
Computer-Aided Prediction of the Interactions of Viral Proteases with Antiviral Drugs: Antiviral Potential of Broad-Spectrum Drugs


Abstract
:1. Introduction
2. Results
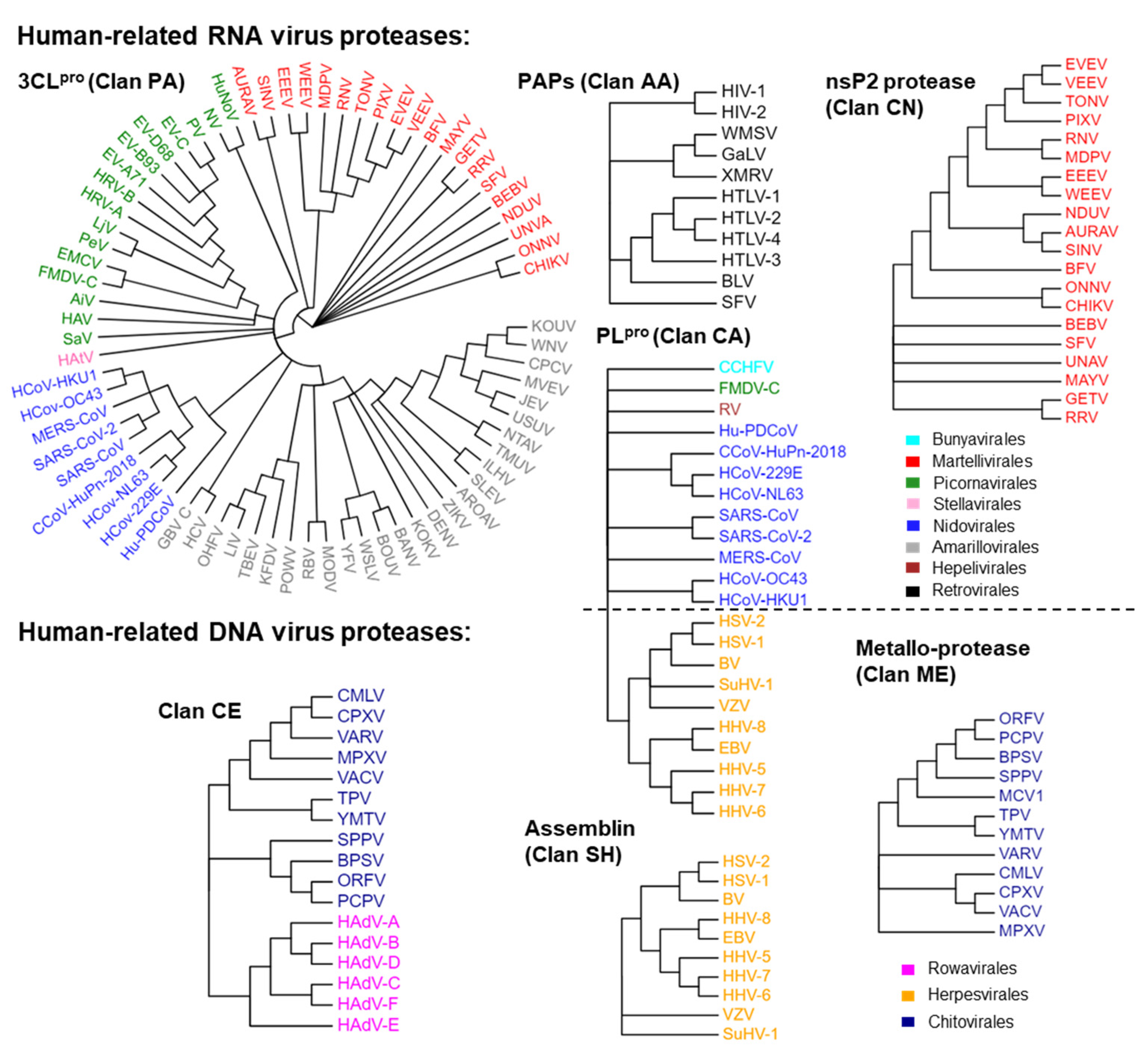
2.1. The Protease Profiles of Human-Infective Viruses
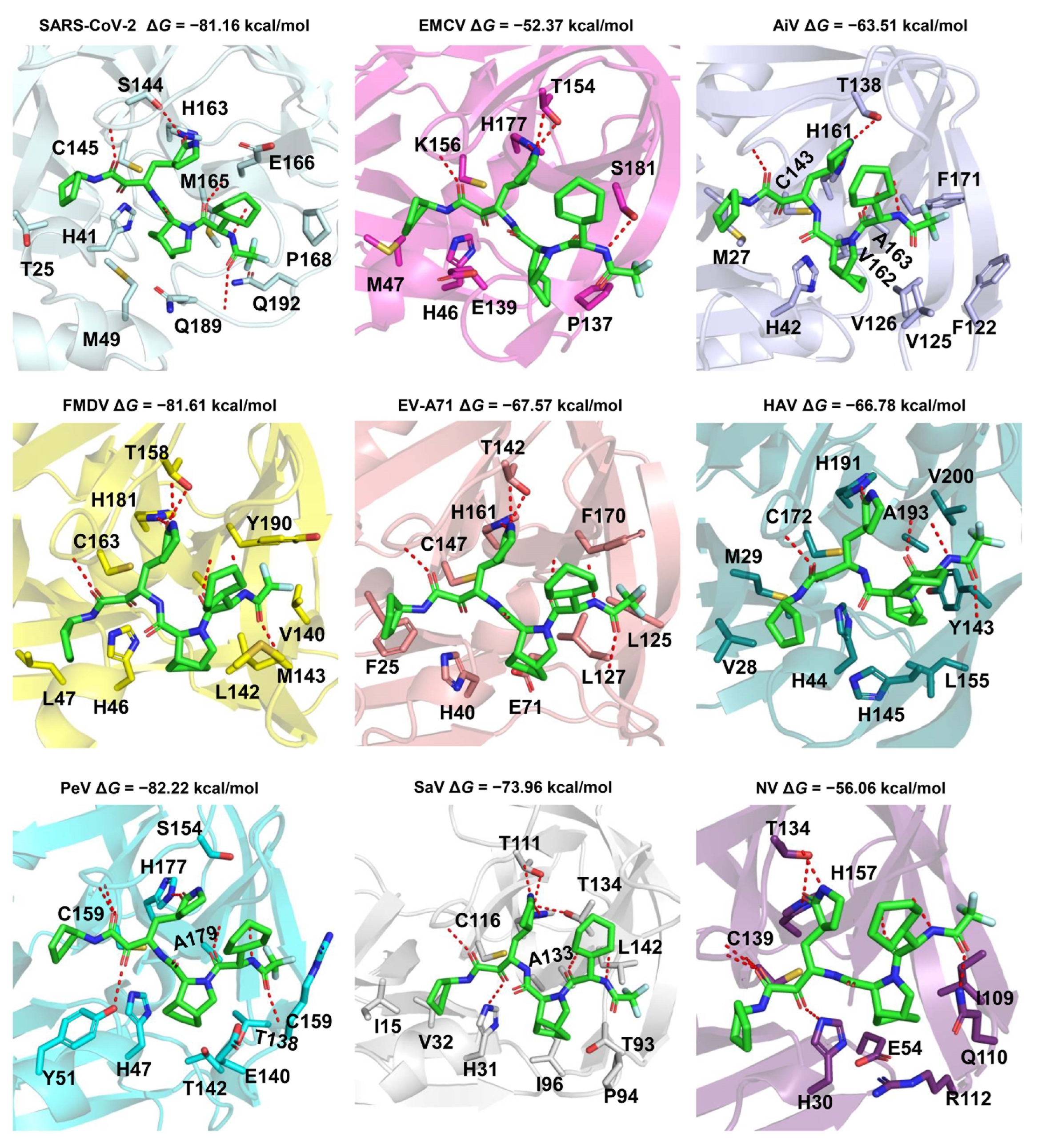
2.2. 3-Chymotrypsin-Like Proteases
2.2.1. The Sequences, Structures, and Substrates of the Class of 3CLpro
2.2.2. The Drug-Protein Complex Structure Prediction and Modeling Method
2.2.3. Drug Repurposing
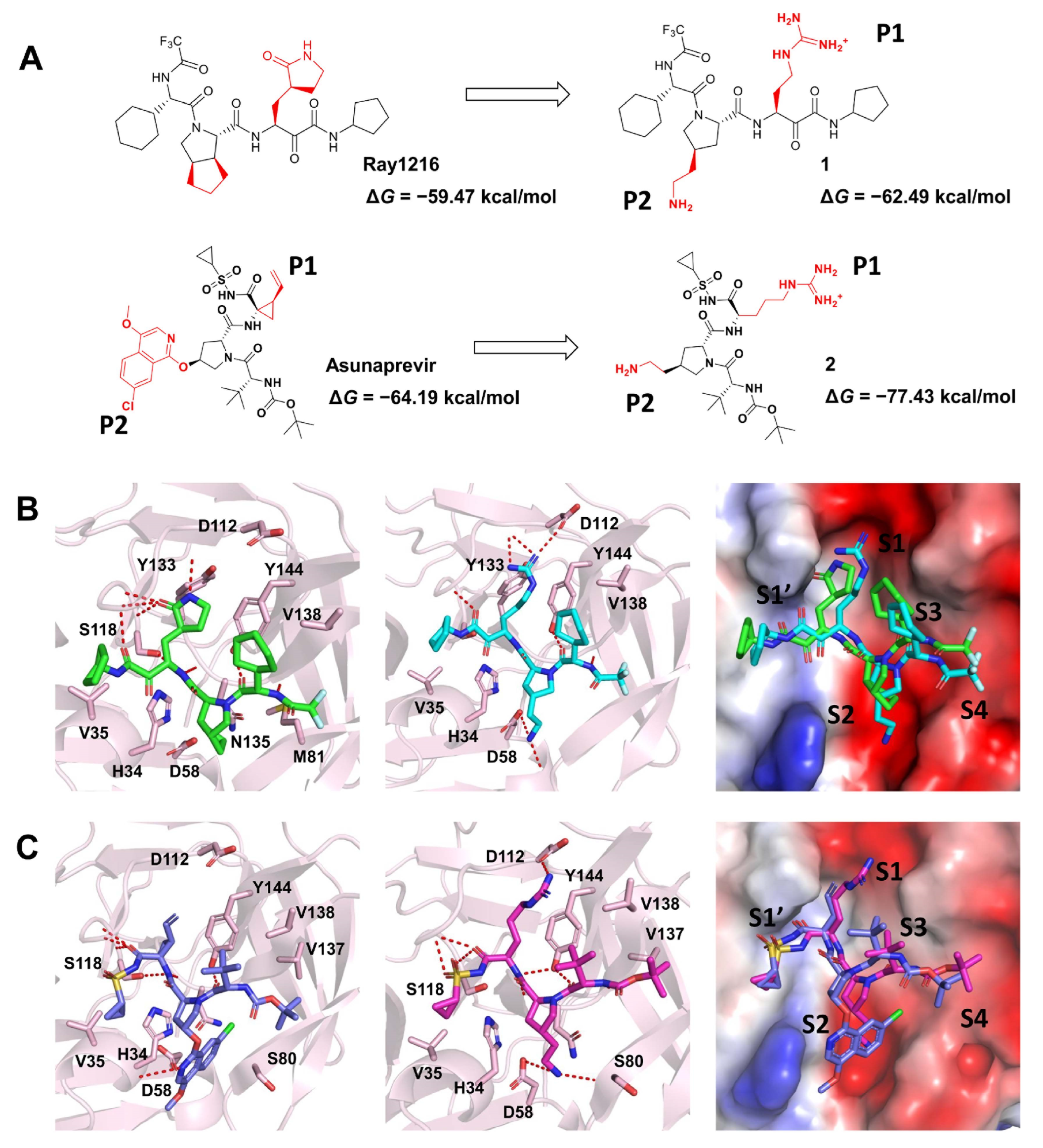
2.2.4. Proposal for a Drug Optimization Strategy
2.3. Pepsin-Like Aspartic Proteases
3. Discussion and Conclusions
4. Materials and Methods
4.1. Data Collection
4.2. The Protease Profiles of Human-Infective Viruses
4.3. Sequence Alignment
4.4. Three-Dimensional Structure Prediction
4.5. The Computational Ligand-Protein Complex Structure Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AiV | Aichi virus | HuNoV | Human norovirus |
| AROAV | Aroa virus | Hu-PDCoV | Human-Porcine Delta coronavirus |
| AURAV | Aura virus | ILHV | Ilheus virus |
| BANV | Banzi virus | JEV | Japanese encephalitis virus |
| BEBV | Bebaru virus | KFDV | Kyasanur Forest disease virus |
| BFV | Barmah Forest virus | KOKV | Kokobera virus |
| BLV | Bovine leukemia virus | KOUV | Koutango virus |
| BOUV | Bouboui virus | LIV | Louping ill virus |
| BPSV | Bovine papular stomatitis virus | LjV | Ljungan virus |
| BToV | Bovine torovirus | MAYV | Mayaro virus |
| BV | Cercopithecine herpesvirus 1 | MCV-1 | Molluscum contagiosum virus |
