
Cytotoxic Activity, Topoisomerase I Inhibition and In Silico Studies of New Sesquiterpene-aryl Ester Derivatives of (-) Drimenol

 , , , ,
, , , ,  , and
, and
Abstract
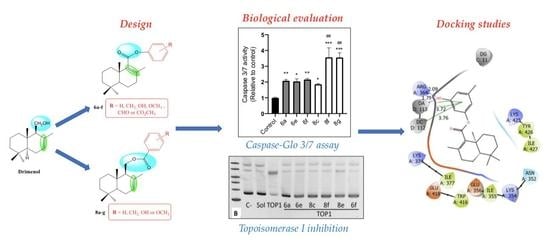
:
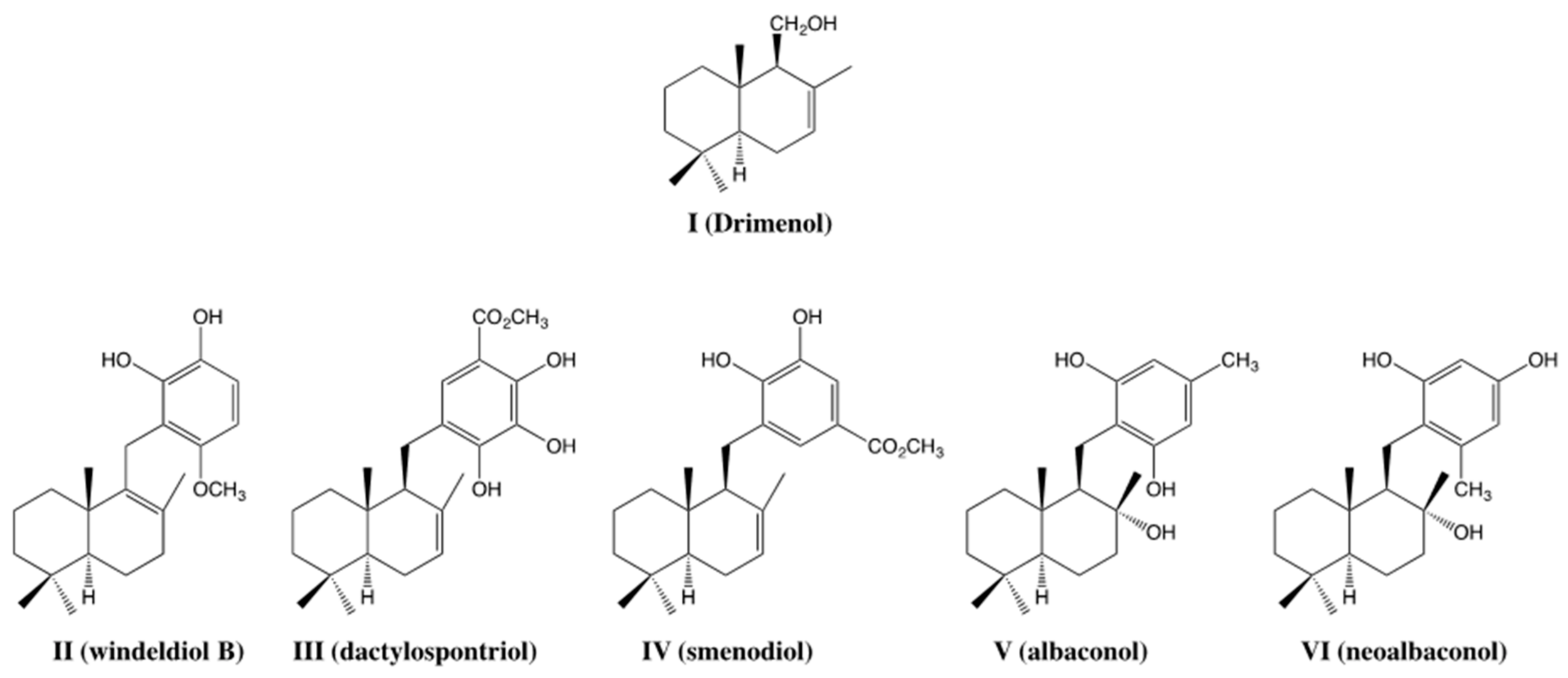
1. Introduction
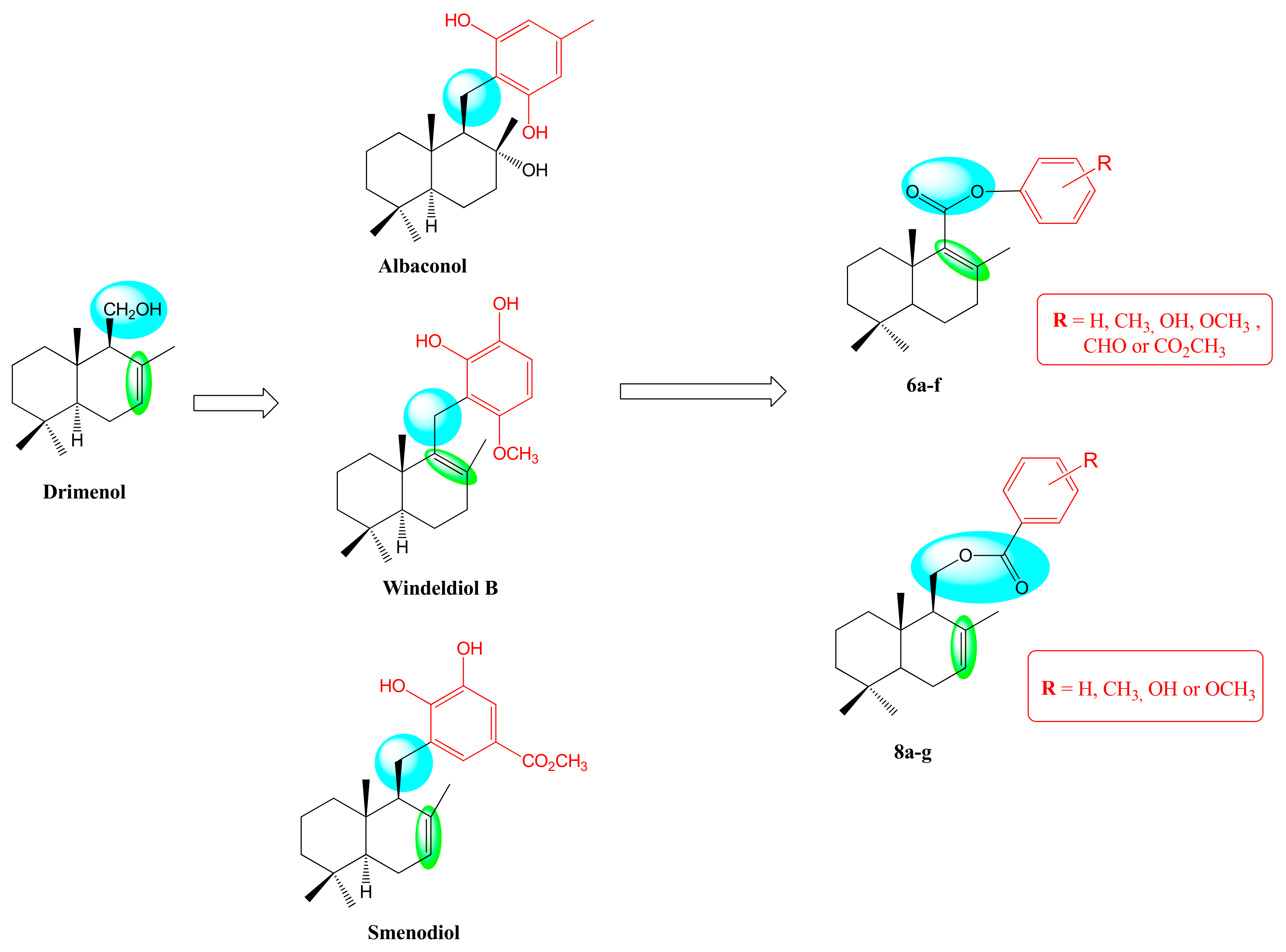
2. Results and Discussion
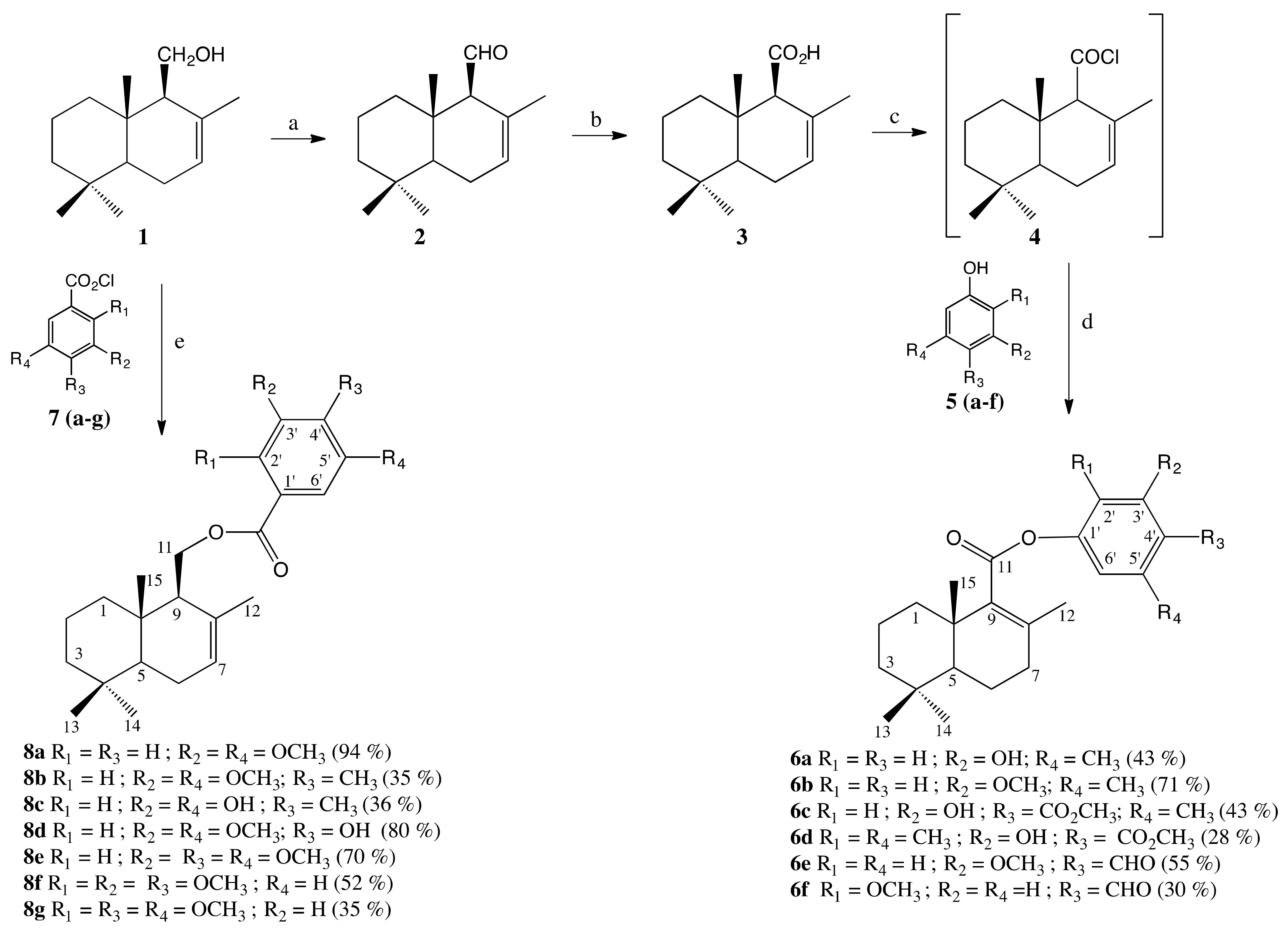
2.1. Synthesis
2.2. Biological Evaluation
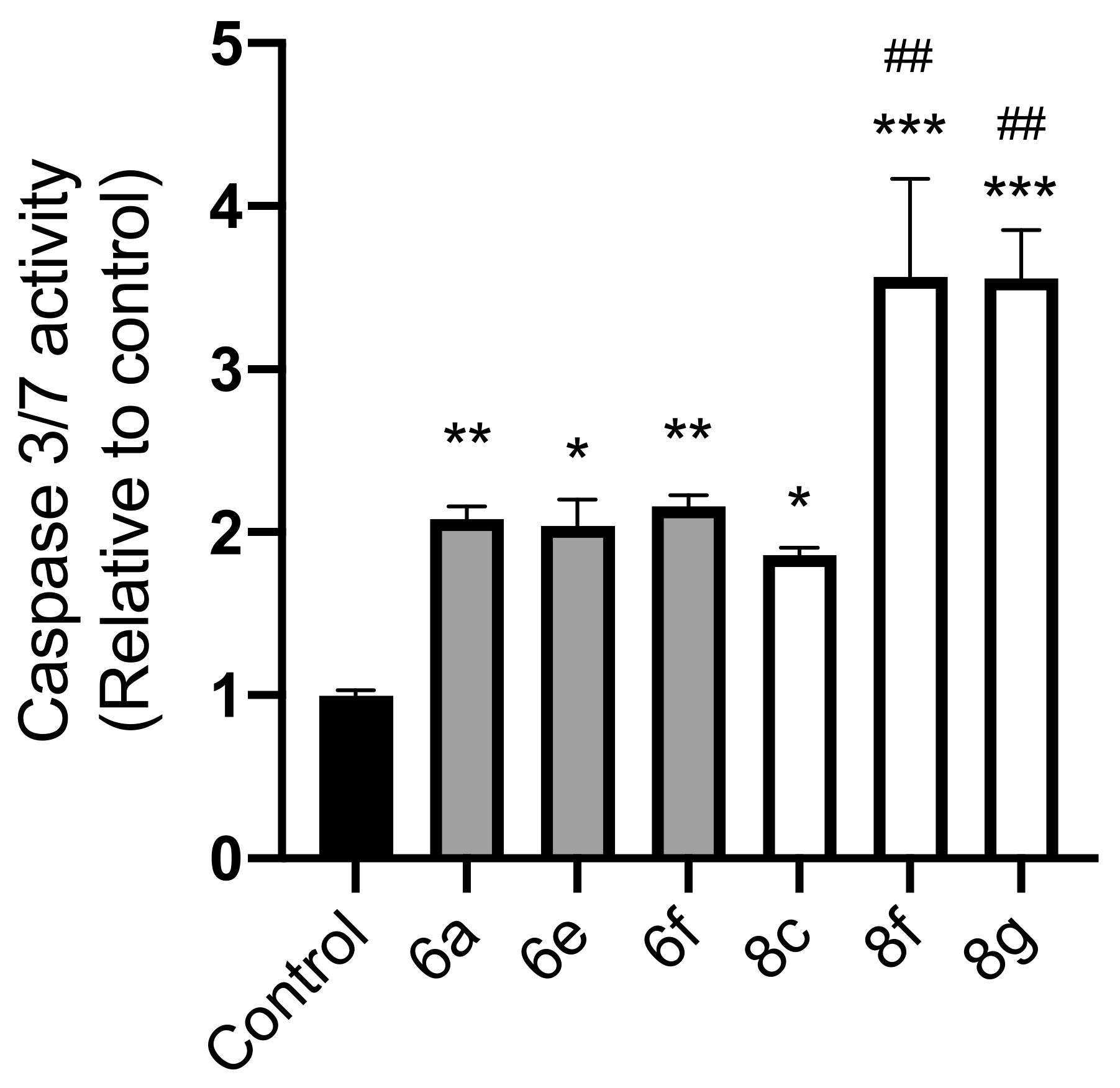
2.3. Caspase-Glo 3/7 Assay
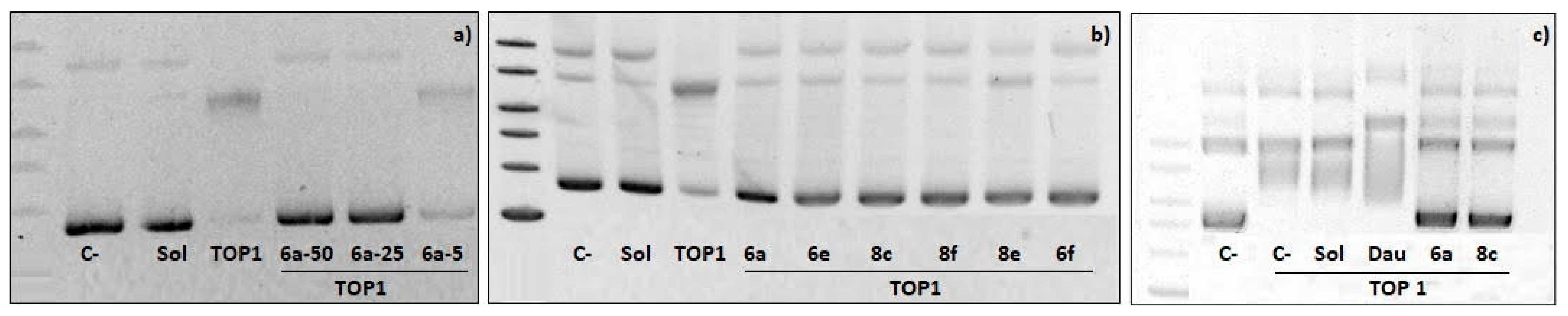
2.4. Topoisomerase I Inhibition
2.5. Docking Studies
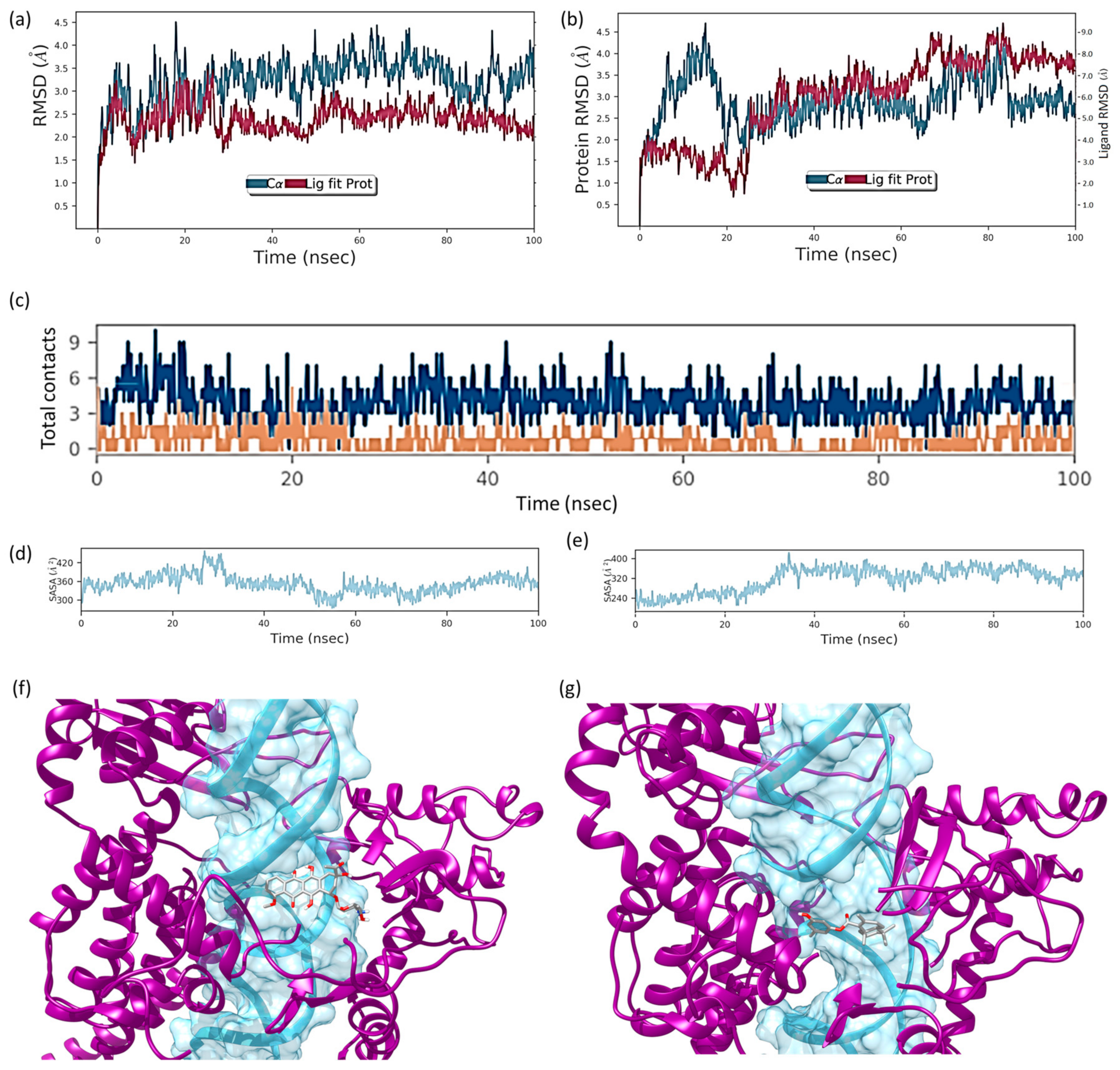
2.6. Molecular Dynamics Simulation
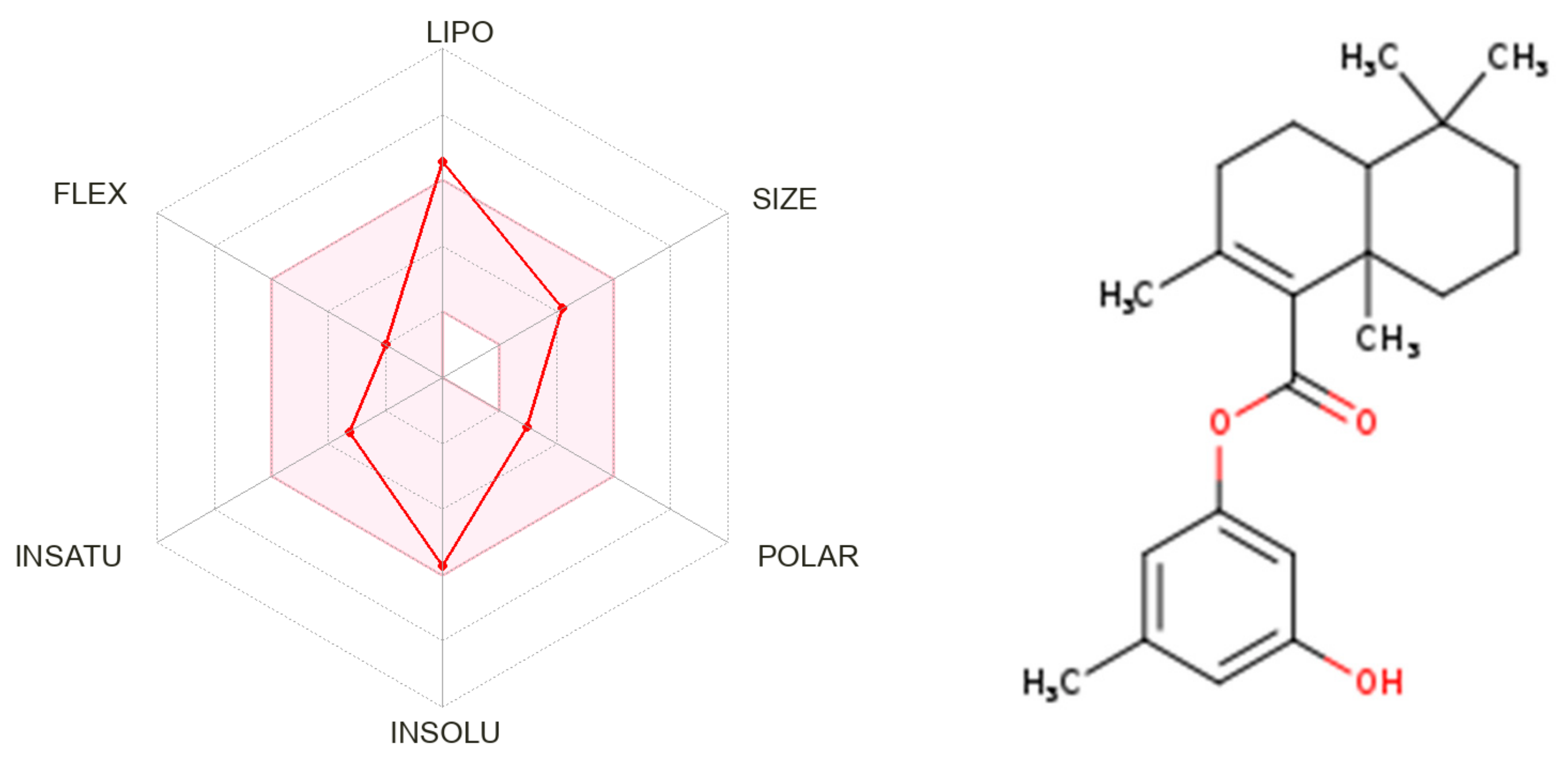
2.7. Physicochemical Properties
3. Experimental Section
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of Drimenal (2)
3.1.3. Synthesis of Drimenic Acid (3)
3.1.4. General Procedure to Obtain 6a–f
3.1.5. General Procedure to Obtain 8a–g
3.2. Biological Evaluation
3.2.1. In Vitro Growth Inhibition Assay
3.2.2. Caspase-Glo 3/7 Assay
3.2.3. Assay of Topoisomerase I Activity
3.3. Molecular Docking and Molecular Dynamics Simulations
3.3.1. Induced Fit Docking
3.3.2. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Santos, T.G.; Dognini, J.; Begnini, I.M.; Rebelo, R.A.; Verdi, M.; de Gasper, A.L.; Dalmarco, E.M. Chemical Characterization of Essential Oils from Drimys angustifolia Miers (Winteraceae) and Antibacterial Activity of their Major Compounds. J. Braz. Chem. Soc. 2013, 24, 164–170. [Google Scholar] [CrossRef]
