



Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study

 ,
,  , , ,
, , ,  , ,
, ,  and
and
Abstract
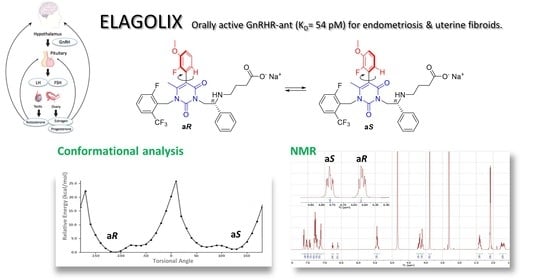
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. NMR Spectroscopy
2.3. HPLC Analyses
2.4. Structure Description
2.5. Conformational Analysis
3. Materials and Methods
3.1. General
3.2. Synthesis of 1 from 3
3.2.1. 1-(2-fluoro-6-(trifluoromethyl)benzyl) urea (4)
3.2.2. 1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine-2,4(1H,3H)-dione (5)
3.2.3. 5-bromo-1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine-2,4(1H,3H)-dione (6)
3.2.4. 5-(2-fluoro-3-methoxyphenyl)-1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine-2,4(1H,3H)-dione (7)
3.2.5. (R)-2-[(tert-butoxy-carbonyl)amino]-2-phenylethyl methane-sulfonate (10)
3.2.6. (R)-3-(amino(phenyl)methyl)-5-(2-fluoro-3-methoxyphenyl)-1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine 2,4(1H,3H)-dione (8)
3.2.7. Ethyl (R)-4-(((5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-(trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl)(phenyl)methyl)amino)butanoate (9)
3.2.8. Elagolix Sodium Salt (1)
3.3. NMR Spectroscopy
3.4. HPLC Analyses
- −
- Chiral HPLC analysis: a Merck-Hitachi (Hitachi Ltd., Tokyo, Japan), equipped with a UV detector model L-4250, pump system model L-6200 and a chromato-integrator model D-2500. The column employed in the analyses was a Phenomenex Lux-Cellulose 1 (Phenomenex, Torrance, CA, USA). The dimension of the column is 250 mm × 4.6 mm, 3 µm. The elution was in isocratic mode with the indicated eluant and flow. All the samples were measured at λ = 254 nm and 25 °C.
- −
- RP-HPLC analysis: Agilent 1100 system (Agilent Technologies, Waldbronn, Germany) equipped with a Zorbax SB-C18 column (150 mm × 3.0 mm, 3.5 µm) for 1 and with a Supelco Discovery C18 (250 mm × 4.6 mm, 5.0 µm) for 7.
3.5. X-ray Analysis
3.6. Conformational Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lamb, Y.N. Elagolix: First Global Approval. Drugs 2018, 78, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Sara, A.R.; Al Hendy, A. Elagolix in the treatment of heavy menstrual bleeding associated with uterine fibroids in premenopausal women. Expert Rev. Clin. Pharmacol. 2021, 14, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Betz, S.F.; Zhu, Y.-F.; Chen, C.; Struthers, R.S. Non-peptide gonadotropin-releasing hormone receptor antagonists. J. Med. Chem. 2008, 51, 3331–3348. [Google Scholar] [CrossRef] [PubMed]
- Tukun, F.-L.; Olberg, D.E.; Riss, P.J.; Haraldsen, I.; Kaass, A.; Klaveness, J. Recent Development of Non-Peptide GnRH Antagonists. Molecules 2017, 22, 2188. [Google Scholar] [CrossRef]
- Tucci, F.C.; Hu, T.; Mesleh, M.F.; Bokser, A.; Allsopp, E.; Gross, T.D.; Guo, Z.; Zhu, Y.-F.; Struthers, R.S.; Ling, N.; et al. Atropisomeric property of 1-(2,6-difluorobenzyl)-3-(2R)-amino-2-phenethyl-5-(2-fluoro-3-methoxyphenyl)-6-methyluracil. Chirality 2005, 17, 559–564. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef]