| CCHFV | Crimean-Congo hemorrhagic fever virus | MDPV | Mosso das Pedras virus |
| CCoV-HuPn-2018 | Canine coronavirus-human pneumonia-2018 | MERS-CoV | Middle East respiratory syndrome |
| CHIKV | Chikungunya virus | MODV | Modoc virus |
| CMLV | Camelpox virus | MPXV | Monkeypox virus |
| CPCV | Cacipacore virus | MVEV | Murray Valley encephalitis virus |
| CPXV | Cowpox virus | NDUV | Ndumu virus |
| DENV | Dengue virus | NTAV | Ntaya virus |
| EBV | Human herpesvirus 4 (Epstein-Barr Virus) | NV | Norwalk virus |
| EEEV | Eastern equine encephalitis virus | OHFV | Omsk hemorrhagic fever virus |
| EMCV | Encephalomyocarditis virus | ONNV | O’nyong-nyong virus |
| EV-A71 | Enterovirus A71 | ORFV | Orf virus |
| EV-B93 | Enterovirus B93 | PCPV | Pseudocowpox virus |
| EV-C | Enterovirus C | PeV | Parechovirus |
| EV-D68 | Enterovirus D68 | PIXV | Pixuna virus |
| EVEV | Everglades virus | POWV | Powassan virus |
| FMDV-C | Foot-and-mouth disease virus C | PV | Poliovirus (EV-C) |
| GaLV | Gibbon ape leukemia virus | RBV | Rio Bravo virus |
| GBV C | GB virus C | RNV | Rio Negro virus |
| GETV | Getah virus | RRV | Ross River virus |
| HAdV-A | Human adenovirus A | RV | Rubella virus |
| HAdV-B | Human adenovirus B | SARS-CoV | Severe acute respiratory syndrome |
| HAdV-C | Human adenovirus C | SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| HAdV-D | Human adenovirus D | SaV | Sapporo virus |
| HAdV-E | Human adenovirus E | SFV | Simian foamy virus |
| HAdV-F | Human adenovirus F | SFV | Semliki Forest virus |
| HAtV | Human astrovirus | SINV | Sindbis virus |
| HAV | Hepatitis A virus | SLEV | St. Louis encephalitis virus |
