



Microwave-Assisted Synthesis and Spectral Properties of Pyrrolidine-Fused Chlorin Derivatives





Abstract
:
1. Introduction
2. Results and Discussion
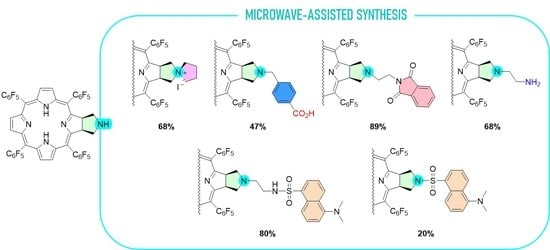
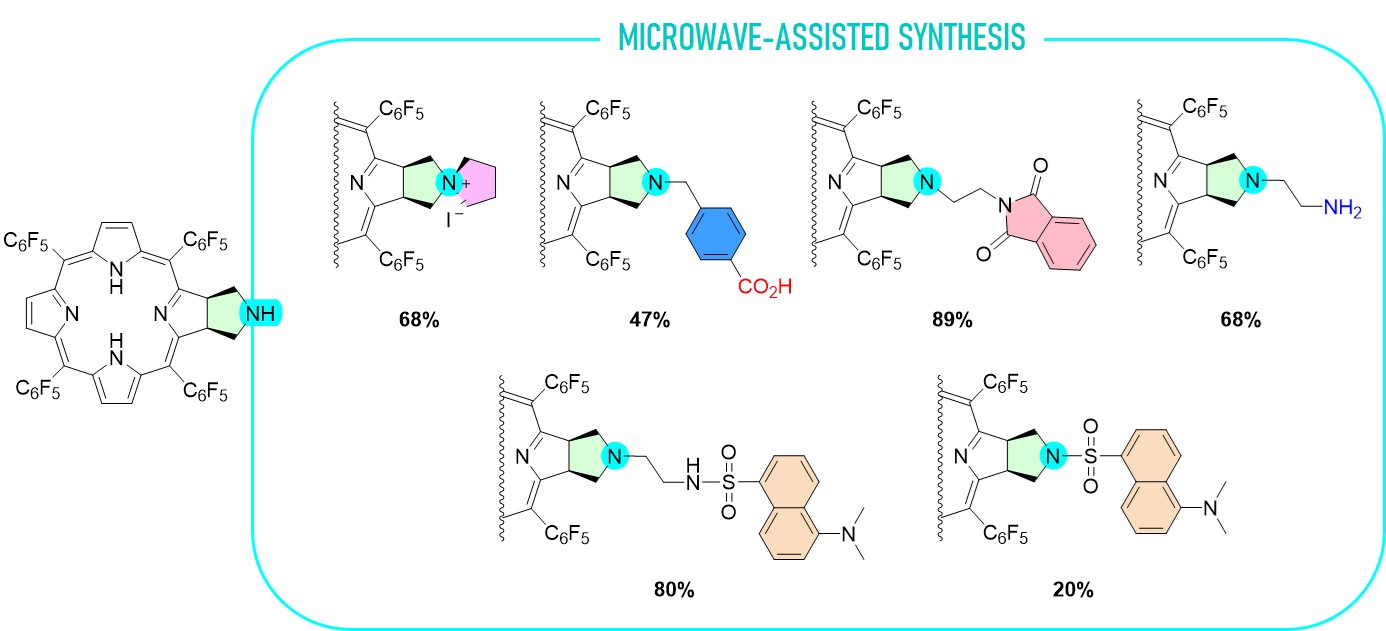
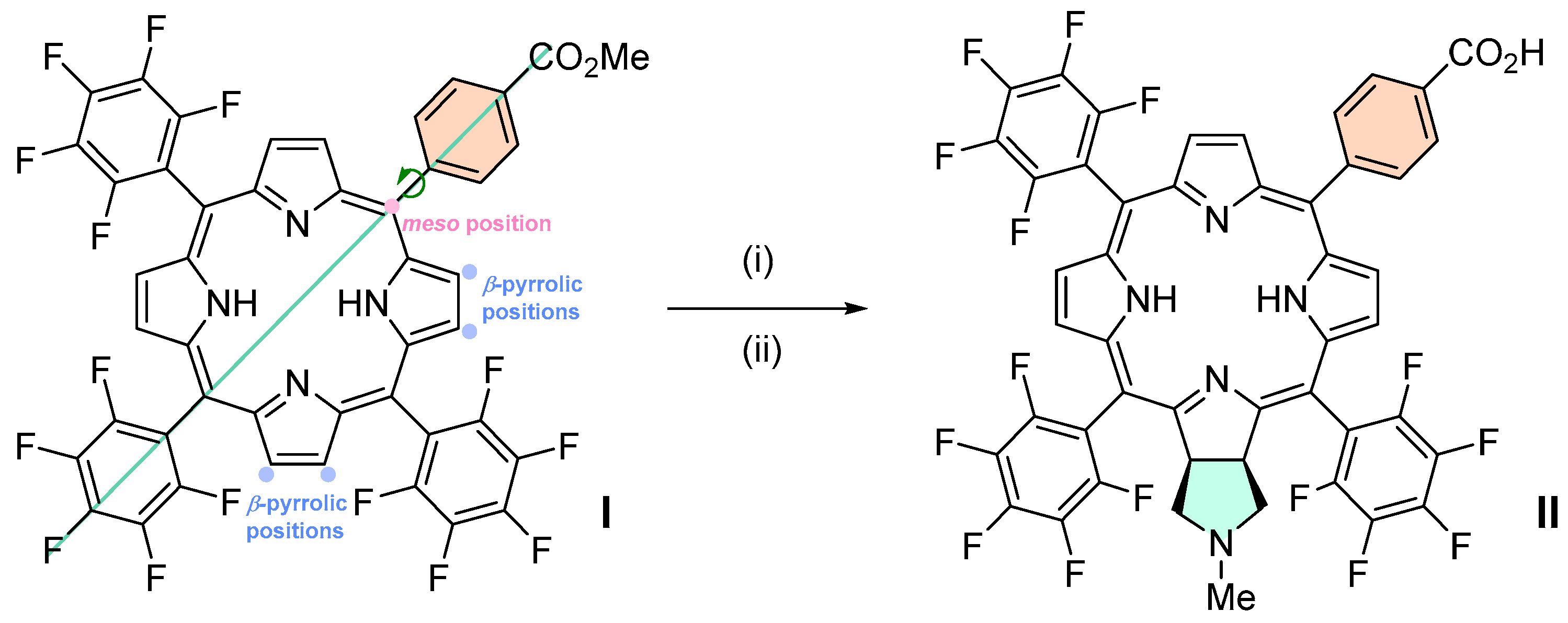
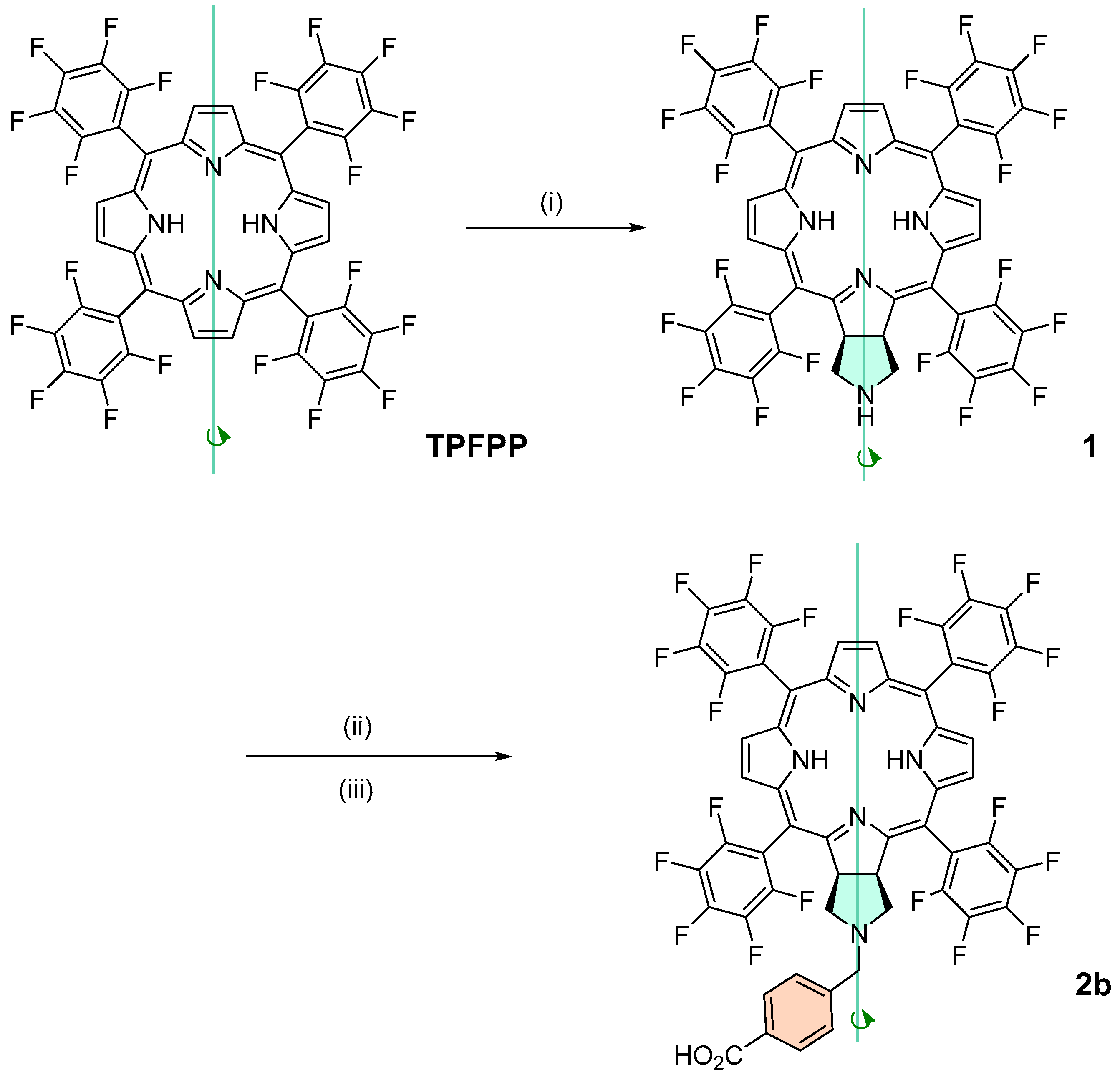
2.1. Synthesis
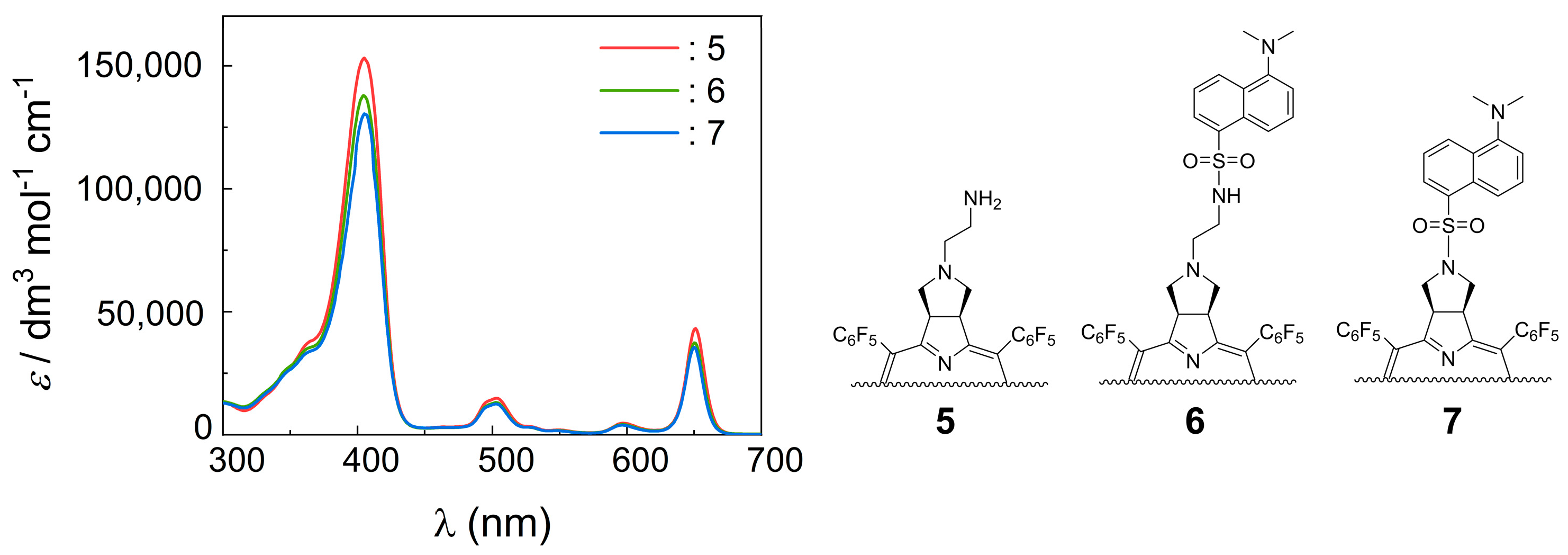
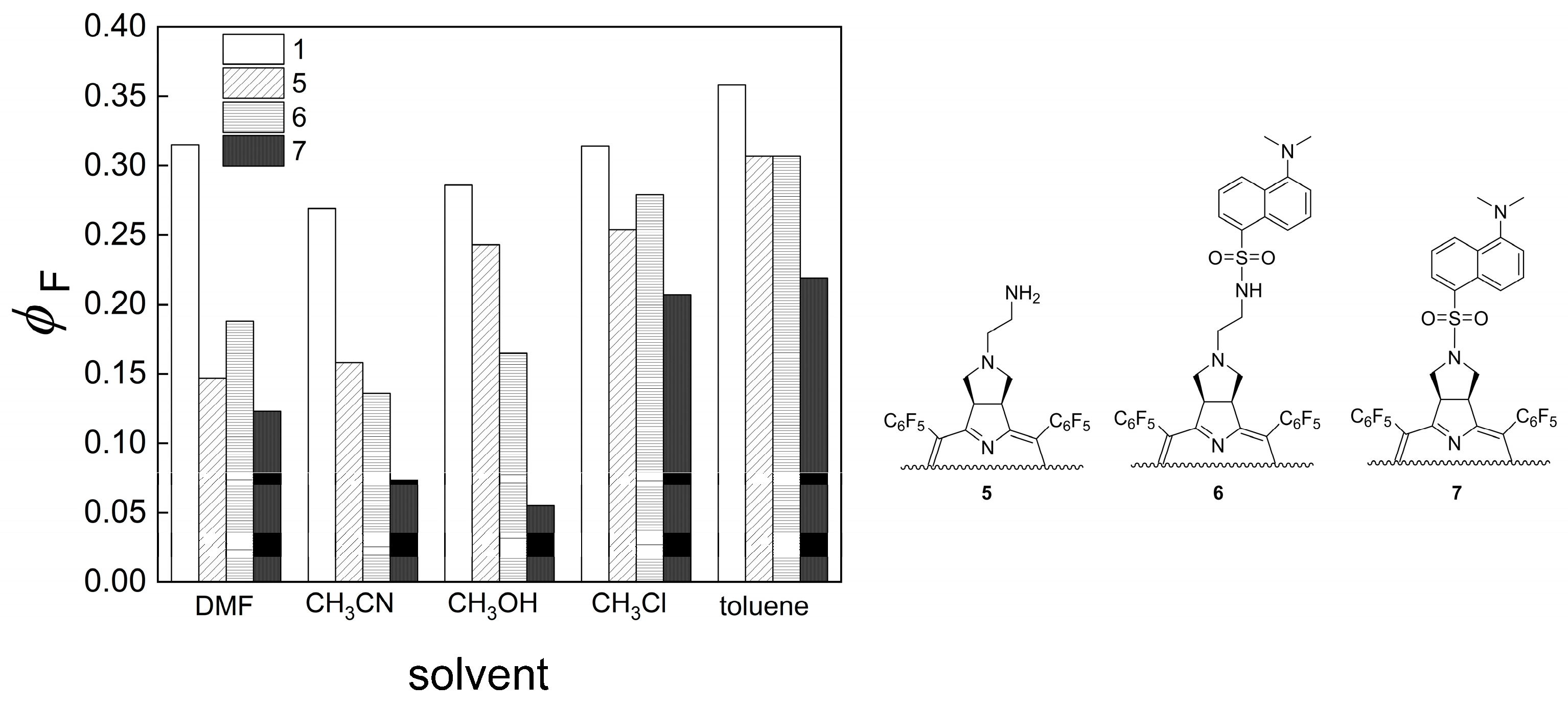
2.2. UV−Vis and Fluorescence Spectroscopy
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis
3.2.1. Synthesis of Chlorin 1 under Conventional Heating
3.2.2. Synthesis of Chlorin 1 under Microwave Heating
3.2.3. Synthesis of Chlorin Zn-1
3.2.4. Synthesis of Chlorin 2a and 2b
3.2.5. Synthesis of Chlorin 2c
3.2.6. Synthesis of Chlorin 3
3.2.7. Synthesis of Chlorin Zn-3
3.2.8. Synthesis of Chlorin 4
3.2.9. Synthesis of Chlorin 5
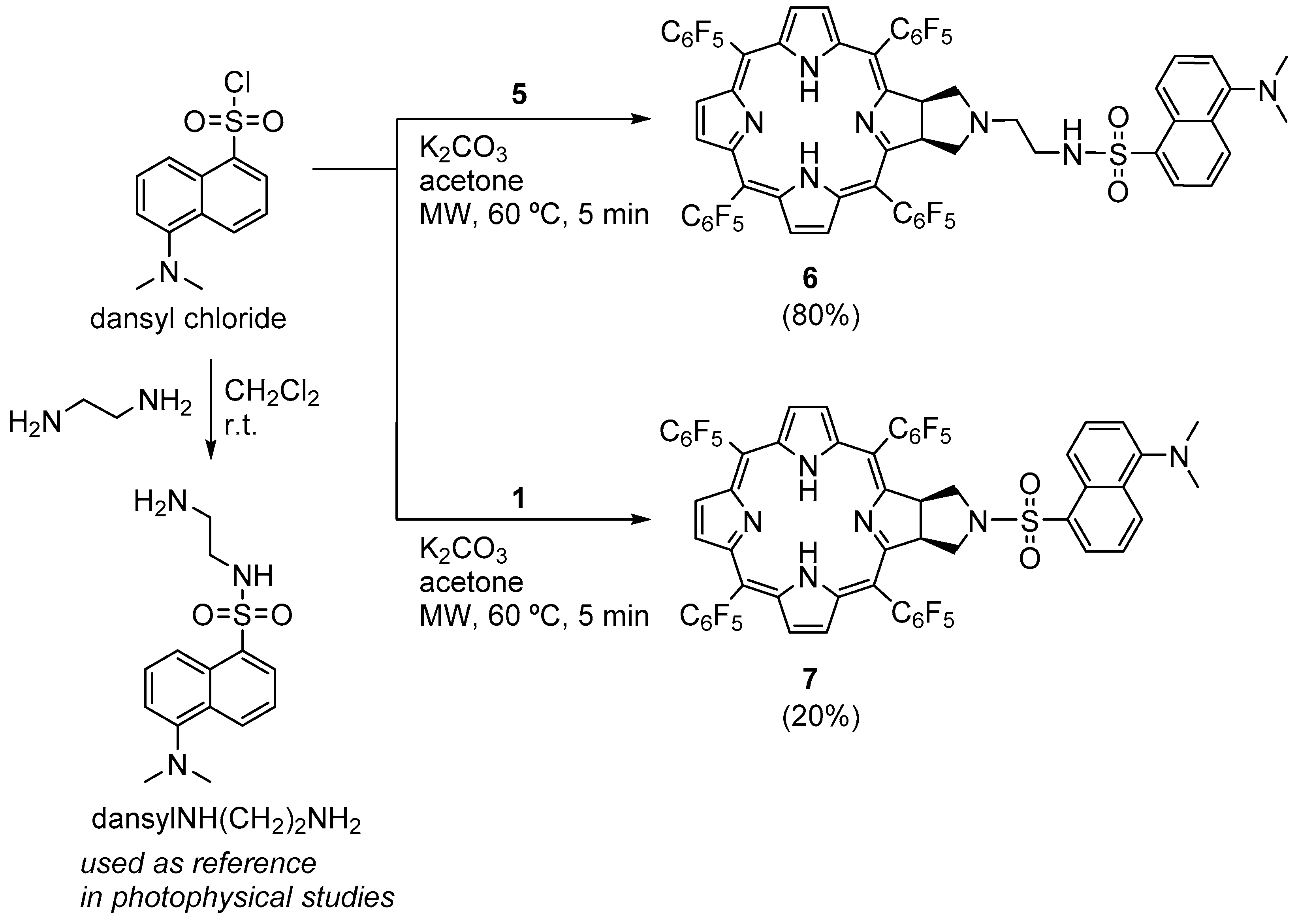
3.2.10. Synthesis of Chlorin−Dansyl Dyad 6
3.2.11. Synthesis of Chlorin−Dansyl Dyad 7
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hamblin, M.R. Photodynamic Therapy for Cancer: What’s Past Is Prologue. Photochem. Photobiol. 2020, 96, 506–516. [Google Scholar] [CrossRef]