- Basilaia, M.; Chen, M.H.; Secka, J.; Gustafson, J.L. Atropisomerism in the Pharmaceutically Relevant Realm. Acc. Chem. Res. 2022, 55, 2904–2919. [Google Scholar] [CrossRef]
- Toenjes, S.T.; Gustafson, J.L. Atropisomerism in medicinal chemistry: Challenges and opportunities. Future Med. Chem. 2018, 10, 409–422. [Google Scholar] [CrossRef]
- Clayden, J.; Moran, W.J.; Edwards, P.J.; LaPlante, S.R. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed Engl. 2009, 48, 6398–6401. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, W.; Li, S.; Shen, L.; Gu, Z.; Zhang, Y.; Li, J.; Chen, S.; Jia, X. Discovery and Assessment of Atropisomers of (±)-Lesinurad. ACS Med. Chem. Lett. 2017, 8, 299–303. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Huai, Q.; Cai, J.; Zoraghi, R.; Francis, S.H.; Corbin, J.D.; Robinson, H.; Xin, Z.; Lin, G.; et al. Multiple conformations of phosphodiesterase-5: Implications for enzyme function and drug development. J. Biol. Chem. 2006, 281, 21469–21479. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, Y.; Huang, C.Q.; Gross, T.D.; Pontillo, J.; Rowbottom, M.W.; Saunders, J.; Struthers, S.; Tucci, F.C.; Xie, Q.; et al. Uracils as potent antagonists of the human gonadotropin-releasing hormone receptor without atropisomers. Bioorg. Med. Chem. Lett. 2005, 15, 2519–2522. [Google Scholar] [CrossRef]
- Wang, Z.; Meng, L.; Liu, X.; Zhang, L.; Yu, Z.; Wu, G. Recent progress toward developing axial chirality bioactive compounds. Eur. J. Med. Chem. 2022, 243, 114700. [Google Scholar] [CrossRef]
- Zhao, L.; Guo, Z.; Chen, Y.; Hu, T.; Wu, D.; Zhu, Y.-F.; Rowbottom, M.; Gross, T.D.; Tucci, F.C.; Struthers, R.S.; et al. 5-Aryluracils as potent GnRH antagonists-Characterization of atropisomers. Bioorg. Med. Chem. Lett. 2008, 18, 3344–3349. [Google Scholar] [CrossRef]
- Yan, W.; Cheng, L.; Wang, W.; Wu, C.; Yang, X.; Du, X.; Ma, L.; Qi, S.; Wei, Y.; Lu, Z.; et al. Structure of the human gonadotropin-releasing hormone receptor GnRH1R reveals an unusual ligand binding mode. Nat. Commun. 2020, 11, 5287. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, Y.; Wu, D.; Chen, C.; Wade, W.; Dwight, W.J.; Huang, C.Q.; Tucci, F.C. Preparation of Pyrimidine-2,4(1H,3H)-dione Derivatives as Gonadotropin-Releasing Hormone Receptor Antagonists. WO Patent WO2005007165, 27 January 2005. [Google Scholar]
- Chen, C.; Wu, D.; Guo, Z.; Xie, Q.; Reinhart, G.J.; Madan, A.; Wen, J.; Chen, T.; Huang, C.Q.; Chen, M.; et al. Discovery of sodium R-(+)-4-{2-5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoromethylbenzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl-1-phenylethylamino}butyrate (elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor. J. Med. Chem. 2008, 51, 7478–7485. [Google Scholar] [CrossRef]
