
Ethanol Coupling Reactions over MgO–Al2O3 Mixed Oxide-Based Catalysts for Producing Biofuel Additives
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Results
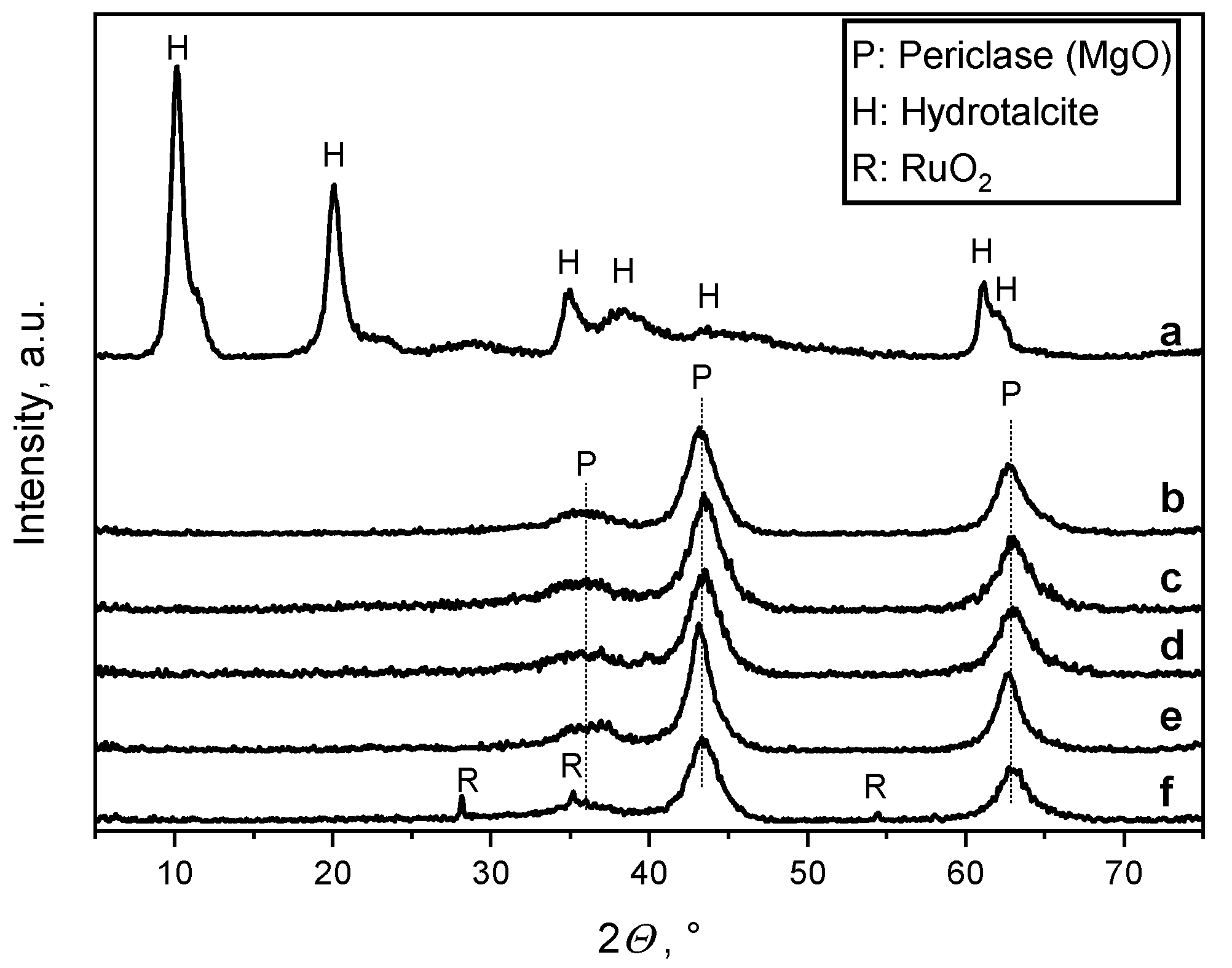
2.1. Catalyst Structure
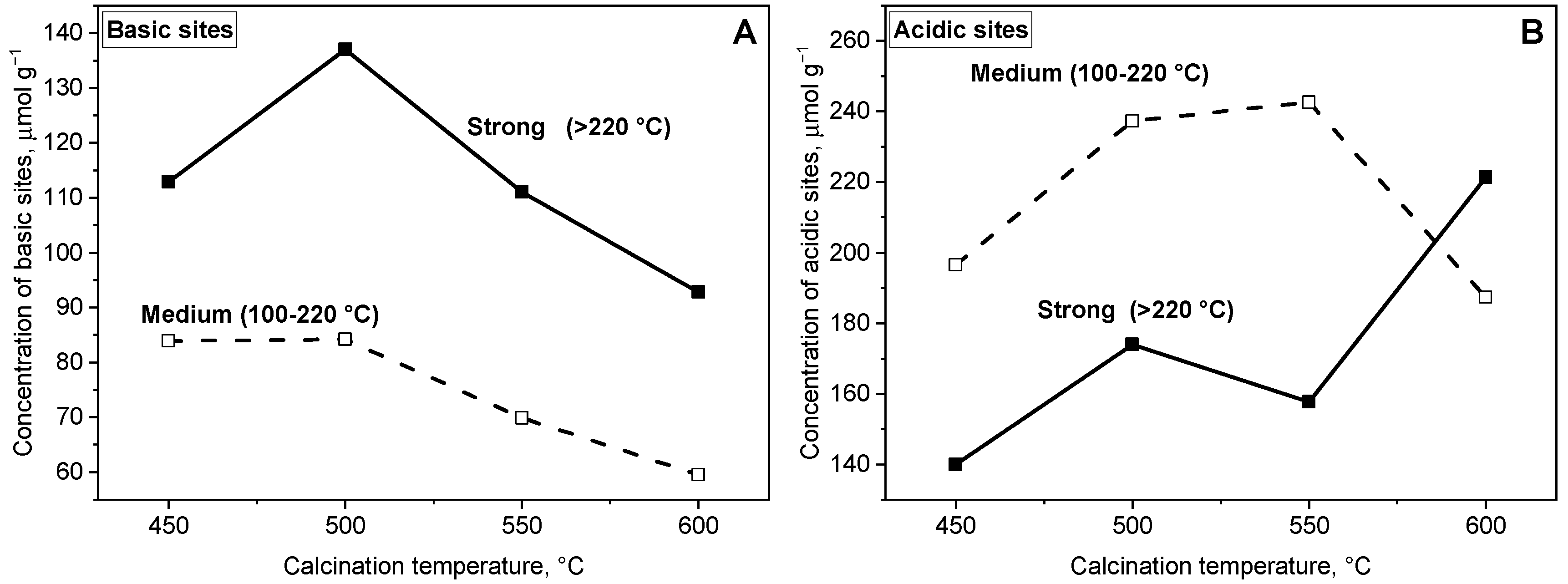
2.2. Acid-Base Properties
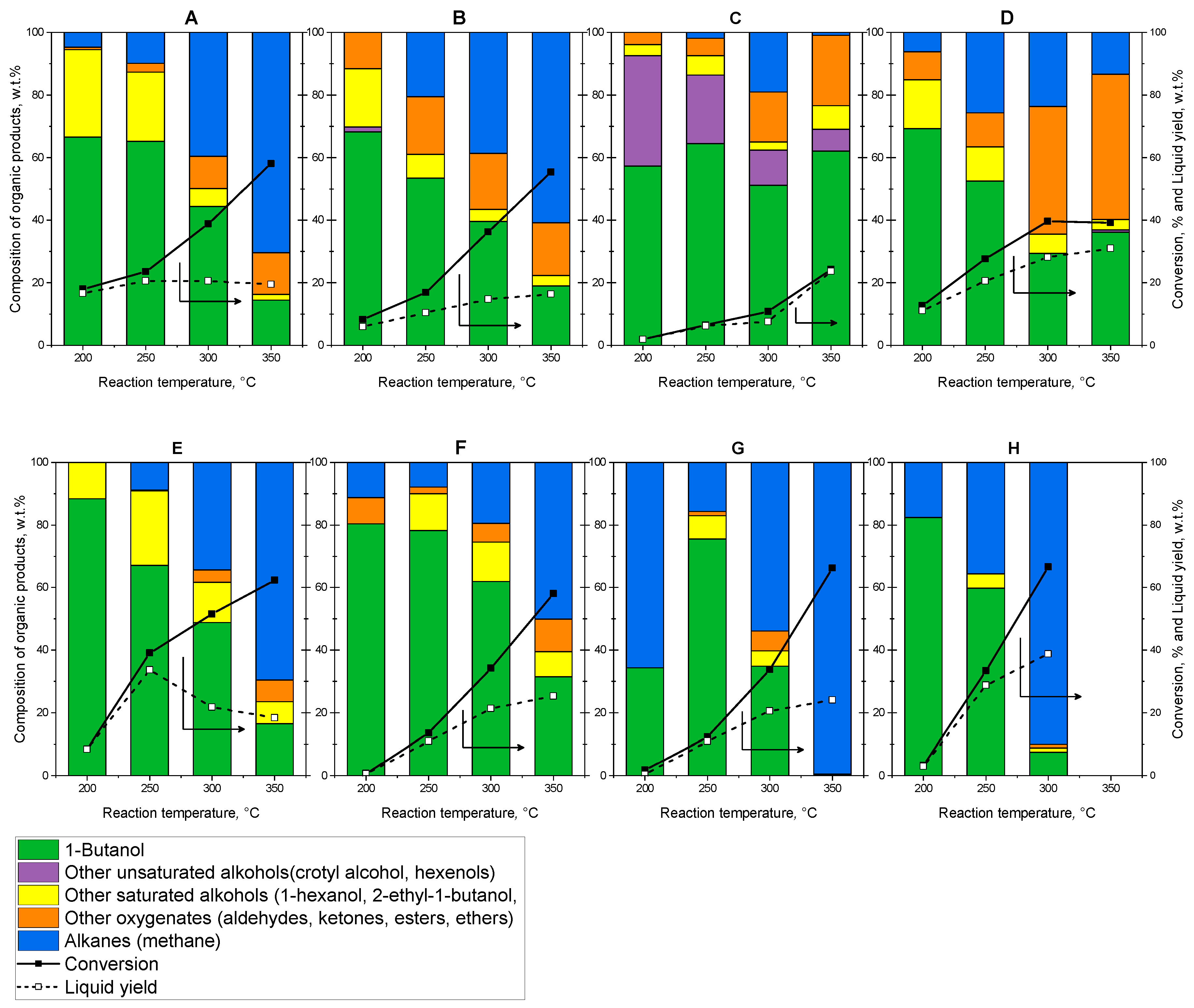
2.3. Catalytic Properties
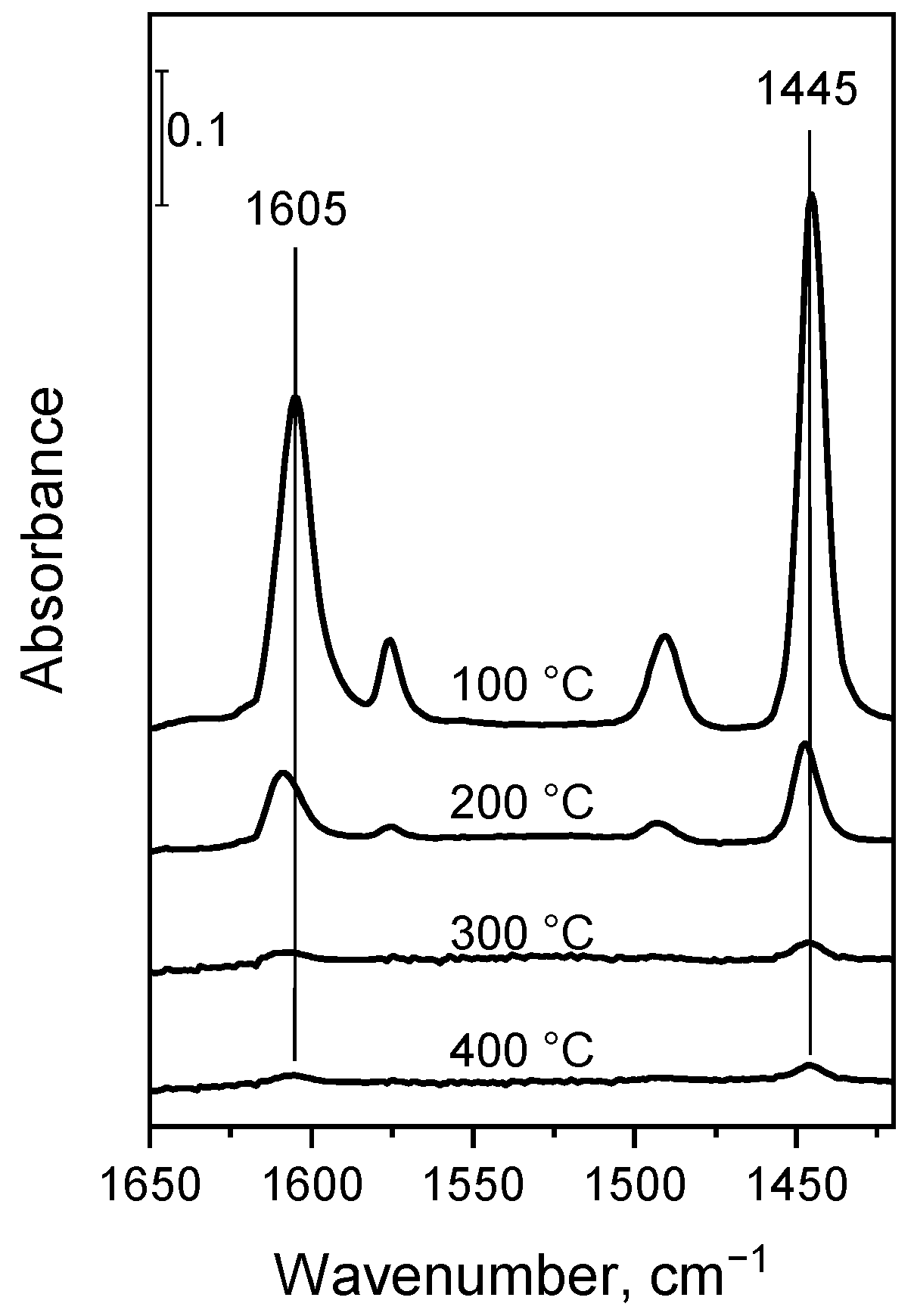
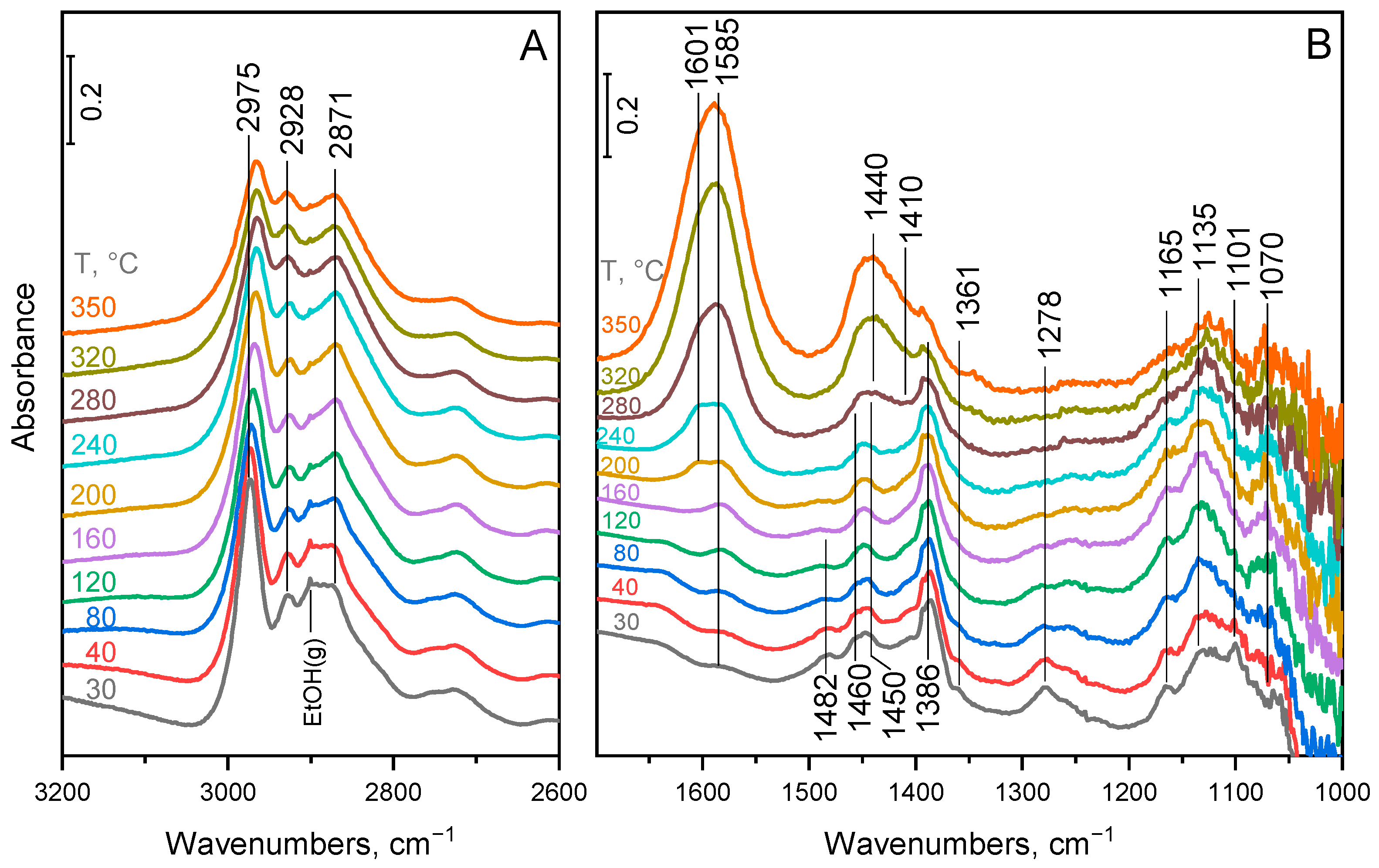
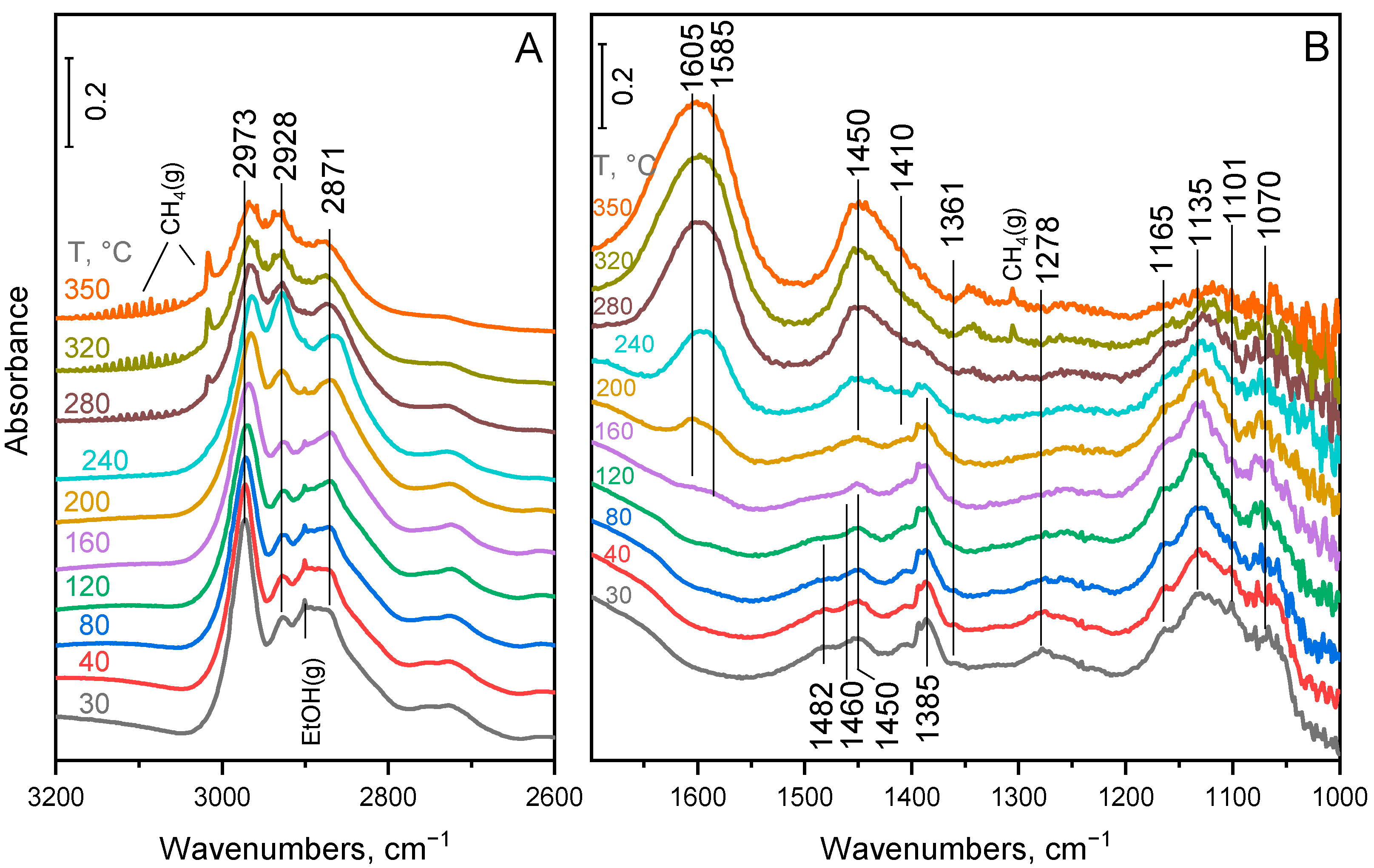
2.4. Surface Species on MgO–Al2O3 and Pd/MgO–Al2O3 Mixed Oxide Catalysts
3. Discussion
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Catalysts Characterization
4.2.1. X-ray Powder Diffraction Measurements
4.2.2. Nitrogen Adsorption Isotherms
4.2.3. CO Pulse Chemisorption
4.2.4. Temperature-Programmed Desorption of CO2 and NH3
4.2.5. Pyridine Adsorption
4.3. Catalytic Tests
4.4. Quasi-Operando DRIFT Spectroscopic Investigations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dagle, R.A.; Winkelman, A.D.; Ramasamy, K.K.; Lebarbier Dagle, V.; Weber, R.S. Ethanol as a Renewable Building Block for Fuels and Chemicals. Ind. Eng. Chem. Res. 2020, 59, 4843–4853. [Google Scholar] [CrossRef]
- Gabriëls, D.; Hernández, W.Y.; Sels, B.; Van Der Voort, P.; Verberckmoes, A. Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization. Catal. Sci. Technol. 2015, 5, 3876–3902. [Google Scholar] [CrossRef]