- Hu, X.; Huang, Y.-Y.; Wang, Y.; Wang, X.; Hamblin, M.R. Antimicrobial Photodynamic Therapy to Control Clinically Relevant Biofilm Infections. Front Microbiol. 2018, 9, 1299. [Google Scholar] [CrossRef]
- Tan, I.B.; Dolivet, G.; Ceruse, P.; Poorten, V.V.; Roest, G.; Rauschning, W. Temoporfin-Mediated Photodynamic Therapy in Patients with Advanced, Incurable Head and Neck Cancer: A Multicenter Study. Head Neck 2010, 32, 1597–1604. [Google Scholar] [CrossRef]
- Kato, H.; Furukawa, K.; Sato, M.; Okunaka, T.; Kusunoki, Y.; Kawahara, M.; Fukuoka, M.; Miyazawa, T.; Yana, T.; Matsui, K.; et al. Phase II Clinical Study of Photodynamic Therapy Using Mono-l-Aspartyl Chlorin E6 and Diode Laser for Early Superficial Squamous Cell Carcinoma of the Lung. Lung Cancer 2003, 42, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.; Gamelas, S.R.D.; Vieira, C.; Faustino, M.A.F.; Tomé, J.P.C.; Almeida, A.; Gomes, A.T.P.C.; Lourenço, L.M.O. Pyrazole-Pyridinium Porphyrins and Chlorins as Powerful Photosensitizers for Photoinactivation of Planktonic and Biofilm Forms of E. Coli. Dye. Pigment. 2021, 193, 109557. [Google Scholar] [CrossRef]
- Sierra-Garcia, I.N.; Cunha, Â.; Lourenço, L.M.O. In Vitro Photodynamic Treatment of Fusarium Oxysporum Conidia through the Action of Thiopyridinium and Methoxypyridinium Chlorins. J. Photochem. Photobiol. A Chem. 2022, 432, 114081. [Google Scholar] [CrossRef]