- Peddireddy, S.R.; Allam, S.K.; Kottur, M.K.; Oruganti, S.; Kandagatla, B. Process for the Preparation of Elagolix Sodium and Its Polymorph. WO Patent WO2017221144, 18 December 2017. [Google Scholar]
- Vlasakova, R.; Cerna, I.; Obadalova, I.; Krejcik, L.; Dammer, O.; Svobodova, J.; Sembera, F. Solid Forms of Elagolix. Wo Patent WO2018224063, 13 December 2018. [Google Scholar]
- Sulake, R.S.; Shinde, S.R.; Siyan, R.S.; Bhise, N.B.; Singh, G.P. Process for the Preparation of Elagolix and Pharmaceutically Acceptable Salts Thereof. WO Patent WO2018198086, 1 November 2018. [Google Scholar]
- Lenna, R.; Fasana, A.; Ortiz, J. Process for the Preparation of the Sodium Salt of Elagolix and Its Intermediates. Wo Patent WO2021044230, 11 March 2022. [Google Scholar]
- Siripragada, M.R.; Pendyam, K.; Kallepally, S.; Avula, S.R.; Gottapu, V.N.; Pappula, V.R. An Improved Process for the Preparation of Elagolix Sodium. WO Patent WO2021064561, 8 April 2021. [Google Scholar]
- Ballete, R.; Jimenez Alonso, O.; Garcia Garcia, E.; Dobarro Rodriguez, A. 3-((R)-2-(Amino-2-phenylethyl)-1-(2-fluoro-6-trifluoromethylbenzyl)-5-iodo-6-methyl-1H-pyrimidine-2,4-dione or a Salt Thereof, Process for its Preparation, and Its in the Synthesis of Elagolix. WO Patent WO2021083554, 6 May 2021. [Google Scholar]
- Zhong, X.; Lv, Q.; Yong, Q.; Hu, W.; Li, D.; Ji, S.; Zhan, L.; Chen, W.; Li, M.; Lin, J.; et al. Forced degradation studies of elagolix sodium with the implementation of high resolution LC-UV-PDA-MSn (n = 1,2,3…) and NMR structural elucidation. J. Pharm. Biomed. Anal. 2023, 224, 115198. [Google Scholar] [CrossRef]
- Ferraboschi, P.; Chiara Sala, M.; Stradi, R.; Ragonesi, L.; Gagliardi, C.; Lanzarotti, P.; Ragg, E.M.; Mori, M.; Meneghetti, F. Full spectroscopic characterization of two crystal pseudopolymorphic forms of the antiandrogen cortexolone 17α-propionate for topic application. Steroids 2017, 128, 95–104. [Google Scholar] [CrossRef]
- Meneghetti, F.; Ferraboschi, P.; Grisenti, P.; Reza Elahi, S.; Mori, M.; Ciceri, S. Crystallographic and NMR Investigation of Ergometrine and Methylergometrine, Two Alkaloids from Claviceps purpurea. Molecules 2020, 25, 331. [Google Scholar] [CrossRef]
- Ciceri, S.; Colombo, D.; Ferraboschi, P.; Grisenti, P.; Iannone, M.; Mori, M.; Meneghetti, F. Vecuronium bromide and its advanced intermediates: A crystallographic and spectroscopic study. Steroids 2021, 176, 108928. [Google Scholar] [CrossRef]
- Gallagher, D.; Treiber, L.; Hughes, R.; Campopiano, O.; Wang, P.; Zhao, Y.; Chou, S.; Ouellette, M.; Hettinger, D. Processes for the Preparation of Uracil Derivatives. WO Patent WO2009062087, 14 May 2009. [Google Scholar]
