compain, G.; Andre, C.; Engilberge, S.; Martiel, I.; Olieric, V.; Wolff, P.; Brillet, K.; Landolfo, M.; Silva da Veiga, C.; et al. Iterative Structure-Based Optimization of Short Peptides Targeting the Bacterial Sliding Clamp. J. Med. Chem. 2021, 64, 17063–17078. [Google Scholar] [CrossRef] [PubMed]
- McCoull, W.; Boyd, S.; Brown, M.R.; Coen, M.; Collingwood, O.; Davies, N.L.; Doherty, A.; Fairley, G.; Goldberg, K.; Hardaker, E.; et al. Optimization of an Imidazo[1,2-a]pyridine Series to Afford Highly Selective Type I1/2 Dual Mer/Axl Kinase Inhibitors with In Vivo Efficacy. J. Med. Chem. 2021, 64, 13524–13539. [Google Scholar] [CrossRef] [PubMed]
- Cristina Mendonca Nogueira, T.; Vinicius Nora de Souza, M. New FDA oncology small molecule drugs approvals in 2020: Mechanism of action and clinical applications. Bioorganic Med. Chem. 2021, 46, 116340. [Google Scholar] [CrossRef]
- Sun, J.; Wang, J.; Wang, X.; Hu, X.; Cao, H.; Bai, J.; Li, D.; Hua, H. Design and synthesis of beta-carboline derivatives with nitrogen mustard moieties against breast cancer. Bioorganic Med. Chem. 2021, 45, 116341. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, S.; Tian, G.; Cheung, H.J.H.; Li, X.; Li, X.D. Concise solid-phase synthesis enables derivatisation of YEATS domain cyclopeptide inhibitors for improved cellular uptake. Bioorganic Med. Chem. 2021, 45, 116342. [Google Scholar] [CrossRef]
- Sato, J.; Kusano, H.; Aoki, T.; Shibuya, S.; Yokoo, K.; Komano, K.; Oguma, T.; Matsumoto, S.; Sato, T.; Yasuo, K.; et al. A novel tricyclic beta-lactam exhibiting potent antibacterial activities against carbapenem-resistant Enterobacterales: Synthesis and structure-activity-relationships. Bioorganic Med. Chem. 2021, 46, 116343. [Google Scholar] [CrossRef]
- Yao, H.; Guo, Q.; Wang, M.; Wang, R.; Xu, Z. Discovery of pyrazole N-aryl sulfonate: A novel and highly potent cyclooxygenase-2 (COX-2) selective inhibitors. Bioorganic Med. Chem. 2021, 46, 116344. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.Y.; Miki, T.; Mihara, H.; Tsutsumi, H. Biofunctional supramolecular hydrogels fabricated from a short self-assembling peptide modified with bioactive sequences for the 3D culture of breast cancer MCF-7 cells. Bioorganic Med. Chem. 2021, 46, 116345. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q.; Su, M.; Luo, F.; Liu, Y.; Wang, D.; Fan, Y. Design, synthesis, and biological evaluation of novel 6-(pyridin-3-yl) quinazolin-4(3H)one derivatives as potential anticancer agents via PI3K inhibition. Bioorganic Med. Chem. 2021, 46, 116346. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Aleem, A.M.; Tena, J.; Rivera-Velazquez, M.; Brah, H.S.; Tripathi, S.; D’Silva, M.; Nadler, J.L.; Kalyanaraman, C.; Jacobson, M.P.; et al. Docking and mutagenesis studies lead to improved inhibitor development of ML355 for human platelet 12-lipoxygenase. Bioorganic Med. Chem. 2021, 46, 116347. [Google Scholar] [CrossRef] [PubMed]
- Toviwek, B.; Phuangsawai, O.; Konsue, A.; Hannongbua, S.; Riley, J.; Mutter, N.; Anderson, M.; Webster, L.; Hallyburton, I.; Read, K.D.; et al. Preparation, biological & cheminformatics-based assessment of N(2),N(4)-diphenylpyrimidine-2,4-diamine as potential Kinase-targeted antimalarials. Bioorganic Med. Chem. 2021, 46, 116348. [Google Scholar]