- Derita, M.; Zacchino, S. Chemotaxonomic Importance of Sesquiterpenes and Flavonoids in Five Argentinian Species of Polygonum Genus. J. Essent. Oil Res. 2011, 23, 11–14. [Google Scholar] [CrossRef]
- Russo, A.; Cardile, V.; Graziano, A.C.E.; Avola, R.; Montenegro, I.; Cuellar, M.; Villena, J.; Madrid, A. Antigrowth activity and induction of apoptosis in human melanoma cells by Drymis winteri forst extract and its active components. Chem. Biol. Interact 2019, 305, 79–85. [Google Scholar] [CrossRef]
- Montenegro, I.; Madrid, A.; Cuellar, M.; Seeger, M.; Alfaro, J.F.; Besoain, X.; Martinez, J.P.; Ramirez, I.; Olguin, Y.; Valenzuela, M. Biopesticide Activity from Drimanic Compounds to Control Tomato Pathogens. Molecules 2018, 23, 2053. [Google Scholar] [CrossRef]
- Derita, M.; Montenegro, I.; Garibotto, F.; Enriz, R.D.; Fritis, M.C.; Zacchino, S.A. Structural Requirements for the Antifungal Activities of Natural Drimane Sesquiterpenes and Analogues, Supported by Conformational and Electronic Studies. Molecules 2013, 18, 2029–2051. [Google Scholar] [CrossRef] [PubMed]