- Larrañaga, M.D.; Lewis, R.J.; Lewis, R.A.; Hawley, G.G. Hawley’s Condensed Chemical Dictionary, 16th ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2016; ISBN 1119312469. [Google Scholar]
- Lewis, R.J. Sax’s Dangerous Properties of Industrial Materials, 12th ed.; Wiley: Hoboken, NJ, USA, 2012; ISBN 9780471701347. [Google Scholar]
- Badertscher, M.; Bühlmann, P.; Pretsch, E. Tables of Spectral Data for Structure Determination of Organic Compounds; Springer: Berlin/Heidelberg, Germany, 1983; ISBN 978-3-662-22455-7. [Google Scholar]
- Marek, R.; Lycka, A. 15N NMR Spectroscopy in Structural Analysis. COC 2002, 6, 35–66. [Google Scholar] [CrossRef]
- D’Acquarica, I.; Gasparrini, F.; Pierini, M.; Villani, C.; Zappia, G. Dynamic HPLC on chiral stationary phases: A powerful tool for the investigation of stereomutation processes. J. Sep. Sci. 2006, 29, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Trapp, O.; Schoetz, G.; Schurig, V. Determination of enantiomerization barriers by dynamic and stopped-flow chromatographic methods. Chirality 2001, 13, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C. Stereolabile chiral compounds: Analysis by dynamic chromatography and stopped-flow methods. Chem. Soc. Rev. 2005, 34, 595–608. [Google Scholar] [CrossRef] [PubMed]
- König, W.A.; Gehrcke, B.; Runge, T.; Wolf, C. Gas chromatographic separation of atropisomeric alkylated and polychlorinated biphenyls using modified cyclodextrins. J. High Resol. Chromatogr. 1993, 16, 376–378. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, Y.-F.; Gross, T.D.; Tucci, F.C.; Gao, Y.; Moorjani, M.; Connors, P.J.; Rowbottom, M.W.; Chen, Y.; Struthers, R.S.; et al. Synthesis and structure-activity relationships of 1-arylmethyl-5-aryl-6-methyluracils as potent gonadotropin-releasing hormone receptor antagonists. J. Med. Chem. 2004, 47, 1259–1271. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable Continuum Model (PCM) Calculations of Solvent Effects on Optical Rotations of Chiral Molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Lan, C.B.; Auclair, K. Ammonium Chloride-Promoted Rapid Synthesis of Monosubstituted Ureas under Microwave Irradiation. Eur. J. Org. Chem. 2021, 2021, 5135–5146. [Google Scholar] [CrossRef]
- Capasso, C.; Winum, J.-Y. Novel method of treating macular degeneration: A patent evaluation (WO2018/107005). Expert Opin. Ther. Pat. 2019, 29, 749–752. [Google Scholar] [CrossRef]
- O’Brien, P.M.; Sliskovic, D.R.; Blankley, C.J.; Roth, B.D.; Wilson, M.W.; Hamelehle, K.L.; Krause, B.R.; Stanfield, R.L. Inhibitors of acyl-CoA:cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 8. Incorporation of amide or amine functionalities into a series of disubstituted ureas and carbamates. Effects on ACAT inhibition in vitro and efficacy in vivo. J. Med. Chem. 1994, 37, 1810–1822. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, Q.; Lin, K.; Zhou, W.; Pan, J.; Chen, L.; Zhou, T. Preparation of Elagolix Intermediate. China Patent CN110498770, 30 April 2021. [Google Scholar]