- Mesquita, M.Q.; Menezes, J.C.J.M.D.S.; Pires, S.M.G.; Neves, M.G.P.M.S.; Simões, M.M.Q.; Tomé, A.C.; Cavaleiro, J.A.S.; Cunha, Â.; Daniel-da-Silva, A.L.; Almeida, A.; et al. Pyrrolidine-Fused Chlorin Photosensitizer Immobilized on Solid Supports for the Photoinactivation of Gram Negative Bacteria. Dye. Pigment. 2014, 110, 123–133. [Google Scholar] [CrossRef]
- Almeida, J.; Zhang, G.; Wang, M.; Queirós, C.; Cerqueira, A.F.R.; Tomé, A.C.; Barone, G.; Vicente, M.G.H.; Hey-Hawkins, E.; Silva, A.M.G.; et al. Synthesis, Characterization, and Cellular Investigations of Porphyrin–and Chlorin–Indomethacin Conjugates for Photodynamic Therapy of Cancer. Org. Biomol. Chem. 2021, 19, 6501–6512. [Google Scholar] [CrossRef]
- Almeida, J.; Aguiar, A.; Leite, A.; Silva, A.M.N.; Cunha-Silva, L.; de Castro, B.; Rangel, M.; Barone, G.; Tomé, A.C.; Silva, A.M.G. 1,3-Dipolar Cycloadditions with Meso-Tetraarylchlorins—Site Selectivity and Mixed Bisadducts. Org. Chem. Front. 2017, 4, 534–544. [Google Scholar] [CrossRef]
- Taniguchi, M.; Lindsey, J.S. Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.G.; Cavaleiro, J.A.S. Porphyrins in Diels–Alder and 1,3-Dipolar Cycloaddition Reactions. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 19, pp. 44–69. ISBN 9780080454078. [Google Scholar]
