
A Short Review of Research Progress on the Synthesis Approaches of Aza-Dibenzocyclooctyne Derivatives

Abstract
:1. Introduction
2. Synthesis Approaches for DIBAC
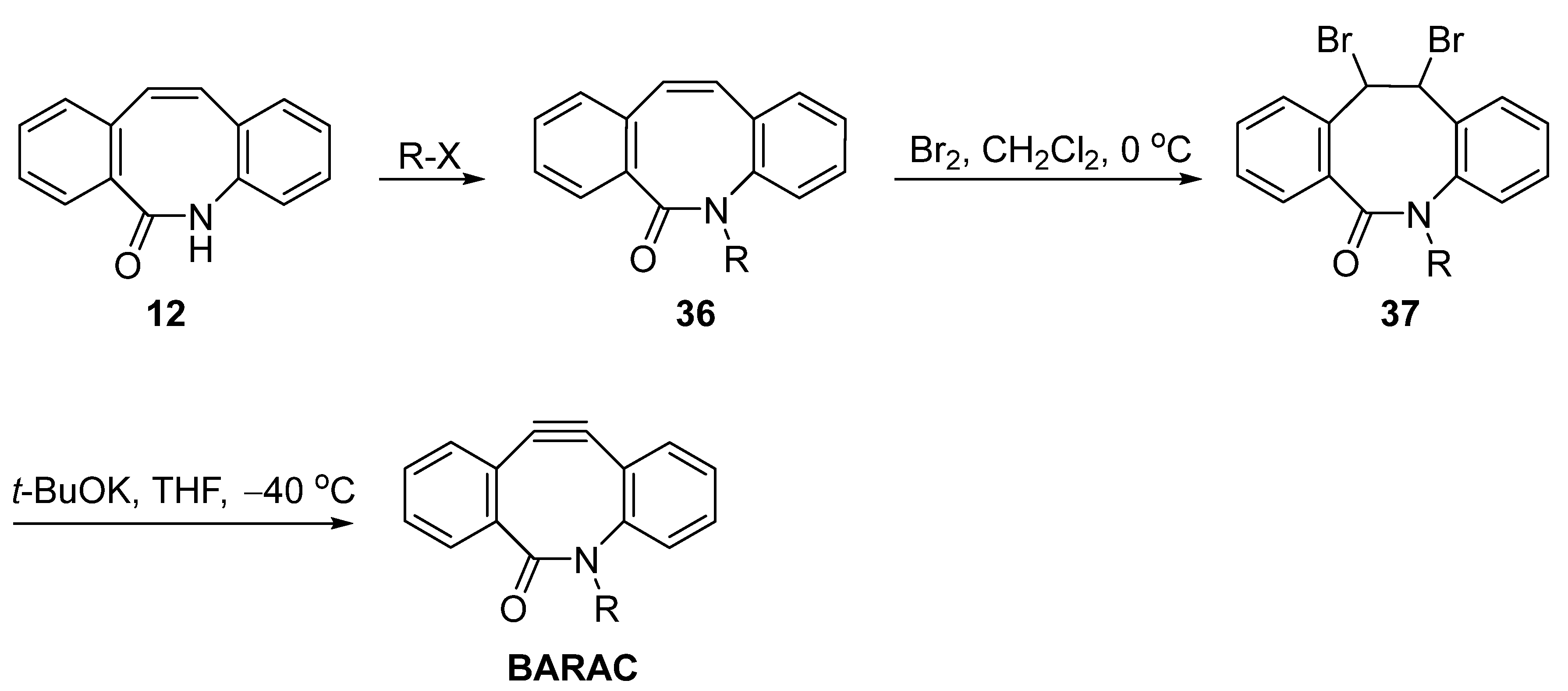
3. Synthesis Approaches for BARAC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Bertozzi, C.R. A Decade of Bioorthogonal Chemistry. Acc. Chem. Res. 2011, 44, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Li, J.; Chen, P.R. Bioorthogonal chemistry in living animals. Natl. Sci. Rev. 2017, 4, 300–302. [Google Scholar] [CrossRef]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Chen, P.R. Unnatural Amino Acid Mediated Protein Bioorthogonal Labeling. Huaxue Xuebao 2012, 70, 1439. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified Staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- Debets, M.F.; van Berkel, S.S.; Schoffelen, S.; Rutjes, F.P.J.T.; van Hest, J.C.M.; van Delft, F.L. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 2010, 46, 97–99. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Versteegen, R.M.; Rossin, R.; ten Hoeve, W.; Janssen, H.M.; Robillard, M.S. Click to release: Instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem. Int. Ed. Engl. 2013, 52, 14112–14116. [Google Scholar] [CrossRef]
- Li, J.; Jia, S.; Chen, P.R. Diels-Alder reaction-triggered bioorthogonal protein decaging in living cells. Nat. Chem. Biol. 2014, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, D.A.; Sherratt, A.R.; Chigrinova, M.; Cheung, L.L.W.; Pezacki, J.P. Strain-promoted cycloadditions involving nitrones and alkynes—Rapid tunable reactions for bioorthogonal labeling. Curr. Opin. Chem. Biol. 2014, 21, 81–88. [Google Scholar] [CrossRef]
- Ramil, C.P.; Lin, Q. Photoclick chemistry: A fluorogenic light-triggered in vivo ligation reaction. Curr. Opin. Chem. Biol. 2014, 21, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-W.; Kamber, D.N.; Prescher, J.A. Building better bioorthogonal reactions. Curr. Opin. Chem. Biol. 2014, 21, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef]
- Nguyen, S.S.; Prescher, J.A. Developing bioorthogonal probes to span a spectrum of reactivities. Nat. Rev. Chem. 2020, 4, 476–489. [Google Scholar] [CrossRef]