- Wu, X.; Fang, G.; Tong, Y.; Jiang, D.; Liang, Z.; Leng, W.; Liu, L.; Tu, P.; Wang, H.; Ni, J.; et al. Catalytic Upgrading of Ethanol to n-Butanol: Progress in Catalyst Development. ChemSusChem 2018, 11, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, J.T.; Davis, R.J. Heterogeneous Catalysts for the Guerbet Coupling of Alcohols. ACS Catal. 2013, 3, 1588–1600. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Apesteguí, C.R.; Ginés, M.J.L.; Iglesia, E. Structural Requirements and Reaction Pathways in Condensation Reactions of Alcohols on MgyAlOx Catalysts. J. Catal. 2000, 190, 261–275. [Google Scholar] [CrossRef]
- Moteki, T.; Flaherty, D.W. Mechanistic Insight to C–C Bond Formation and Predictive Models for Cascade Reactions among Alcohols on Ca- and Sr-Hydroxyapatites. ACS Catal. 2016, 6, 4170–4183. [Google Scholar] [CrossRef]
- Kibby, C. Dehydrogenation of alcohols and hydrogen transfer from alcohols to ketones over hydroxyapatite catalysts. J. Catal. 1973, 31, 65–73. [Google Scholar] [CrossRef]
- Ho, C.R.; Shylesh, S.; Bell, A.T. Mechanism and Kinetics of Ethanol Coupling to Butanol over Hydroxyapatite. ACS Catal. 2016, 6, 939–948. [Google Scholar] [CrossRef]