- Cerqueira, A.F.R.; Snarskis, G.; Zurauskas, J.; Guieu, S.; Paz, F.A.A.; Tomé, A.C. Site-Selective Modification of a Porpholactone—Selective Synthesis of 12,13- and 17,18-Dihydroporpholactones. Molecules 2020, 25, 2642. [Google Scholar] [CrossRef]
- Almeida, J.; Silva, A.M.N.; Rebelo, S.L.H.; Cunha-Silva, L.; Rangel, M.; de Castro, B.; Leite, A.; Silva, A.M.G. Synthesis and Coordination Studies of 5-(4′-Carboxyphenyl)-10,15,20-Tris(Pentafluorophenyl)Porphyrin and Its Pyrrolidine-Fused Chlorin Derivative. New J. Chem. 2018, 42, 8169–8179. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. 1,3-Dipolar Cycloaddition Reactions of Porphyrins with Azomethine Ylides. J. Org. Chem. 2005, 70, 2306–2314. [Google Scholar] [CrossRef]
- Gonzales, J.; Bhupathiraju, N.V.S.D.K.; Hart, D.; Yuen, M.; Sifuentes, M.P.; Samarxhiu, B.; Maranan, M.; Berisha, N.; Batteas, J.; Drain, C.M. One-Pot Synthesis of Four Chlorin Derivatives by a Divergent Ylide. J. Org. Chem. 2018, 83, 6307–6314. [Google Scholar] [CrossRef] [PubMed]
- Altona, C.; de Graaff, R.A.G.; Leeuwestein, C.H.; Romers, C. (S)-(–)-Spiro(4,4)Nonane-1,6-Dione, X-Ray Analysis and Valence-Force Calculations. J. Chem. Soc. D 1971, 20, 1305–1307. [Google Scholar] [CrossRef]