- Murtagh, J.; Frimannsson, D.O.; O’Shea, D.F. Azide conjugatable and pH responsive near-infrared fluorescent imaging probes. Org. Lett. 2009, 11, 5386–5389. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, Y.; Shi, H.; Hu, Y.; Feng, L.; Luo, Z.; Zhou, M.; He, J.; Zhou, Z.; Zhang, Y.; et al. Redox-mediated disassembly to build activatable trimodal probe for molecular imaging of biothiols. ACS Nano 2016, 10, 10075–10085. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. 1, 3-dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Breugst, M.; Reissig, H.-U. The Huisgen Reaction: Milestones of the 1, 3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. Engl. 2020, 59, 12293–12307. [Google Scholar] [CrossRef]
- Padwa, A.; Pearson, W.H. (Eds.) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products; Wiley-Interscience: New York, NY, USA, 2003. [Google Scholar]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper (I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Wittig, G.; Krebs, A. Zur Existenz niedergliedriger cycloalkine, I. Chem. Ber. 1961, 94, 3260–3275. [Google Scholar] [CrossRef]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kang, D.W.; Leucke, H.F.; Bond, M.R.; Ghosh, S.; Love, D.C.; Ahn, J.-S.; Kang, D.-O.; Hanover, J.A. Optimizing the selectivity of DIFO-based reagents for intracellular bioorthogonal application. Carbohydr. Res. 2013, 377, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Mbua, N.E.; Guo, J.; Wolfert, M.A.; Steet, R.; Boons, G.-J. Strain-promoted alkyne–azide cycloadditions (SPAAC) reveal new features of glycoconjugate biosynthesis. ChemBioChem 2011, 12, 1912–1921. [Google Scholar] [CrossRef]
- Friscourt, F.; Ledin, P.A.; Mbua, N.E.; Flanagan-Steet, H.R.; Wolfert, M.A.; Steet, R.; Boons, G.-J. Polar dibenzocyclooctynes for selective labeling of extracellular glycoconjugates of living cells. J. Am. Chem. Soc. 2012, 134, 5381–5389. [Google Scholar] [CrossRef]
- Ning, X.; Guo, J.; Wolfert, M.A.; Boons, G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. Engl. 2008, 47, 2253–2255. [Google Scholar] [CrossRef]
- Debets, M.F.; Prins, J.S.; Merkx, D.; van Berkel, S.S.; van Delft, F.L.; van Hest, J.C.M.; Rutjes, F.P.J.T. Synthesis of DIBAC analogues with excellent SPAAC rate constants. Org. Biomol. Chem. 2014, 12, 5031–5037. [Google Scholar] [CrossRef]
- Jewett, J.C.; Sletten, E.M.; Bertozzi, C.R. Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 2010, 132, 3688–3690. [Google Scholar] [CrossRef]
- Kuzmin, A.; Poloukhtine, A.; Wolfert, M.A.; Popik, V.V. Surface functionalization using catalyst-free azide–alkyne cycloaddition. Bioconjug. Chem. 2010, 21, 2076–2085. [Google Scholar] [CrossRef]