- Ndou, A. Dimerisation of ethanol to butanol over solid-base catalysts. Appl. Catal. A Gen. 2003, 251, 337–345. [Google Scholar] [CrossRef]
- Scalbert, J.; Thibault-Starzyk, F.; Jacquot, R.; Morvan, D.; Meunier, F. Ethanol condensation to butanol at high temperatures over a basic heterogeneous catalyst: How relevant is acetaldehyde self-aldolization? J. Catal. 2014, 311, 28–32. [Google Scholar] [CrossRef]
- Yang, C.; Meng, Z.Y. Bimolecular Condensation of Ethanol to 1-Butanol Catalyzed by Alkali Cation Zeolites. J. Catal. 1993, 142, 37–44. [Google Scholar] [CrossRef]
- Meunier, F.C.; Scalbert, J.; Thibault-Starzyk, F. Unraveling the mechanism of chemical reactions through thermodynamic analyses: A short review. Appl. Catal. A Gen. 2015, 504, 220–227. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Díez, V.K.; Xu, M.; Iglesia, E.; Apesteguía, C.R. Structure and Surface and Catalytic Properties of Mg-Al Basic Oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef]
- Shen, J.; Tu, M.; Hu, C. Structural and Surface Acid/Base Properties of Hydrotalcite-Derived MgAlO Oxides Calcined at Varying Temperatures. J. Solid State Chem. 1998, 137, 295–301. [Google Scholar] [CrossRef]
- Kuśtrowski, P.; Sułkowska, D.; Chmielarz, L.; Rafalska-Łasocha, A.; Dudek, B.; Dziembaj, R. Influence of thermal treatment conditions on the activity of hydrotalcite-derived Mg–Al oxides in the aldol condensation of acetone. Microporous Mesoporous Mater. 2005, 78, 11–22. [Google Scholar] [CrossRef]