- SMART & SAINT Software Reference Manual, Version 6.45; Bruker Analytical X-Ray Systems, Inc.: Madison, WI, USA, 2003.
- Sheldrick, G.M. SADABS, Version 2008/1; Bruker AXS Inc.: Karlsruhe, Germany, 2008.
- Sheldrick, G.M. SHELXL-2018; Universität Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Nardelli, M. Parst: A system of fortran routines for calculating molecular structure parameters from results of crystal structure analyses. Comput. Chem. 1983, 7, 95–98. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H | 5 | 6 | 7 | 8 | 9 | 1 |
|---|---|---|---|---|---|---|
| 1 | / | / | / | / | / | / |
| 2 | / | / | / | / | / | / |
| 3 | 8.97 (brs) | Exchanged with CD3OD | 8.52 (brs) | / | / | / |
| 4 | / | / | / | / | / | / |
| 5 | 5.59 (s) | / | / | / | / | / |
| 6 | / | / | / | / | / | / |
| 7 | 5.36 (s) | 5.38 (s) | 5.47 (s) | 5.50 (m) | 5.47 (m) | 5.43 (m) |
| 8 | / | / | / | / | / | / |
| 9 | / | / | / | / | / | / |
| 10 | 7.25 (dd, J = 12.3 and 8.5 Hz) | 7.20 (dd, J= 11.7 and 8.3 Hz) | 7.28 (dd, J = 11.8 and 8.3 Hz) | 7.26 (m, overlapped with 28) | 7.29 (m, overlapped with 28) | 7.44 (m) |
| 11 | 7.42 (m) | 7.37 (m) | 7.42 (m) | 7.41 (m, overlapped with 26 and 26’) | 7.41 (m) | 7.53 (td, J = 8.1, 5.0 Hz) |
| 12 | 7.54 (d, J = 7.9 Hz) | 7.48 (d, J = 7.8 Hz) | 7.54 (d, J = 7.9 Hz) | 7.55 (d, J = 7.9 Hz) | 7.54 (d, J = 7.9 Hz) | 7.62 (d, J = 7.9 Hz) |
| 13 | / | / | / | / | / | / |
| 14 | / | / | / | / | / | / |
| 15 | 2.15 (s) | 2.35 (s) | 2.05 (s) | 2.07 (s) | 2.07 and 2.06 * | 2.085 (s) and 2.076 (s) * |
| 16 | / | / | / | / | / | / |
| 17 | / | / | / | / | / | / |
| 18 | / | / | / | / | / | / |
| 19 | / | / | 6.97 (m) | 6.97 (td, J = 8.2 and 1.3 Hz) | 6.97 (tt, J = 8.1 and 1.6 Hz) | 7.10 (n.d., overlapped with 20) |
| 20 | / | / | 7.10 (dd, J = 9.0 and 7.9 Hz) | 7.11 (tdd, J = 8.0, 2.8, and 1.3 Hz) | 7.11 (tdd, J = 8.0, 4.6, and 1.3 Hz) | 7.11 and 7.14 (n.d., overlapped with 19) |
| 21 | / | / | 6.81 (m) | 6.85 (td, J = 6.2 and 1.5 Hz) and 6.78 (td, J = 6.2 and 1.4 Hz) * | 6.83 (ddd, J = 7.7, 6.0, and 1.6 Hz) and 6.76 (ddd, J = 7.7, 6.0, and 1.6 Hz) * | 6.76 (m) and 6.61 (m) * |
| 22 | / | / | 3.88 (s) | 3.89 (s) | 3.889 (s) and 3.886 (s) * | 3.883 (s) and 3.880 (s) * |
| 23 | / | / | / | 4.29 (m o ddd, J = 16.2, 13.1, 9.8 Hz, Ha) and 4.11 (m, Hb) | 4.28 (m, Ha) and 4.04(m, Hb) | 4.26–4.07 (m) |
| 24 | / | / | / | 4.42 (dd, J = 9.8 and 4.5 Hz) | 4.12 (m) | 4.11 (n.d.) |
| 25 | / | / | / | / | / | / |
| 26 and 26′ | / | / | / | 7.41 (m, overlapped with 11) | 7.37 (m) | 7.29 (n.d., overlapped with 28) |
| 27 and 27′ | / | / | / | 7.32 (m) | 7.30 (m, overlapped with 10) | 7.27 (n.d., overlapped with 26 and 26’) |
| 28 | / | / | / | 7.27 (m, overlapped with 10) | 7.24 (m) | 7.22 (m) |