| HCoV 229E | Human coronavirus 229E | SPPV | Sealpox virus |
| HCoV HKU1 | Human coronavirus HKU1 | SuHV-1 | Suid herpesvirus 1 |
| HCoV NL63 | Human coronavirus NL63 | TBEV | Tick-borne encephalitis virus |
| HCoV OC43 | Human coronavirus OC43 | TMUV | Tembusu virus |
| HCV | Hepatitis C Virus | TONV | Tonate virus |
| HHV-5/HCMV | Human herpesvirus 5 | TPV | Tanapox virus |
| HHV-6 | Human herpesvirus 6 | UNVA | Una virus |
| HHV-7 | Human herpesvirus 7 | USUV | Usutu virus |
| HHV-8 | Human herpesvirus 8 | VACV | Vaccinia virus |
| HIV-1 | Human immunodeficiency virus 1 | VARV | Variola virus |
| HIV-2 | Human immunodeficiency virus 2 | VEEV | Venezuelan equine encephalitis virus |
| HRV-A | Human rhinovirus A | WEEV | Western equine encephalitis virus |
| HRV-B | Human rhinovirus B | WMSV | Woolly monkey sarcoma virus |
| HSV-1 | Human herpesvirus 1 | WNV | West Nile virus |
| HSV-2 | Human herpesvirus 2 | WSLV | Wesselsbron virus |
| HSV-3 (VZV) | Human herpesvirus 3 (Varicella-zostervirus) | XMRV | Xenotropic murine leukemia virus-related virus |
| HTLV-1 | Primate T-lymphotropic virus 1 | YFV | Yellow fever virus |
| HTLV-2 | Primate T-lymphotropic virus 2 | YMTV | Yaba monkey tumor virus |
| HTLV-3 | Primate T-lymphotropic virus 3 | ZIKV | Zika virus |
| HTLV-4 | Primate T-lymphotropic virus 4 |
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Statement on the Fifteenth Meeting of the IHR (2005) Emergency Committee on the COVID-19 Pandemic. Available online: https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic (accessed on 1 November 2023).