- Ramasamy, K.K.; Gray, M.; Job, H.; Santosa, D.; Li, X.S.; Devaraj, A.; Karkamkar, A.; Wang, Y. Role of Calcination Temperature on the Hydrotalcite Derived MgO–Al2O3 in Converting Ethanol to Butanol. Top. Catal. 2016, 59, 46–54. [Google Scholar] [CrossRef]
- León, M.; Díaz, E.; Ordóñez, S. Ethanol catalytic condensation over Mg–Al mixed oxides derived from hydrotalcites. Catal. Today 2011, 164, 436–442. [Google Scholar] [CrossRef]
- Carvalho, D.L.; Borges, L.E.P.; Appel, L.G.; Ramírez de la Piscina, P.; Homs, N. In situ infrared spectroscopic study of the reaction pathway of the direct synthesis of n-butanol from ethanol over MgAl mixed-oxide catalysts. Catal. Today 2013, 213, 115–121. [Google Scholar] [CrossRef]
- Metzker, G.; Mora Vargas, J.A.; de Lima, L.P.; Perrone, O.M.; Siqueira, M.R.; Varanda, L.C.; Boscolo, M. First row transition metals on the ethanol Guerbet reaction: Products distribution and structural behavior of mixed metal oxides as catalysts. Appl. Catal. A Gen. 2021, 623, 118272. [Google Scholar] [CrossRef]
- Ramasamy, K.K.; Gray, M.; Job, H.; Smith, C.; Wang, Y. Tunable catalytic properties of bi-functional mixed oxides in ethanol conversion to high value compounds. Catal. Today 2016, 269, 82–87. [Google Scholar] [CrossRef]