| 29 | / | / | / | 2.13 | 1.63 (brs) | / |
| 30 | / | / | / | / | 2.46 (m, Ha) and 2.38 (m, Hb) | 2.40 (m) |
| 31 | / | / | / | / | 1.68 (m) | 1.75 (m, Ha) and 1.68 (m, Hb) |
| 32 | / | / | / | / | 2.29 (m) | 2.09 (m, overlapped with 15) |
| 33 | / | / | / | / | / | / |
| CH2CH3 | / | / | / | / | 4.084 (q, J = 7.1Hz,) and 4.082 (q, J = 7.1Hz) * | / |
| CH2CH3 | / | / | / | / | 1.21 (t, J = 7.1 Hz) | / |
| 13C | 5 | 6 | 7 | 8 | 9 | 1 |
|---|---|---|---|---|---|---|
| 1 | / | / | / | / | / | / |
| 2 | 151.7 | 150.8 | 151.0 | 152.3 | 152.3 and 152.2 * | 153.3 and 153.2 * |
| 3 | / | / | / | / | / | / |
| 4 | 162.4 | 159.6 | 161.2 | 161.8 and 161.6 * | 161.7 and 161.6 * | 163.5 and 163.3 * |
| 5 | 102.5 | 99.2 | 108.8 | 108.3 and 108.2 * | 108.3 and 108.2 * | 109.3 and 109.2 * |
| 6 | 153.6 | 151.6 | 151.9 | 149.9 and 149.8 * | 149.7 and 149.6 * | 152.6 and 152.5 * |
| 7 | 41.1 (m) | 42.5 (m) | 41.8 (m) | 42.7 (brs) | 42.7 (brs) | 44.1 (brs) |
| 8 | 121.8 (d, J = 11.6 Hz) | 121.2 (d, J = 11.3 Hz) | 121.8 (d, J = 11.1 Hz) | 122.0 (d, J= 11.4 Hz) | 122.1 (d, J = 11.3 Hz) | 123.6 (overlapped with 12 and 16) |
| 9 | 161.4 (d, J = 250.0 Hz) | 161.1 (d, J = 249.6 Hz) | 161.4 (d, J = 250.0 Hz) | 161.3 (d, J = 249.5 Hz) | 161.3 (d, J = 250.0 Hz) | 162.8 (d, J =248.0 Hz) |
| 10 | 120.9 (d, J = 24.0 Hz) | 120.9 (d, J = 24.0 Hz) | 121.0 (d, J = 24.4 Hz) | 120.9 (d, J = 24.1 Hz) | 120.9 (d, J = 24.4 Hz) | 122.15 (d, J = 24.3 Hz) and 122.19 (d, J = 24.3 Hz) * |
| 11 | 129.5 (d, J = 10.0 Hz) | 129.6 (d, J = 9.7 Hz) | 129.5 (d, J = 9.8 Hz) | 129.3 (d, J = 9.8 Hz) | 129.3 (d, J = 9.6 Hz) | 130.9 and 130.8 * |
| 12 | 122.6 (m) | 122.5 (m) | 122.6 (m) | 122.6 (m) | 122.5 (m) | 123.7 (overlapped with 8 and 16) |
| 13 | 129.6 (dd, J = 30.0 and 3.6 Hz; partially hidden by 11) | 129.3 (dd, J = 30.9 and 3.7 Hz; | 129.6 (dd, J = 30.9 and 4.0 Hz) | 129.5 (dd, J = 31.0 and 3.1, partially overlapped with 11) | 129.3 (dd, J = 30.2 and 3.0 Hz) | 130.6 (dd, J = 31.1 and 3.3 Hz) |
| 14 | 123.4 (dd, J = 273.6 and 4.0 Hz) | 123.3 (dd, J = 274.3 and 4.2 Hz) | 123.5 (dd, J = 273.8 and 3.9 Hz) | 123.5 (dd, J = 273.6 and 3.8 Hz) | 123.5 (dd, J = 271.1 and 3.7 Hz) | 125.1 (dd, J = 273.3 and 3.8 Hz) |
| 15 | 20.1 | 20.1 | 17.9 | 17.8 | 17.8 | 18.1 |
| 16 | / | / | 121.5 (d, J = 13.6 Hz) | 122.3 (d, J = 13.7 Hz) and 122.2 (d, J = 13.7 Hz) * | 122.37 (d, J = 13.8 Hz) and 122.35 (d, J = 13.8 Hz) | 123.8 (overlapped with 8 and 12) |
| 17 | / | / | 149.9 (d, J = 246.4 Hz) | 150.2 (d, J = 246.3 Hz) and 150.1 (d, J = 246.3 Hz) * | 150.23 (d, J = 245.5) and 150.17 (d, J = 245.5) | 151.8 (d, J = 245.4 Hz) |
| 18 | / | / | 148.0 (d, J = 11.0 Hz) | 148.0 (d, J = 11.0 Hz) | 148.0 (d, J = 11.0 Hz) | 149.5 (d, J = 11.2 Hz) |
| 19 | / | / | 113.5 | 113.3 | 113.2 | 114.8 |
| 20 | / | / | 123.8 | 123.8 (overlapped with 21) | 123.80 (s) and 123.76 (s) * | 125.1 (s) and 125.0 (s) * |