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Mahieux, R.; Gessain, A. The human HTLV-3 and HTLV-4 retroviruses: New members of the HTLV family. Pathol. Biol. 2009, 57, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 2021, 600, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.M.; Wolthers, K.C.; Wertheim-van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.-H.; Lee, J.S.-Y.; Saif, L.J.; Gray, G.C. Novel canine coronavirus isolated from a hospitalized patient with pneumonia in east malaysia. Clin. Infect. Dis. 2021, 74, 446–454. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Yuen, K.-Y. More and more coronaviruses: Human coronavirus HKU1. Viruses 2009, 1, 57–71. [Google Scholar] [CrossRef]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Hijikata, M.; Mizushima, H.; Tanji, Y.; Komoda, Y.; Hirowatari, Y.; Akagi, T.; Kato, N.; Kimura, K.; Shimotohno, K. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl. Acad. Sci. USA 1993, 90, 10773–10777. [Google Scholar] [CrossRef] [PubMed]
- Raney, K.D.; Sharma, S.D.; Moustafa, I.M.; Cameron, C.E. Hepatitis C virus non-structural protein 3 (HCV NS3): A multifunctional antiviral target. J. Biol. Chem. 2010, 285, 22725–22731. [Google Scholar] [CrossRef] [PubMed]
- Brik, A.; Wong, C.-H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Su, H.; Shang, W.; Zhou, F.; Zhang, Y.; Zhao, W.; Zhang, Q.; Xie, H.; Jiang, L.; Nie, T.; et al. Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir. Nat. Commun. 2023, 14, 6463. [Google Scholar] [CrossRef]
- Chen, X.; Huang, X.; Ma, Q.; Kuzmič, P.; Zhou, B.; Xu, J.; Liu, B.; Jiang, H.; Zhang, W.; Yang, C.; et al. Inhibition mechanism and antiviral activity of an α-ketoamide based SARS-CoV-2 main protease inhibitor. bioRxiv 2023. [Google Scholar] [CrossRef]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1207–1217. [Google Scholar] [CrossRef]
- Lin, C.; Kwong, D.A.; Perni, B.R. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of Hepatitis C virus NS3.4A serine protease. Infect. Disord.—Drug Targets 2006, 6, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Arasappan, A.; Bennett, F.; Bogen, S.L.; Venkatraman, S.; Blackman, M.; Chen, K.X.; Hendrata, S.; Huang, Y.; Huelgas, R.M.; Nair, L.; et al. Discovery of narlaprevir (SCH 900518): A potent, second generation HCV NS3 serine protease inhibitor. ACS Med. Chem. Lett. 2010, 1, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Gardiner, D.F.; Lawitz, E.; Martorell, C.; Everson, G.T.; Ghalib, R.; Reindollar, R.; Rustgi, V.; McPhee, F.; Wind-Rotolo, M.; et al. Preliminary study of two antiviral agents for Hepatitis C genotype 1. N. Engl. J. Med. 2012, 366, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Andrews, S.W.; Condroski, K.R.; Buckman, B.; Serebryany, V.; Wenglowsky, S.; Kennedy, A.L.; Madduru, M.R.; Wang, B.; Lyon, M.; et al. Discovery of Danoprevir (ITMN-191/R7227), a highly selective and potent inhibitor of Hepatitis C Virus (HCV) NS3/4A protease. J. Med. Chem. 2014, 57, 1753–1769. [Google Scholar] [CrossRef]
- Gentile, I.; Borgia, F.; Buonomo, R.A.; Zappulo, E.; Castaldo, G.; Borgia, G. ABT-450: A novel protease inhibitor for the treatment of Hepatitis C Virus infection. Curr. Med. Chem. 2014, 21, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-I.; Lenz, O.; Fanning, G.; Verbinnen, T.; Delouvroy, F.; Scholliers, A.; Vermeiren, K.; Rosenquist, Å.; Edlund, M.; Samuelsson, B.; et al. In Vitro activity and preclinical profile of TMC435350, a potent Hepatitis C Virus protease inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. [Google Scholar] [CrossRef]
- McCauley, J.A.; McIntyre, C.J.; Rudd, M.T.; Nguyen, K.T.; Romano, J.J.; Butcher, J.W.; Gilbert, K.F.; Bush, K.J.; Holloway, M.K.; Swestock, J.; et al. Discovery of Vaniprevir (MK-7009), a macrocyclic Hepatitis C Virus NS3/4a protease inhibitor. J. Med. Chem. 2010, 53, 2443–2463. [Google Scholar] [CrossRef]
- Harper, S.; McCauley, J.A.; Rudd, M.T.; Ferrara, M.; DiFilippo, M.; Crescenzi, B.; Koch, U.; Petrocchi, A.; Holloway, M.K.; Butcher, J.W.; et al. Discovery of MK-5172, a macrocyclic Hepatitis C Virus NS3/4a protease inhibitor. ACS Med. Chem. Lett. 2012, 3, 332–336. [Google Scholar] [CrossRef]
- Taylor, J.G.; Zipfel, S.; Ramey, K.; Vivian, R.; Schrier, A.; Karki, K.K.; Katana, A.; Kato, D.; Kobayashi, T.; Martinez, R.; et al. Discovery of the pan-genotypic hepatitis C virus NS3/4A protease inhibitor voxilaprevir (GS-9857): A component of Vosevi®. Bioorganic Med. Chem. Lett. 2019, 29, 2428–2436. [Google Scholar] [CrossRef]