- Tsai, W.C.; Gilbert, N.C.; Ohler, A.; Armstrong, M.; Perry, S.; Kalyanaraman, C.; Yasgar, A.; Rai, G.; Simeonov, A.; Jadhav, A.; et al. Kinetic and structural investigations of novel inhibitors of human epithelial 15-lipoxygenase-2. Bioorganic Med. Chem. 2021, 46, 116349. [Google Scholar] [CrossRef]
- Wang, B.; Feng, W.; Wang, J.; Dong, Y.; Liu, Y.; Yao, Y.; Zhang, J.; Shi, W.; Liu, L.; Zhang, H.; et al. Discovery of potent and selective Bcl-2 inhibitors with acyl sulfonamide skeleton. Bioorganic Med. Chem. 2021, 47, 116350. [Google Scholar] [CrossRef] [PubMed]
- Biteau, N.G.; Roy, V.; Lambry, J.C.; Becker, H.F.; Myllykallio, H.; Agrofoglio, L.A. Synthesis of acyclic nucleoside phosphonates targeting flavindependent thymidylate synthase in Mycobacterium tuberculosis. Bioorganic Med. Chem. 2021, 46, 116351. [Google Scholar] [CrossRef]
- Ma, Z.; Jiang, L.; Li, B.; Liang, D.; Feng, Y.; Liu, L.; Jiang, C. Discovery of benzimidazole derivatives as potent and selective aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitors with glucose consumption improving activity. Bioorganic Med. Chem. 2021, 46, 116352. [Google Scholar] [CrossRef]
- Chang, T.C.; Tanaka, K. In vivo organic synthesis by metal catalysts. Bioorganic Med. Chem. 2021, 46, 116353. [Google Scholar] [CrossRef]
- Kumar, S.; Mittal, A.; Mittal, A. A review upon medicinal perspective and designing rationale of DPP-4 inhibitors. Bioorganic Med. Chem. 2021, 46, 116354. [Google Scholar] [CrossRef]
- Schwarthoff, S.; Tischer, N.; Sager, H.; Schatz, B.; Rohrbach, M.M.; Raztsou, I.; Robaa, D.; Gaube, F.; Arndt, H.D.; Winckler, T. Evaluation of gamma-carboline-phenothiazine conjugates as simultaneous NMDA receptor blockers and cholinesterase inhibitors. Bioorganic Med. Chem. 2021, 46, 116355. [Google Scholar] [CrossRef]
- Mahajan, S.; Choudhary, S.; Kumar, P.; Tomar, S. Antiviral strategies targeting host factors and mechanisms obliging +ssRNA viral pathogens. Bioorganic Med. Chem. 2021, 46, 116356. [Google Scholar] [CrossRef]
- Yudi Utomo, R.; Asawa, Y.; Okada, S.; Ban, H.S.; Yoshimori, A.; Bajorath, J.; Nakamura, H. Development of curcumin-based amyloid beta aggregation inhibitors for Alzheimer’s disease using the SAR matrix approach. Bioorganic Med. Chem. 2021, 46, 116357. [Google Scholar] [CrossRef]
- Liu, X.J.; Xu, L.; Pang, X.J.; Ying Yuan, X.; Yu, G.X.; Li, Y.R.; Guan, Y.F.; Zhang, Y.B.; Song, J.; Zhang, Q.R.; et al. Progress in the development of small molecular inhibitors of the Bruton’s tyrosine kinase (BTK) as a promising cancer therapy. Bioorganic Med. Chem. 2021, 47, 116358. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.T.; Bame, E.; Bajrami, B.; Black, C.; Bohnert, T.; Boiselle, C.; Burdette, D.; Burns, J.C.; Delva, L.; Donaldson, D.; et al. Discovery and Preclinical Characterization of BIIB091, a Reversible, Selective BTK Inhibitor for the Treatment of Multiple Sclerosis. J. Med. Chem. 2022, 65, 1206–1224. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ren, R.; Sun, X.; Zou, Y.; Shi, Y.; Di, B.; Niu, M.M. Discovery of a Dual Tubulin and Poly(ADP-Ribose) Polymerase-1 Inhibitor by Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, and Biological Evaluation. J. Med. Chem. 2021, 64, 15702–15715. [Google Scholar] [CrossRef]
- Cui, H.; Carlson, A.S.; Schleiff, M.A.; Divakaran, A.; Johnson, J.A.; Buchholz, C.R.; Zahid, H.; Vail, N.R.; Shi, K.; Aihara, H.; et al. 4-Methyl-1,2,3-Triazoles as N-Acetyl-Lysine Mimics Afford Potent BET Bromodomain Inhibitors with Improved Selectivity. J. Med. Chem. 2021, 64, 10497–10511. [Google Scholar] [CrossRef]