- Edouarzin, E.; Horn, C.; Paudyal, A.; Zhang, C.; Lu, J.; Tong, Z.; Giaever, G.; Nislow, C.; Veerapandian, R.; Hua, D.H.; et al. Broad-spectrum antifungal activities and mechanism of drimane sesquiterpenoids. Microb. Cell 2020, 7, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Delgado, V.; Saitz, C.; Armstrong, V. Drimenol: A versatile synthon for compounds with trans-drimane skeleton. Nat. Prod. Commun. 2011, 6, 477–490. [Google Scholar] [CrossRef]
- Marcos, I.S.; Conde, A.; Moro, R.F.; Basabe, P.; Diez, D.; Urones, J.G. Quinone/Hydroquinone Sesquiterpenes. Mini-Rev. Org. Chem. 2010, 7, 230–254. [Google Scholar] [CrossRef]
- Coval, S.J.; Conover, M.A.; Mierzwa, R.; King, A.; Puar, M.S. Wiedendiol-a and Wiedendiol-B, Cholesteryl Ester Transfer Protein Inhibitors from the Marine Sponge Xestosponglia-Wiedenmayeri. Bioorg. Med. Chem. Lett. 1995, 5, 605–610. [Google Scholar] [CrossRef]
- Rodriguez, J.; Quinoa, E.; Riguera, R.; Peters, B.M.; Abrell, L.M.; Crews, P. The Structures and Stereochemistry of Cytotoxic Sesquiterpene Quinones from Dactylospongia elegans. Tetrahedron 1992, 48, 6667–6680. [Google Scholar] [CrossRef]
- Venkateswarlu, Y.; Faulkner, D.J.; Steiner, J.L.R.; Corcoran, E.; Clardy, J. Smenochromenes, unusual macrocyclic sesquiterpene hydroquinone derivatives from a Seychelles sponge of the genus Smenospongia. J. Org. Chem. 1991, 56, 6271–6274. [Google Scholar] [CrossRef]
- Jiso, A.; Demuth, P.; Bachowsky, M.; Haas, M.; Seiwert, N.; Heylmann, D.; Rasenberger, B.; Christmann, M.; Dietrich, L.; Brunner, T.; et al. Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer. Cancers 2021, 13, 3282. [Google Scholar] [CrossRef] [PubMed]