- Scheid, A.J.; Barbosa-Coutinho, E.; Schwaab, M.; Salau, N.P.G. Mechanism and Kinetic Modeling of Ethanol Conversion to 1-Butanol over Mg and Al Oxide Derived from Hydrotalcites. Ind. Eng. Chem. Res. 2019, 58, 12981–12995. [Google Scholar] [CrossRef]
- Quesada, J.; Faba, L.; Díaz, E.; Ordóñez, S. Role of the surface intermediates in the stability of basic mixed oxides as catalyst for ethanol condensation. Appl. Catal. A Gen. 2017, 542, 271–281. [Google Scholar] [CrossRef]
- Foger, K. Dispersed Metal Catalysts. In Catalysis: Science and Technology; Springer: Berlin/Heidelberg, Germany, 1984; Volume 6, pp. 227–305. [Google Scholar]
- Bergeret, G.; Gallezot, P. Particle Size and Dispersion Measurements. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 738–765. [Google Scholar]
- Rynkowski, J.M.; Paryjczak, T.; Lenik, M. On the nature of oxidic nickel phases in NiO/γ-Al2O3 catalysts. Appl. Catal. A Gen. 1993, 106, 73–82. [Google Scholar] [CrossRef]
- Busca, G. The surface of transitional aluminas: A critical review. Catal. Today 2014, 226, 2–13. [Google Scholar] [CrossRef]
- Larkin, P. IR and Raman Spectra-Structure Correlations. In Infrared and Raman Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2011; pp. 73–115. [Google Scholar]