- Balducci, E.; Bellucci, L.; Petricci, E.; Taddei, M.; Tafi, A. Microwave-Assisted Intramolecular Huisgen Cycloaddition of Azido Alkynes Derived from α-Amino Acids. J. Org. Chem. 2009, 74, 1314–1321. [Google Scholar] [CrossRef]
- Costa, J.I.T.T.; Tomé, A.C.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.S. 5,10,15,20-Tetrakis(Pentafluorophenyl)Porphyrin: A Versatile Platform to Novel Porphyrinic Materials. J. Porphyr. Phthalocyanines 2011, 15, 1116–1133. [Google Scholar] [CrossRef]
- Gutsche, C.S.; Ortwerth, M.; Gräfe, S.; Flanagan, K.J.; Senge, M.O.; Reissig, H.-U.; Kulak, N.; Wiehe, A. Nucleophilic Aromatic Substitution on Pentafluorophenyl-Substituted Dipyrranes and Tetrapyrroles as a Route to Multifunctionalized Chromophores for Potential Application in Photodynamic Therapy. Chem. A Eur. J. 2016, 22, 13953–13964. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hu, M.; Zhan, P.; Peng, X. Energy Transfer Cassettes Based on Organic Fluorophores: Construction and Applications in Ratiometric Sensing. Chem. Soc. Rev. 2013, 42, 29–43. [Google Scholar] [CrossRef]
- Kim, J.; Lim, S.-H.; Yoon, Y.; Thangadurai, T.D.; Yoon, S. A Fluorescent Ammonia Sensor Based on a Porphyrin Cobalt(II)–Dansyl Complex. Tetrahedron Lett. 2011, 52, 2645–2648. [Google Scholar] [CrossRef]