- Ng Teresa, I.; Tripathi, R.; Reisch, T.; Lu, L.; Middleton, T.; Hopkins Todd, A.; Pithawalla, R.; Irvin, M.; Dekhtyar, T.; Krishnan, P.; et al. In Vitro antiviral activity and resistance profile of the next-generation Hepatitis C Virus NS3/4A protease inhibitor Glecaprevir. Antimicrob. Agents Chemother. 2017, 62, 10–1128. [Google Scholar] [CrossRef]
- Zephyr, J.; Nageswara Rao, D.; Vo, S.V.; Henes, M.; Kosovrasti, K.; Matthew, A.N.; Hedger, A.K.; Timm, J.; Chan, E.T.; Ali, A.; et al. Deciphering the molecular mechanism of HCV protease inhibitor Fluorination as a general approach to avoid drug resistance. J. Mol. Biol. 2022, 434, 167503. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.C.; Sarin, P.S.; Gelmann, E.P.; Robert-Guroff, M.; Richardson, E.; Kalyanaraman, V.S.; Mann, D.; Sidhu, G.D.; Stahl, R.E.; Zolla-Pazner, S.; et al. Isolation of Human T-Cell Leukemia Virus in Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 865–867. [Google Scholar] [CrossRef] [PubMed]
- James, J.S. Saquinavir (Invirase): First protease inhibitor approved--reimbursement, information hotline numbers. AIDS Treat. News 1995, 237, 1–2. [Google Scholar]
- Kempf, D.J.; Sham, H.L.; Marsh, K.C.; Flentge, C.A.; Betebenner, D.; Green, B.E.; McDonald, E.; Vasavanonda, S.; Saldivar, A.; Wideburg, N.E.; et al. Discovery of Ritonavir, a potent inhibitor of HIV protease with high oral bioavailability and clinical efficacy. J. Med. Chem. 1998, 41, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Vacca, J.P.; Dorsey, B.D.; Schleif, W.A.; Levin, R.B.; McDaniel, S.L.; Darke, P.L.; Zugay, J.; Quintero, J.C.; Blahy, O.M.; Roth, E. L-735,524: An orally bioavailable human immunodeficiency virus type 1 protease inhibitor. Proc. Natl. Acad. Sci. USA 1994, 91, 4096–4100. [Google Scholar] [CrossRef]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F.; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (Nelfinavir Mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Clair, M.S.; Millard, J.; Rooney, J.; Tisdale, M.; Parry, N.; Sadler, B.M.; Blum, M.R.; Painter, G. In vitro antiviral activity of 141W94 (VX-478) in combination with other antiretroviral agents. Antivir. Res. 1996, 29, 53–56. [Google Scholar] [CrossRef]
- Sham, H.L.; Kempf, D.J.; Molla, A.; Marsh, K.C.; Kumar, G.N.; Chen, C.-M.; Kati, W.; Stewart, K.; Lal, R.; Hsu, A.; et al. ABT-378, a highly potent inhibitor of the Human Immunodeficiency Virus protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224. [Google Scholar] [CrossRef]
- Robinson, B.S.; Riccardi, K.A.; Gong, Y.-f.; Guo, Q.; Stock, D.A.; Blair, W.S.; Terry, B.J.; Deminie, C.A.; Djang, F.; Colonno, R.J.; et al. BMS-232632, a highly potent Human Immunodeficiency Virus protease inhibitor that can be used in combination with other available antiretroviral agents. Antimicrob. Agents Chemother. 2000, 44, 2093–2099. [Google Scholar] [CrossRef]
- Flexner, C.; Bate, G.; Kirkpatrick, P. Tipranavir. Nat. Rev. Drug Discov. 2005, 4, 955–956. [Google Scholar] [CrossRef]
- Molina, J.-M.; Hill, A. Darunavir (TMC114): A new HIV-1 protease inhibitor. Expert Opin. Pharmacother. 2007, 8, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Sun, S.-Q.; Guo, H.-C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, Y.; Zheng, M. Enterovirus A71 antivirals: Past, present, and future. Acta Pharm. Sin. B 2022, 12, 1542–1566. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K.; Binford, S.L.; Brothers, M.A.; Jackson, R.L.; Ford, C.E.; Diem, M.D.; Maldonado, F.; Dragovich, P.S.; Zhou, R.; Prins, T.J.; et al. In Vitro antiviral activity of AG7088, a potent inhibitor of Human Rhinovirus 3C protease. Antimicrob. Agents Chemother. 1999, 43, 2444–2450. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Galasiti Kankanamalage, A.C.; Chang, K.-O.; Groutas, W.C. Recent advances in the discovery of Norovirus therapeutics. J. Med. Chem. 2015, 58, 9438–9450. [Google Scholar] [CrossRef] [PubMed]
- Behnam, M.A.M.; Nitsche, C.; Boldescu, V.; Klein, C.D. The medicinal chemistry of Dengue Virus. J. Med. Chem. 2016, 59, 5622–5649. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A review of virus–host interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef]
- Melancon, P.; Garoff, H. Processing of the Semliki Forest virus structural polyprotein: Role of the capsid protease. J. Virol. 1987, 61, 1301–1309. [Google Scholar] [CrossRef]
- Skoging, U.; Liljeström, P. Role of the C-terminal tryptophan residue for the structure-function of the alphavirus capsid protein11Edited by J. Karn. J. Mol. Biol. 1998, 279, 865–872. [Google Scholar] [CrossRef]