- Zhi-Hui, D.; Ze-Jun, D.; Ji-Kai, L. Albaconol, A Novel Prenylated Resorcinol (=Benzene-1,3-diol) from Basidiomycetes Albatrellus confluens. Helv. Chim. Acta 2001, 84, 259–262. [Google Scholar] [CrossRef]
- Deng, Q.; Yu, X.; Xiao, L.; Hu, Z.; Luo, X.; Tao, Y.; Yang, L.; Liu, X.; Chen, H.; Ding, Z.; et al. Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis. 2013, 4, e804. [Google Scholar] [CrossRef]
- Qing, C.; Liu, M.H.; Yangi, W.M.; Zhang, Y.L.; Wang, L.; Liu, J.K. Effects of albaconol from the basidiomycete Albatrellus confluens on DNA topoisomerase II-mediated DNA cleavage and relaxation. Planta Med. 2004, 70, 792–796. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Krogh, B.O.; Shuman, S. Catalytic Mechanism of DNA Topoisomerase IB. Mol. Cell 2000, 5, 1035–1041. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; López, J.T.; Cortés, M.J. (−)-3β-Acetoxydrimenin from the leaves of Drimys winteri. Phytochemistry 1985, 25, 253–254. [Google Scholar] [CrossRef]
- Aricu, A.N.; Kuchkova, K.I.; Secara, E.S.; Barba, A.N.; Dragalin, I.P.; Ungur, N.D.; Mel’nik, E.; Kravtsov, V.K. Synthesis and Structure of Drimane Sesquiterpenoids Containing Pyrimidine, Pyrazine, 1,2,4-triazole, and Carbazole Rings. Chem. Nat. Compd. 2018, 54, 455–460. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Collins, J.M.; Doroshow, J.H. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol. Cancer 2010, 9, 1451–1460. [Google Scholar] [CrossRef]