| 21 | / | / | 123.9 (d, J = 4.6 Hz) | 123.9 and 123.8 (overlapped with 20) * | 123.92 (s) and 123.85 (s) * | 125.0 and 124.9 * |
| 22 | / | / | 56.3 | 56.2 | 56.2 | 56.8 |
| 23 | / | / | / | 49.10 and 49.14 * | 47.7 and 47.6 * | 48.1 and 47.9 * |
| 24 | / | / | / | 54.1 and 54.2 * | 60.9 and 60.8 * | 62.2 and 62.0 * |
| 25 | / | / | / | 143.7 and 143.6 * | 141.92 (s) and 141.89 (s) * | 142.1 and 141.9 * |
| 26 and 26’ | / | / | / | 126.4 and 126.3 * | 127.2 and 127.1 (overlapped with 28) * | 128.7 |
| 27 and 27’ | / | / | / | 128.5 and 128.4 * | 128.4 and 128.3 * | 129.4 |
| 28 | / | / | / | 127.3 and 127.2 * | 127.2 (overlapped with 26 and 26′) | 128.5 |
| 29 | / | / | / | / | / | / |
| 30 | / | / | / | / | 46.51 (s) and 46.47 (s) * | 48.7 |
| 31 | / | / | / | 25.4 | 27.86 (s) and 27.83 (s) * | |
| 32 | / | / | / | / | 32.0 | 37.03 (s) and 37.00 (s) * |
| 33 | / | / | / | / | 173.7 60.1 | 182.3 |
| CH2CH3 | / | / | / | / | / | |
| CH2CH3 | / | / | / | / | 14.2 | / |
| 15N | 5 | 6 | 7 | 8 | 9 | 1 |
|---|---|---|---|---|---|---|
| 1 | 136.7 | 137.9 | 134.3 | 134.3 | 135.5 | 137.1 |
| 3 | 154.3 b | 153.4 b | 151.9 b | 160.1 | 161.3 | 160.5 |
| 29 | / | / | / | 31.2 | 43.6 | 46.3 |
| 13C | HMBC (13C→1H) |
|---|---|
| C-2 | H-7 and H-23 |
| C-4 | H-23 and H-15 (weak) |
| C-5 | H-21 and H-15 |
| C-6 | H-15 |
| C-8 | H-7, H-10, and H-12 |
| C-9 | H-7, H-10, and H-11 |
| C-13 | H-7 and H-11 |
| C-14 | H-12 |
| C-16 | H-20 |
| C-17 | H-19 and H-21 |
| C-18 | H-20 and H-22 |
| C-25 | H-23, H-24, H-27, and H-27’ |
| C-33 | H-31 and H-32 |
| Conformers | τ1 (°) | τ2 (°) | τ3 (°) | τ4 (°) | Gas Phase ΔE (kcal/mol) | Gas Phase (%) |
|---|---|---|---|---|---|---|
| 1A | −81.6 | −48.5 | −132.0 | 49.1 | 4.35 | 0.0 |
| 1B | −70.0 | 147.8 | −130.0 | 49.7 | 6.84 | 0.0 |
| 1C | 96.0 | −54.0 | −125.0 | 70.0 | 0.67 | 20.5 |
| 1D | 111.8 | 134.3 | −133.0 | 52.5 | 4.60 | 0.0 |
| 1E | −80.5 | −47.0 | −61.3 | −52.4 | 4.67 | 0.0 |
| 1F | −69.4 | 150.3 | −55.0 | −55.5 | 7.09 | 0.0 |
| 1G | 113.0 | 136.0 | −60.5 | −55.6 | 5.01 | 0.0 |
| 1H | 93.2 | −62.1 | −64.1 | −55.6 | 1.90 | 2.6 |
| 1I | 94.5 | −53.6 | 61.3 | −142.6 | 2.26 | 1.4 |
| 1J | 118.5 | 147.4 | 59.4 | −140.8 | 4.86 | 0.0 |
| 1K | −74.0 | −40.3 | 67.8 | −141.2 | 4.78 | 0.0 |
| 1L | −69.1 | 148.9 | 60.5 | −142.9 | 3.69 | 0.1 |
| 1M | 92.6 | −61.9 | −110.0 | −134.5 | 0.00 | 63.9 |
| 1N | 113.6 | 146.5 | −77.5 | 147.4 | 3.98 | 0.1 |
| 1O | −85.9 | −51.6 | −115.4 | −136.4 | 3.58 | 0.2 |
| 1P | −70.9 | 148.7 | −75.9 | 146.3 | 5.33 | 0.0 |
| 1Q | −81.0 | −46.4 | −75.4 | 142.8 | 3.64 | 0.1 |
| 1R | −71.1 | 148.9 | −75.2 | 147.0 | 5.40 | 0.0 |
| 1S | 92.7 | −55.0 | −76.1 | 139.9 | 1.09 | 10.1 |
| 1T | 113.9 | 146.9 | −77.6 | 147.5 | 3.93 | 0.1 |
| 1U | −86.3 | −49.9 | 56.0 | 53.0 | 4.53 | 0.0 |
| 1V | −70.1 | 145.9 | 51.1 | 55.2 | 3.67 | 0.1 |