- Adronov, A.; Chadwick, R.; Van Gyzen, S.; Liogier, S. Scalable synthesis of strained cyclooctyne derivatives. Synthesis 2014, 46, 669–677. [Google Scholar] [CrossRef]
- Eaton, P.E.; Carlson, G.R.; Lee, J.T. Phosphorus pentoxide-methanesulfonic acid. Convenient alternative to polyphosphoric acid. J. Org. Chem. 1973, 38, 4071–4073. [Google Scholar] [CrossRef]
- Jäger, M.; Görls, H.; Günther, W.; Schubert, U.S. Pd-Catalyzed Ring Assembly by Vinylation and Intramolecular Heck Coupling: A Versatile Strategy Towards Functionalized Azadibenzocyclooctynes. Chem.-Eur. J. 2013, 19, 2150–2157. [Google Scholar] [PubMed]
- Slugovc, C.; Perner, B.; Stelzer, F.; Mereiter, K. “Second Generation” Ruthenium Carbene Complexes with a cis-Dichloro Arrangement. Organometallics 2004, 23, 3622–3626. [Google Scholar] [CrossRef]
- Denmark, S.E.; Butler, C.R. Vinylation of aryl bromides using an inexpensive vinylpolysiloxane. Org. Lett. 2006, 8, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Starke, F.; Walther, M.; Pietzsch, H.-J. A Novel Dibenzoazacyclooctyne Precursor in Regioselective Copper-Free Click Chemistry. Arkivoc 2010, 2010, 350–359. [Google Scholar] [CrossRef]
- Sakata, Y.; Nabekura, R.; Hazama, Y.; Hany, M.; Nishiyama, T.; Kii, I.; Hosoya, T. Synthesis of Functionalized Dibenzoazacyclooctynes by a Decomplexation Method for Dibenzo-Fused Cyclooctyne–Cobalt Complexes. Org. Lett. 2023, 25, 1051–1055. [Google Scholar] [CrossRef]
- Hioki, Y.; Itoh, M.; Mori, A.; Okano, K. One-Pot Deprotonative Synthesis of Biarylazacyclooctynones. Synlett 2020, 31, 189–193. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DBCO Compound | Solvent | k (10−3 M−1 s−1) |
|---|---|---|
| OCT | CD3CN | 2.4 |
| DIFO DIBO | CD3CN CH3OH | 76 57 |
| DIBAC BARAC | CD3OD CD3CN | 310 960 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Liu, L.; Cheng, L. A Short Review of Research Progress on the Synthesis Approaches of Aza-Dibenzocyclooctyne Derivatives. Molecules 2023, 28, 3715. https://doi.org/10.3390/molecules28093715
He Y, Liu L, Cheng L. A Short Review of Research Progress on the Synthesis Approaches of Aza-Dibenzocyclooctyne Derivatives. Molecules. 2023; 28(9):3715. https://doi.org/10.3390/molecules28093715
Chicago/Turabian StyleHe, Yinming, Li Liu, and Liang Cheng. 2023. "A Short Review of Research Progress on the Synthesis Approaches of Aza-Dibenzocyclooctyne Derivatives" Molecules 28, no. 9: 3715. https://doi.org/10.3390/molecules28093715