- Lin, F.W.; Xu, X.L.; Wu, J.; Wan, L.S.; Xu, Z.K. Cobalt-Porphyrin/Dansyl Piperazine Complex Coated Filter Paper for “Turn on” Fluorescence Sensing of Ammonia Gas. RSC Adv. 2015, 5, 99361–99363. [Google Scholar] [CrossRef]
- Lim, M.H.; Lippard, S.J. Fluorescence-Based Nitric Oxide Detection by Ruthenium Porphyrin Fluorophore Complexes. Inorg. Chem. 2004, 43, 6366–6370. [Google Scholar] [CrossRef] [PubMed]
- Tecilla, P.; Dixon, R.P.; Slobodkin, G.; Alavi, D.S.; Waldeck, D.H.; Hamilton, A.D. Hydrogen-Bonding Self-Assembly of Multichromophore Structures. J. Am. Chem. Soc. 1990, 112, 9408–9410. [Google Scholar] [CrossRef]
- Leite, A.; Silva, A.M.G.; Coutinho, C.; Cunha-Silva, L.; de Castro, B.; Rangel, M. Design of a Water Soluble Fluorescent 3-Hydroxy-4-Pyridinone Ligand Active at Physiological PH Values. J. Fluoresc. 2016, 26, 1773–1785. [Google Scholar] [CrossRef]
- Mocanu, S.; Ionita, G.; Ionescu, S.; Tecuceanu, V.; Enache, M.; Leonties, A.R.; Stavarache, C.; Matei, I. New Environment-Sensitive Bis-Dansyl Molecular Probes Bearing Alkyl Diamine Linkers: Emissive Features and Interaction with Cyclodextrins. Chem. Phys. Lett. 2018, 713, 226–234. [Google Scholar] [CrossRef]