- Quiñones, N.; Hernández, S.; Espinoza Catalán, L.; Villena, J.; Brito, I.; Cabrera, A.R.; Salas, C.O.; Cuellar, M.A. (-)-Shikimic Acid as a Chiral Building Block for the Synthesis of New Cytotoxic 6-Aza-Analogues of Angucyclinones. Molecules 2018, 23, 1422. [Google Scholar] [CrossRef]
- Taborga, L.; Espinoza, L.; Moller, A.; Carrasco, H.; Cuellar, M.; Villena, J. Antiproliferative effect and apoptotic activity of linear geranylphenol derivatives from phloroglucinol and orcinol. Chem.-Biol. Interact. 2016, 247, 22–29. [Google Scholar] [CrossRef]
- Villena, J.; Montenegro, I.; Said, B.; Werner, E.; Flores, S.; Madrid, A. Ultrasound assisted synthesis and cytotoxicity evaluation of known 2′,4′-dihydroxychalcone derivatives against cancer cell lines. Food Chem. Toxicol. 2021, 148, 111969. [Google Scholar] [CrossRef]
- Montenegro, I.; Tomasoni, G.; Bosio, C.; Quiñones, N.; Madrid, A.; Carrasco, H.; Olea, A.; Martinez, R.; Cuellar, M.; Villena, J. Study on the Cytotoxic Activity of Drimane Sesquiterpenes and Nordrimane Compounds against Cancer Cell Lines. Molecules 2014, 19, 18993–19006. [Google Scholar] [CrossRef]