| 1W | 93.5 | −53.5 | 49.9 | 53.8 | 2.77 | 0.6 |
| 1X | 114.2 | 142.7 | 51.3 | 53.6 | 5.32 | 0.0 |
| Code | τ1 | τ2 | τ3 | τ4 | τ5 | Water ΔE (kcal/mol) | Water (%) |
|---|---|---|---|---|---|---|---|
| 7 (crystal structure) | −105.5 | −144.7 | 110.4 | ||||
| 1M−A | 87.7° | −63.3° | −104.7° | −133.9° | −111.8° | 0.00 | 83.6 |
| 1M−C | 91.3° | −60.2° | −106.7° | −132.6° | 75.2° | 2.07 | 2.6 |
| 1M−D | 92.9° | −60.3° | −110.0° | −135.0° | 126.3° | 1.06 | 13.8 |
| Conformer | t6 | Distance CH22 – H19 (Å) | Gas Phase ΔE (kcal/mol) | Gas Phase (%) | Methanol ΔE (kcal/mol) | Methanol (%) |
|---|---|---|---|---|---|---|
| 1M-A | 0 | 2.56 | 0.00 | 44.5 | 0 | 86.0 |
| 1M-A (I) | −66 | 3.60 | 0.29 | 27.5 | 1.62 | 5.6 |
| 1M-A (II) | +66 | 3.60 | 0.27 | 28.0 | 1.38 | 8.4 |
| 1M-D | 0 | 2.56 | 0.22 | 30.5 | 0.00 | 84.5 |
| 1M-D (I) | −66 | 3.60 | 0.32 | 25.6 | 1.36 | 8.5 |
| 1M-D (II) | +66 | 3.60 | 0.00 | 43.9 | 1.48 | 7.0 |
| Crystal Data | |
|---|---|
| Chemical formula | C20H15F5N2O3 |
| Mr | 426.34 |
| Crystal system, space group | Orthorhombic, P bca |
| a, b, c (Å) | 11.165 (2), 11.073 (2), 30.367(6) |
| V (Å3) | 3754.2(13)) |
| Z | 8 |
| F (000) | 1744 |
| Density (g/cm3) | 1.509 |
| Temperature (K) | 298 |
| Radiation type | Mo-Kα (λ = 0.71073 Å) |
| µ (mm−1) | 1.135 |
| Crystal size (mm) | 0.06 × 0.03 × 0.02 |
| Data collection | |
| Diffractometer | Bruker-Axs Smart-Apex CCD |
| Tmin, Tmax | 0.893, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 29,138, 4175, 1715 |
| Rint | 0.0298 |
| Structure refinement | |
| R, wR2, S | 0.0437 (I > 2σ(I)) and 0.1119 (all), 0.0973 (I > 2σ(I)) and 0.1126 (all), 0.760 (all) |
| No. of parameters | 271 |
| No. of restraints | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciceri, S.; Colombo, D.; Fassi, E.M.A.; Ferraboschi, P.; Grazioso, G.; Grisenti, P.; Iannone, M.; Castellano, C.; Meneghetti, F. Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study. Molecules 2023, 28, 3861. https://doi.org/10.3390/molecules28093861
Ciceri S, Colombo D, Fassi EMA, Ferraboschi P, Grazioso G, Grisenti P, Iannone M, Castellano C, Meneghetti F. Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study. Molecules. 2023; 28(9):3861. https://doi.org/10.3390/molecules28093861
Chicago/Turabian StyleCiceri, Samuele, Diego Colombo, Enrico M. A. Fassi, Patrizia Ferraboschi, Giovanni Grazioso, Paride Grisenti, Marco Iannone, Carlo Castellano, and Fiorella Meneghetti. 2023. "Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study" Molecules 28, no. 9: 3861. https://doi.org/10.3390/molecules28093861
APA StyleCiceri, S., Colombo, D., Fassi, E. M. A., Ferraboschi, P., Grazioso, G., Grisenti, P., Iannone, M., Castellano, C., & Meneghetti, F. (2023). Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study. Molecules, 28(9), 3861. https://doi.org/10.3390/molecules28093861