- Gonsalves, A.M.d.A.R.; Varejão, J.M.T.B.; Pereira, M.M. Some New Aspects Related to the Synthesis of Meso-substituted Porphyrins. J. Heterocycl. Chem. 1991, 28, 635–640. [Google Scholar] [CrossRef]
- Wang, J.; Shang, J.; Xiang, Y.; Tong, A. General Method for Post-Synthetic Modification of Oligonucleotides Based on Oxidative Amination of 4-Thio-2′-Deoxyuridine. Bioconjug. Chem. 2021, 32, 721–728. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | X−R | Product (Yield %) |
| 1 | 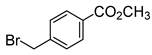 |  |
| 2 |  |  |
| 3 |  | 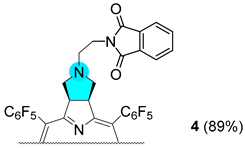 |
| 4 |  | 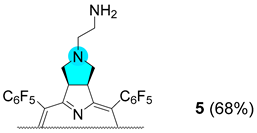 |
| Chlorin | Absorption | Emission | |
|---|---|---|---|
| λ max, nm (ε/dm3 mol−1 cm−1) | λ max, nm (ϕ F) | ||
| 1 | 404 (145 × 103) | 650 (55 × 103) | 655 (0.315) |
| 2b | 405 (120 × 103) | 650 (60 × 103) | 655 (0.101) |
| 3 | 401 (162 × 103) | 647 (41 × 103) | 651 (0.333) |
| Zn-3 | 417 (200 × 103) | 622 (34 × 103) | 625 (0.076) |
| 5 | 405 (153 × 103) | 650 (43 × 103) | 655 (0.147) |
| 6 | 405 (144 × 103) | 650 (37 × 103) | 655 (0.188) |
| 7 | 405 (132 × 103) | 650 (35 × 103) | 655 (0.123) |
| Solvent | P.I. | ϕF (λmax) Chlorin 1 | ϕF (λmax) Chlorin 5 | ϕF (λmax) Dyad 6 | ϕF (λmax) Dyad 7 |
|---|---|---|---|---|---|
| DMF | 6.4 | 0.315 (655) | 0.147 (655) | 0.188 (655) | 0.123 (655) |