- Panchenko, V.N.; Paukshtis, E.A.; Murzin, D.Y.; Simakova, I.L. Solid Base Assisted n -Pentanol Coupling over VIII Group Metals: Elucidation of the Guerbet Reaction Mechanism by DRIFTS. Ind. Eng. Chem. Res. 2017, 56, 13310–13321. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; An, Z.; Zhu, Y.; Shu, X.; Song, H.; Xiang, X.; He, J. Acid–Base Promoted Dehydrogenation Coupling of Ethanol on Supported Ag Particles. Ind. Eng. Chem. Res. 2020, 59, 3342–3350. [Google Scholar] [CrossRef]
- Podobiński, J.; Śliwa, M.; Datka, J. Ethoxy Groups on ZrO2, CuO, and CuO/ZrO2 Studied by IR Spectroscopy. Molecules 2022, 27, 4790. [Google Scholar] [CrossRef]
- Molina-Ramírez, S.; Cortés-Reyes, M.; Herrera, C.; Larrubia, M.A.; Alemany, L.J. Catalytic upgrading of ethanol to n-butanol over a novel Ca-Fe modified mixed oxide Mg-Al catalyst from hydrotalcite-base precursor. Catal. Today 2022, 394–396, 365–375. [Google Scholar] [CrossRef]
- Vecchietti, J.; Pérez-Bailac, P.; Lustemberg, P.G.; Fornero, E.L.; Pascual, L.; Bosco, M.V.; Martínez-Arias, A.; Ganduglia-Pirovano, M.V.; Bonivardi, A.L. Shape-Controlled Pathways in the Hydrogen Production from Ethanol Steam Reforming over Ceria Nanoparticles. ACS Catal. 2022, 12, 10482–10498. [Google Scholar] [CrossRef]