- Cyr, L.; Langler, R.; Lavigne, C. Cell cycle arrest and apoptosis responses of human breast epithelial cells to the synthetic organosulfur compound p-methoxyphenyl p-toluenesulfonate. Anticancer Res 2008, 28, 2753–2763. [Google Scholar]
- Sordet, O.; Khan, Q.A.; Kohn, K.W.; Pommier, Y. Apoptosis induced by topoisomerase inhibitors. Curr. Med. Chem. Anticancer Agents 2003, 3, 271–290. [Google Scholar] [CrossRef]
- David Foglesong, P.; Reckord, C.; Swink, S. Doxorubicin inhibits human DNA topoisomerase I. Cancer Chemother. Pharmacol. 1992, 30, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, S.; Song, Z.; Li, W.; Zhu, F.; Zhang, J.; Li, S. Synthesis and bio-inspired optimization of drimenal: Discovery of chiral drimane fused oxazinones as promising antifungal and antibacterial candidates. Eur. J. Med. Chem. 2018, 143, 558–567. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) a | |||||
|---|---|---|---|---|---|
| Compounds | MCF-7 | HT-29 | PC-3 | MCF-10 | |
| 6a | 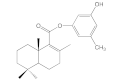 | 33.7 ± 1.3 | 56.9 ± 4.0 | 85.5 ± 7.1 | 67.2 ± 6.3 |
| 6b | 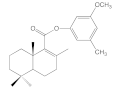 | >100 | >100 | >100 | 67.2 ± 15.3 |
| 6c |  | >100 | >100 | >100 | >100 |
| 6d |  | >100 | >100 | >100 | >100 |
| 6e | 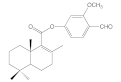 | 38.3 ± 2.6 | 38.1 ± 2.0 | 52.9 ± 8.2 | 50.0 ± 8.9 |
| 6f | 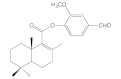 | 22.4 ± 1.0 | 58.0 ± 3.9 | 30.9 ± 2.5 | 16.5 ± 0.9 |