| MeCN | 5.8 | 0.269 (655) | 0.158 (652) | 0.136 (652) | 0.073 (652) |
| MeOH | 5.1 | 0.286 (654) | 0.243 (652) | 0.165 (654) | 0.055 (651) |
| CHCl3 | 4.1 | 0.314 (655) | 0.254 (654) | 0.279 (653) | 0.207 (657) |
| toluene | 2.4 | 0.358 (658) | 0.307 (656) | 0.307 (657) | 0.219 (654) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, J.; Tomé, A.C.; Rangel, M.; Silva, A.M.G. Microwave-Assisted Synthesis and Spectral Properties of Pyrrolidine-Fused Chlorin Derivatives. Molecules 2023, 28, 3833. https://doi.org/10.3390/molecules28093833
Almeida J, Tomé AC, Rangel M, Silva AMG. Microwave-Assisted Synthesis and Spectral Properties of Pyrrolidine-Fused Chlorin Derivatives. Molecules. 2023; 28(9):3833. https://doi.org/10.3390/molecules28093833
Chicago/Turabian StyleAlmeida, José, Augusto C. Tomé, Maria Rangel, and Ana M. G. Silva. 2023. "Microwave-Assisted Synthesis and Spectral Properties of Pyrrolidine-Fused Chlorin Derivatives" Molecules 28, no. 9: 3833. https://doi.org/10.3390/molecules28093833
APA StyleAlmeida, J., Tomé, A. C., Rangel, M., & Silva, A. M. G. (2023). Microwave-Assisted Synthesis and Spectral Properties of Pyrrolidine-Fused Chlorin Derivatives. Molecules, 28(9), 3833. https://doi.org/10.3390/molecules28093833