- Thomas, S.; Rai, J.; John, L.; Günther, S.; Drosten, C.; Pützer, B.M.; Schaefer, S. Functional dissection of the alphavirus capsid protease: Sequence requirements for activity. Virol. J. 2010, 7, 327. [Google Scholar] [CrossRef]
- Yi, J.; Peng, J.; Yang, W.; Zhu, G.; Ren, J.; Li, D.; Zheng, H. Picornavirus 3C—A protease ensuring virus replication and subverting host responses. J. Cell Sci. 2021, 134, jcs253237. [Google Scholar] [CrossRef] [PubMed]
- Speroni, S.; Rohayem, J.; Nenci, S.; Bonivento, D.; Robel, I.; Barthel, J.; Luzhkov, V.B.; Coutard, B.; Canard, B.; Mattevi, A. Structural and biochemical analysis of human pathogenic Astrovirus serine protease at 2.0 Å resolution. J. Mol. Biol. 2009, 387, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534–16539. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Gao, X.; Tang, K.; Cai, J.-P.; Hu, M.; Luo, P.; Wen, L.; Ye, Z.-W.; Luo, C.; Tsang, J.O.-L.; et al. Targeting papain-like protease for broad-spectrum coronavirus inhibition. Protein Cell 2022, 13, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Elkin, B.A.; Knaap, R.C.M.; Kikkert, M.; Mark, B.L. Structure and function of viral deubiquitinating enzymes. J. Mol. Biol. 2017, 429, 3441–3470. [Google Scholar] [CrossRef]
- Mielech, A.M.; Chen, Y.; Mesecar, A.D.; Baker, S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014, 194, 184–190. [Google Scholar] [CrossRef]
- Durie, I.A.; Dzimianski, J.V.; Daczkowski, C.M.; McGuire, J.; Faaberg, K.; Pegan, S.D. Structural insights into the interaction of papain-like protease 2 from the alphacoronavirus porcine epidemic diarrhea virus and ubiquitin. Acta Crystallogr. D Struct. Biol. 2021, 77, 943–953. [Google Scholar] [CrossRef]
- Cheong, E.Z.K.; Quek, J.P.; Xin, L.; Li, C.; Chan, J.Y.; Liew, C.W.; Mu, Y.; Zheng, J.; Luo, D. Crystal structure of the Rubella virus protease reveals a unique papain-like protease fold. J. Biol. Chem. 2022, 298, 102250. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Bailey-Elkin, B.A.; Mark, B.L.; Götte, M. Independent inhibition of the polymerase and deubiquitinase activities of the Crimean-Congo Hemorrhagic Fever Virus full-length L-protein. PLoS Neglected Trop. Dis. 2020, 14, e0008283. [Google Scholar] [CrossRef]
- Zühlsdorf, M.; Hinrichs, W. Assemblins as maturational proteases in herpesviruses. J. Gen. Virol. 2017, 98, 1969–1984. [Google Scholar] [CrossRef]
- Hall, D.L.; Darke, P.L. Activation of the Herpes Simplex Virus Type 1 Protease (∗). J. Biol. Chem. 1995, 270, 22697–22700. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.E.K.; Tuske, S.; Das, K.; Ruiz, F.X.; Bauman, J.D.; Boyer, P.L.; DeStefano, J.J.; Hughes, S.H.; Arnold, E. Crystal structure of a Retroviral polyprotein: Prototype Foamy Virus Protease-Reverse Transcriptase (PR-RT). Viruses 2021, 13, 1495. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-S.; Li, H.-C.; Lin, S.-I.; Yang, C.-H.; Chien, W.-Y.; Syu, C.-L.; Lo, S.-Y. Cleavage of dicer protein by I7 protease during vaccinia virus infection. PLoS ONE 2015, 10, e0120390. [Google Scholar] [CrossRef] [PubMed]
- Ansarah-Sobrinho, C.; Moss, B. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 2004, 78, 6335–6343. [Google Scholar] [CrossRef] [PubMed]
- Ruzindana-Umunyana, A.; Sircar, S.; Weber, J.M. The effect of mutant peptide cofactors on Adenovirus protease activity and virus infection. Virology 2000, 270, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hedengren-Olcott, M.; Byrd, C.M.; Watson, J.; Hruby, D.E. The Vaccinia Virus G1L putative Metalloproteinase is essential for viral replication in vivo. J. Virol. 2004, 78, 9947–9953. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Shindyalov, I.N.; Bourne, P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998, 11, 739–747. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Joseph, M.H.; Georgios, A. MM-GB(PB)SA calculations of protein-ligand binding free energies. In Molecular Dynamics; Lichang, W., Ed.; IntechOpen: Rijeka, Croatia, 2012; Chapter 9. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.A.; Knox, S.R.; Sweeney, T.R.; Yang, J.; Roqué-Rosell, N.; Belsham, G.J.; Leatherbarrow, R.J.; Curry, S. Insights into cleavage specificity from the crystal structure of Foot-and-Mouth Disease Virus 3C protease complexed with a peptide substrate. J. Mol. Biol. 2010, 395, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Qi, J.; Chen, Z.; Xu, X.; Gao, F.; Lin, D.; Qian, W.; Liu, H.; Jiang, H.; Yan, J.; et al. Enterovirus 71 and Coxsackievirus A16 3C proteases: Binding to Rupintrivir and their substrates and anti-Hand, Foot, and Mouth Disease Virus drug design. J. Virol. 