- de Souza, E.F.; Pacheco, H.P.; Miyake, N.; Davis, R.J.; Toniolo, F.S. Computational and Experimental Mechanistic Insights into the Ethanol-to-Butanol Upgrading Reaction over MgO. ACS Catal. 2020, 10, 15162–15177. [Google Scholar] [CrossRef]
- Yuan, B.; Zhang, J.; An, Z.; Zhu, Y.; Shu, X.; Song, H.; Xiang, X.; Wang, W.; Jing, Y.; Zheng, L.; et al. Atomic Ru catalysis for ethanol coupling to C4+ alcohols. Appl. Catal. B Environ. 2022, 309, 121271. [Google Scholar] [CrossRef]
- van den Brand, J.; Blajiev, O.; Beentjes, P.C.J.; Terryn, H.; de Wit, J.H.W. Interaction of Anhydride and Carboxylic Acid Compounds with Aluminum Oxide Surfaces Studied Using Infrared Reflection Absorption Spectroscopy. Langmuir 2004, 20, 6308–6317. [Google Scholar] [CrossRef] [PubMed]
- Milev, A.S.; Kannangara, G.S.K.; Wilson, M.A. Template-Directed Synthesis of Hydroxyapatite from a Lamellar Phosphonate Precursor. Langmuir 2004, 20, 1888–1894. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, Tuning, and Exploiting the Basicity of Hydrotalcites for Applications in Heterogeneous Catalysis. Chem. A Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Sasaki, E.; Watanabe, E.; Hyodo, S.; Nishijima, H. Development of dehydrogenation catalyst for hydrogen generation in organic chemical hydride method. Int. J. Hydrogen Energy 2006, 31, 1348–1356. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CO Adsorbed a | DMe b | v c | a c | dMe c |