| 8a |  | >100 | >100 | >100 | 44.8 ± 3.1 |
| 8b | 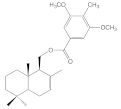 | >100 | >100 | >100 | 21.2 ± 3.1 |
| 8c | 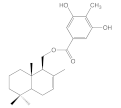 | 35.9 ± 6.4 | >100 | >100 | >100 |
| 8d | 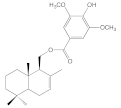 | 18.4 ± 0.5 | 44.7 ± 6.7 | 33.3 ± 2.9 | 16.5 ± 0.5 |
| 8e | 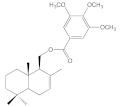 | 51.0 ± 10.0 | 51.5 ± 3.2 | 76.9 ± 10.2 | 32.1 ± 8.3 |
| 8f | 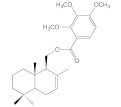 | 6.2 ± 0.5 | 26.2 ± 5.4 | 7.1 ± 1.1 | 6.8 ± 0.9 |
| 8g |  | 28.7 ± 3.5 | 31.3 ± 2.0 | 27.7 ± 2.6 | 16.3 ± 0.6 |
| Drimenol | >100 b | n.d. | >100 b | n.d. | |
| Daunorubicin | 0.33 ± 0.02 c | 15.11 ± 0.5 c | 0.41 ± 0.04 c | 9.32 ± 1.01 c | |
| 5-FU | 22.3 ± 0.2 c | 2.9 ± 0.7 c | 16.4 ± 0.6 c | <0.5 d | |
| Compound | Selectivity Index (SI) a | ||
|---|---|---|---|
| MCF-10/MCF-7 | MCF-10/HT-29 | MCF-10/PC-3 | |
| 6a | 2.0 | 1.2 | 0.8 |
| 6e | 1.3 | 1.3 | 0.9 |
| 6f | 0.7 | 0.3 | 0.5 |
| 8c | 2.8 | n.d. | n.d. |
| 8d | 0.9 | 0.4 | 0.5 |
| 8e | 0.6 | 0.6 | 0.4 |
| 8f | 1.1 | 0.3 | 0.9 |
| 8g | 0.6 | 0.5 | 0.6 |
| Daunorubicin | 28 | 0.6 | 23 |
| 5-FU | 0.02 | 0.2 | 0.03 |
| Compound | MW (Da) | HBA | HBD | cLogP | TPSA (A2) | NRB |
|---|---|---|---|---|---|---|
| Desirable value | ≤500 | ≤10 | ≤5 | ≤5 | ≤140 | ≤10 |