2011, 85, 10319–10331. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Cherney, M.M.; Bergmann, E.M.; Zhang, J.; Huitema, C.; Pettersson, H.; Eltis, L.D.; Vederas, J.C.; James, M.N.G. An episulfide cation (thiiranium ring) trapped in the active site of HAV 3C proteinase inactivated by peptide-based Ketone inhibitors. J. Mol. Biol. 2006, 361, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Galasiti Kankanamalage, A.C.; Kim, Y.; Rathnayake, A.D.; Damalanka, V.C.; Weerawarna, P.M.; Doyle, S.T.; Alsoudi, A.F.; Dissanayake, D.M.P.; Lushington, G.H.; Mehzabeen, N.; et al. Structure-based exploration and exploitation of the S4 subsite of norovirus 3CL protease in the design of potent and permeable inhibitors. Eur. J. Med. Chem. 2017, 126, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Morazzani, E.; Compton, J.R.; Harmon, M.; Soloveva, V.; Glass, P.J.; Garcia, A.D.; Marugan, J.J.; Legler, P.M. In silico screening of inhibitors of the Venezuelan Equine Encephalitis Virus nonstructural protein 2 cysteine protease. Viruses 2023, 15, 1503. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Sako, Y.; Nikaido, E.; Ueda, T.; Kozono, I.; Ichihashi, Y.; Nakahashi, A.; Onishi, M.; Yamatsu, Y.; Kato, T.; et al. Peptide-to-small molecule: Discovery of non-covalent, active-site inhibitors of β-Herpesvirus proteases. ACS Med. Chem. Lett. 2023, 14, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Grosche, P.; Sirockin, F.; Mac Sweeney, A.; Ramage, P.; Erbel, P.; Melkko, S.; Bernardi, A.; Hughes, N.; Ellis, D.; Combrink, K.D.; et al. Structure-based design and optimization of potent inhibitors of the adenoviral protease. Bioorganic Med. Chem. Lett. 2015, 25, 438–443. [Google Scholar] [CrossRef]
- Mac Sweeney, A.; Grosche, P.; Ellis, D.; Combrink, K.; Erbel, P.; Hughes, N.; Sirockin, F.; Melkko, S.; Bernardi, A.; Ramage, P.; et al. Discovery and structure-based optimization of Adenain inhibitors. ACS Med. Chem. Lett. 2014, 5, 937–941. [Google Scholar] [CrossRef]
- McGrath, W.J.; Ding, J.; Didwania, A.; Sweet, R.M.; Mangel, W.F. Crystallographic structure at 1.6-Å resolution of the human adenovirus proteinase in a covalent complex with its 11-amino-acid peptide cofactor: Insights on a new fold. Biochim. Et Biophys. Acta (BBA)—Proteins Proteom. 2003, 1648, 1–11. [Google Scholar] [CrossRef]
- Sinha, S.; Tam, B.; Wang, S.M. Applications of molecular dynamics simulation in protein study. Membranes 2022, 12, 844. [Google Scholar] [CrossRef]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Bennett, F.; Huang, Y.; Hendrata, S.; Lovey, R.; Bogen, S.L.; Pan, W.; Guo, Z.; Prongay, A.; Chen, K.X.; Arasappan, A.; et al. The introduction of P4 substituted 1-methylcyclohexyl groups into Boceprevir®: A change in direction in the search for a second generation HCV NS3 protease inhibitor. Bioorganic Med. Chem. Lett. 2010, 20, 2617–2621. [Google Scholar] [CrossRef]
- Romano, K.P.; Ali, A.; Aydin, C.; Soumana, D.; Özen, A.; Deveau, L.M.; Silver, C.; Cao, H.; Newton, A.; Petropoulos, C.J.; et al. The molecular basis of drug resistance against Hepatitis C Virus NS3/4A protease inhibitors. PLoS Pathog. 2012, 8, e1002832. [Google Scholar] [CrossRef]
- Soumana, D.I.; Ali, A.; Schiffer, C.A. Structural analysis of Asunaprevir resistance in HCV NS3/4A protease. ACS Chem. Biol. 2014, 9, 2485–2490. [Google Scholar] [CrossRef]
- Soumana, D.I.; Kurt Yilmaz, N.; Ali, A.; Prachanronarong, K.L.; Schiffer, C.A. Molecular and dynamic mechanism underlying drug resistance in genotype 3 Hepatitis C NS3/4A protease. J. Am. Chem. Soc. 2016, 138, 11850–11859. [Google Scholar] [CrossRef]
- Cummings, M.D.; Lindberg, J.; Lin, T.-I.; de Kock, H.; Lenz, O.; Lilja, E.; Felländer, S.; Baraznenok, V.; Nyström, S.; Nilsson, M.; et al. Induced-fit binding of the macrocyclic noncovalent inhibitor TMC435 to its HCV NS3/NS4A protease target. Angew. Chem. Int. Ed. 2010, 49, 1652–1655. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, N.E.; Ohanessian, M.; Biswas, S.; McGee, T.D., Jr.; Mahon, B.P.; Ostrov, D.A.; Garcia, J.; Tang, Y.; McKenna, R.; Roitberg, A.; et al. Defective hydrophobic sliding mechanism and active site expansion in HIV-1 protease drug resistant variant Gly48Thr/Leu89Met: Mechanisms for the loss of Saquinavir binding potency. Biochemistry 2015, 54, 422–433. [Google Scholar] [CrossRef]