|---|---|---|---|---|---|
| mmol/gcat. | % | nm3 | nm2 | nm | |
| 1% Pd/MgO–Al2O3 | 11.53 | 45.2 | 0.0147 | 0.0793 | 2.5 |
| 1% Pt/MgO–Al2O3 | 19.33 | 20.6 | 0.0151 | 0.0807 | 5.4 |
| 1% Ru/MgO–Al2O3 | 3.35 | 3.4 | 0.01365 | 0.0635 | 37.9 |
| 5% Ni/MgO–Al2O3 | 6.28 | 14.2 | 0.01095 | 0.0651 | 7.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vikár, A.; Lónyi, F.; Makoye, A.; Nagy, T.; Novodárszki, G.; Barthos, R.; Szabó, B.; Valyon, J.; Mihályi, M.R.; Deka, D.; et al. Ethanol Coupling Reactions over MgO–Al2O3 Mixed Oxide-Based Catalysts for Producing Biofuel Additives. Molecules 2023, 28, 3788. https://doi.org/10.3390/molecules28093788
Vikár A, Lónyi F, Makoye A, Nagy T, Novodárszki G, Barthos R, Szabó B, Valyon J, Mihályi MR, Deka D, et al. Ethanol Coupling Reactions over MgO–Al2O3 Mixed Oxide-Based Catalysts for Producing Biofuel Additives. Molecules. 2023; 28(9):3788. https://doi.org/10.3390/molecules28093788
Chicago/Turabian StyleVikár, Anna, Ferenc Lónyi, Amosi Makoye, Tibor Nagy, Gyula Novodárszki, Róbert Barthos, Blanka Szabó, József Valyon, Magdolna R. Mihályi, Dhanapati Deka, and et al. 2023. "Ethanol Coupling Reactions over MgO–Al2O3 Mixed Oxide-Based Catalysts for Producing Biofuel Additives" Molecules 28, no. 9: 3788. https://doi.org/10.3390/molecules28093788