- Luo, W.; Huang, Z.; Xu, D.; Yang, M.; Zhu, Y.; Shen, L.; Chen, S.; Tao, X.; Bin, W.; Hu, Y.; et al. Discovery and preclinical evaluations of JBD0131, a novel nitrodihydro-imidazooxazole anti-tuberculosis agent. Bioorganic Med. Chem. Lett. 2022, 72, 128871. [Google Scholar] [CrossRef]
- Mykhailenko, O.; Bezruk, I.; Ivanauskas, L.; Georgiyants, V. Comparative analysis of apocarotenoids and phenolic constituents of Crocus sativus stigmas from 11 countries: Ecological impact. Arch. Der Pharm. 2022, 355, e2100468. [Google Scholar] [CrossRef]
- Wang, R. Current perspectives on naturally occurring saponins as anticancer agents. Arch. Der Pharm. 2022, 355, e2100469. [Google Scholar] [CrossRef]
- Maher, M.; Zaher, A.F.; Mahmoud, Z.; Kassab, A.E. Recent green approaches for the synthesis of pyrazolo[3,4-d]pyrimidines: A mini review. Arch. Der Pharm. 2022, 355, e2100470. [Google Scholar] [CrossRef]
- Aggul, A.G.; Uzun, N.; Kuzu, M.; Taslimi, P.; Gulcin, I. Some phenolic natural compounds as carbonic anhydrase inhibitors: An in vitro and in silico study. Arch. Der Pharm. 2022, 355, e2100476. [Google Scholar] [CrossRef] [PubMed]
- Solangi, M.; Khan, K.M.; Chigurupati, S.; Saleem, F.; Qureshi, U.; Ul-Haq, Z.; Jabeen, A.; Felemban, S.G.; Zafar, F.; Perveen, S.; et al. Isatin thiazoles as antidiabetic: Synthesis, in vitro enzyme inhibitory activities, kinetics, and in silico studies. Arch. Der Pharm. 2022, 355, e2100481. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the PROTAC | Clinical Trial No. | Date of Entry into Clinical Trials | Highest Clinical Phase | Target Protein | E3 Ligase Used |
|---|---|---|---|---|---|
| ARV-110 | NCT03888612 | March, 2019 | Phase II | AR | Cereblon |
| CC-94676 | NCT04428788 | June, 2020 | Phase I | AR | _ |
| ARV-766 | NCT05067140 | October, 2021 | Phase I | AR | Cereblon |
| Parameter | SMIs | PROTACs |
|---|---|---|
| Druggability | Only limited targets are druggable | Extended scope for druggable targets |
| Selectivity | Lack of selectivity | Highly selective |
| Dosage | Require high dose; thus they are less potent | Minimal dose and highly protein |
| MOA | Competitive active site inhibition | Proteasomal degradation of target protein |
| Drug–target interactions |
|
|
| Lipinski RO5 | Most of them obeys RO5 | Most of the PROTACs do not obey Lipinski rules |
| Synthetic feasibility | Usually easy to synthesize | Usually strenuous to synthesize |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yedla, P.; Babalghith, A.O.; Andra, V.V.; Syed, R. PROTACs in the Management of Prostate Cancer. Molecules 2023, 28, 3698. https://doi.org/10.3390/molecules28093698
Yedla P, Babalghith AO, Andra VV, Syed R. PROTACs in the Management of Prostate Cancer. Molecules. 2023; 28(9):3698. https://doi.org/10.3390/molecules28093698
Chicago/Turabian StyleYedla, Poornachandra, Ahmed O. Babalghith, Vindhya Vasini Andra, and Riyaz Syed. 2023. "PROTACs in the Management of Prostate Cancer" Molecules 28, no. 9: 3698. https://doi.org/10.3390/molecules28093698
APA StyleYedla, P., Babalghith, A. O., Andra, V. V., & Syed, R. (2023). PROTACs in the Management of Prostate Cancer. Molecules, 28(9), 3698. https://doi.org/10.3390/molecules28093698