- Liu, Z.; Yedidi, R.S.; Wang, Y.; Dewdney, T.G.; Reiter, S.J.; Brunzelle, J.S.; Kovari, I.A.; Kovari, L.C. Insights into the mechanism of drug resistance: X-ray structure analysis of multi-drug resistant HIV-1 protease ritonavir complex. Biochem. Biophys. Res. Commun. 2013, 431, 232–238. [Google Scholar] [CrossRef]
- Coman, R.M.; Robbins, A.H.; Fernandez, M.A.; Gilliland, C.T.; Sochet, A.A.; Goodenow, M.M.; McKenna, R.; Dunn, B.M. The contribution of naturally occurring polymorphisms in altering the biochemical and structural characteristics of HIV-1 subtype C protease. Biochemistry 2008, 47, 731–743. [Google Scholar] [CrossRef]
- Tie, Y.; Wang, Y.-F.; Boross, P.I.; Chiu, T.-Y.; Ghosh, A.K.; Tozser, J.; Louis, J.M.; Harrison, R.W.; Weber, I.T. Critical differences in HIV-1 and HIV-2 protease specificity for clinical inhibitors. Protein Sci. 2012, 21, 339–350. [Google Scholar] [CrossRef]
- Stoll, V.; Qin, W.; Stewart, K.D.; Jakob, C.; Park, C.; Walter, K.; Simmer, R.L.; Helfrich, R.; Bussiere, D.; Kao, J.; et al. X-ray crystallographic structure of ABT-378 (Lopinavir) bound to HIV-1 protease. Bioorganic Med. Chem. 2002, 10, 2803–2806. [Google Scholar] [CrossRef]
- Klei, H.E.; Kish, K.; Lin, P.-F.M.; Guo, Q.; Friborg, J.; Rose, R.E.; Zhang, Y.; Goldfarb, V.; Langley, D.R.; Wittekind, M.; et al. X-ray crystal structures of Human Immunodeficiency Virus Type 1 protease mutants complexed with Atazanavir. J. Virol. 2007, 81, 9525–9535. [Google Scholar] [CrossRef]
- Wong-Sam, A.; Wang, Y.-F.; Zhang, Y.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. Drug resistance mutation L76V alters nonpolar interactions at the flap–core interface of HIV-1 protease. ACS Omega 2018, 3, 12132–12140. [Google Scholar] [CrossRef]
- Kožíšek, M.; Lepšík, M.; Grantz Šašková, K.; Brynda, J.; Konvalinka, J.; Řezáčová, P. Thermodynamic and structural analysis of HIV protease resistance to darunavir—Analysis of heavily mutated patient-derived HIV-1 proteases. FEBS J. 2014, 281, 1834–1847. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2017, 46, D624–D632. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.-H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Zhou, Z.-J.; Qiu, Y.; Pu, Y.; Huang, X.; Ge, X.-Y. BioAider: An efficient tool for viral genome analysis and its application in tracing SARS-CoV-2 transmission. Sustain. Cities Soc. 2020, 63, 102466. [Google Scholar] [CrossRef]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sharma, R.; Kesari, P.; Kumar, P.; Tomar, S. Structure-function insights into chikungunya virus capsid protein: Small molecules targeting capsid hydrophobic pocket. Virology 2018, 515, 223–234. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clans | Description | Proteases | Catalytic Residues | Functions |
|---|---|---|---|---|
| Clan PA | Proteases of mixed nucleophiles, superfamily A (Cysteine or Serine proteases) | 3CLpro | His-Cys His-Asn/Glu-Cys His-Asn/Glu-Ser | Cleave the polyprotein Structural protein |
| Clan CA | Cysteine peptidase, superfamily A | PLpro | Cys-His-Asn/Asp | Cleave the polyprotein Structural protein DUBs activity |
| Clan CE | Cysteine peptidase, superfamily E | Poxviridae core protease Adenoviridae adenain | His-Glu/Asp-Cys | Structural protein Viral assembly |
| Clan CN | Cysteine peptidase, superfamily N | Alphavirus nsP2 | Cys-His | Cleave the polyprotein |
| Clan SH | Serine peptidase, superfamily H | Herpesvirus assemblin | His-Ser-His | Structural protein Viral assembly |
| Clan AA | Aspartic peptidase, superfamily A | PAPs | Asp-Asp | Cleave the polyprotein Structural protein |
| Clan ME | Metallo-peptidase, superfamily E | Poxviridae Metallo-protease | His-Xaa-Xaa-Glu-His | Virion maturation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, P.; Li, S.; Wang, S.; Zhang, X.; Bai, F. Computer-Aided Prediction of the Interactions of Viral Proteases with Antiviral Drugs: Antiviral Potential of Broad-Spectrum Drugs. Molecules 2024, 29, 225. https://doi.org/10.3390/molecules29010225
Ren P, Li S, Wang S, Zhang X, Bai F. Computer-Aided Prediction of the Interactions of Viral Proteases with Antiviral Drugs: Antiviral Potential of Broad-Spectrum Drugs. Molecules. 2024; 29(1):225. https://doi.org/10.3390/molecules29010225
Chicago/Turabian StyleRen, Pengxuan, Shiwei Li, Shihang Wang, Xianglei Zhang, and Fang Bai. 2024. "Computer-Aided Prediction of the Interactions of Viral Proteases with Antiviral Drugs: Antiviral Potential of Broad-Spectrum Drugs" Molecules 29, no. 1: 225. https://doi.org/10.3390/molecules29010225