| 6a | 342.47 | 3 | 1 | 5.01 | 46.53 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araque, I.; Ramírez, J.; Vergara, R.; Mella, J.; Aránguiz, P.; Espinoza, L.; Vera, W.; Montenegro, I.; Salas, C.O.; Villena, J.; et al. Cytotoxic Activity, Topoisomerase I Inhibition and In Silico Studies of New Sesquiterpene-aryl Ester Derivatives of (-) Drimenol. Molecules 2023, 28, 3959. https://doi.org/10.3390/molecules28093959
Araque I, Ramírez J, Vergara R, Mella J, Aránguiz P, Espinoza L, Vera W, Montenegro I, Salas CO, Villena J, et al. Cytotoxic Activity, Topoisomerase I Inhibition and In Silico Studies of New Sesquiterpene-aryl Ester Derivatives of (-) Drimenol. Molecules. 2023; 28(9):3959. https://doi.org/10.3390/molecules28093959
Chicago/Turabian StyleAraque, Ileana, Javiera Ramírez, Rut Vergara, Jaime Mella, Pablo Aránguiz, Luis Espinoza, Waleska Vera, Iván Montenegro, Cristian O. Salas, Joan Villena, and et al. 2023. "Cytotoxic Activity, Topoisomerase I Inhibition and In Silico Studies of New Sesquiterpene-aryl Ester Derivatives of (-) Drimenol" Molecules 28, no. 9: 3959. https://doi.org/10.3390/molecules28093959
APA StyleAraque, I., Ramírez, J., Vergara, R., Mella, J., Aránguiz, P., Espinoza, L., Vera, W., Montenegro, I., Salas, C. O., Villena, J., & Cuellar, M. A. (2023). Cytotoxic Activity, Topoisomerase I Inhibition and In Silico Studies of New Sesquiterpene-aryl Ester Derivatives of (-) Drimenol. Molecules, 28(9), 3959. https://doi.org/10.3390/molecules28093959