
Air- and Water-Stable Heteroleptic Copper (I) Complexes Bearing Bis(indazol-1-yl)methane Ligands: Synthesis, Characterisation, and Computational Studies
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterisation of Compounds
2.1.1. Synthesis and Characterisation of Bis(indazol-1-yl)methane Analogues Ligands
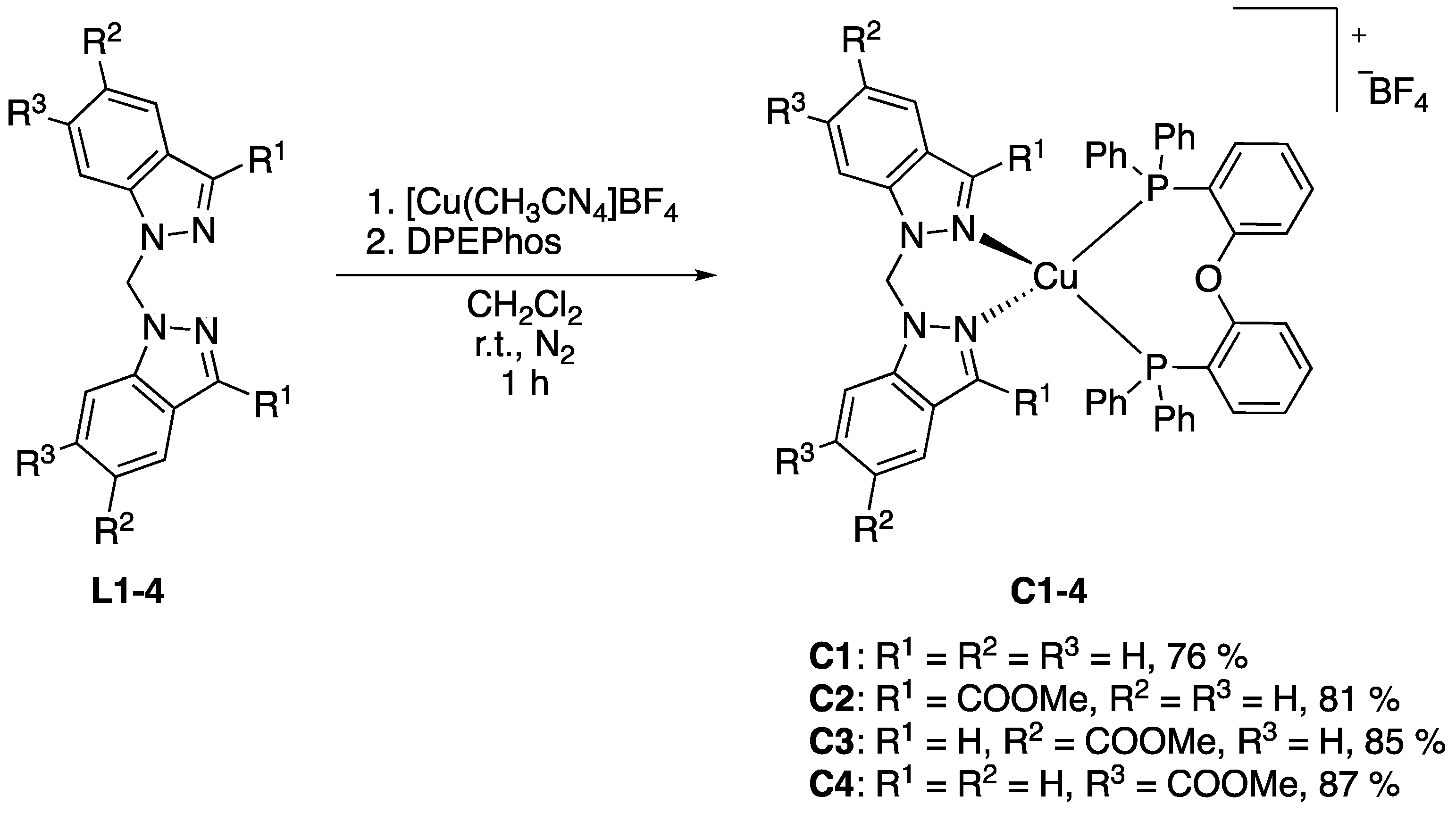
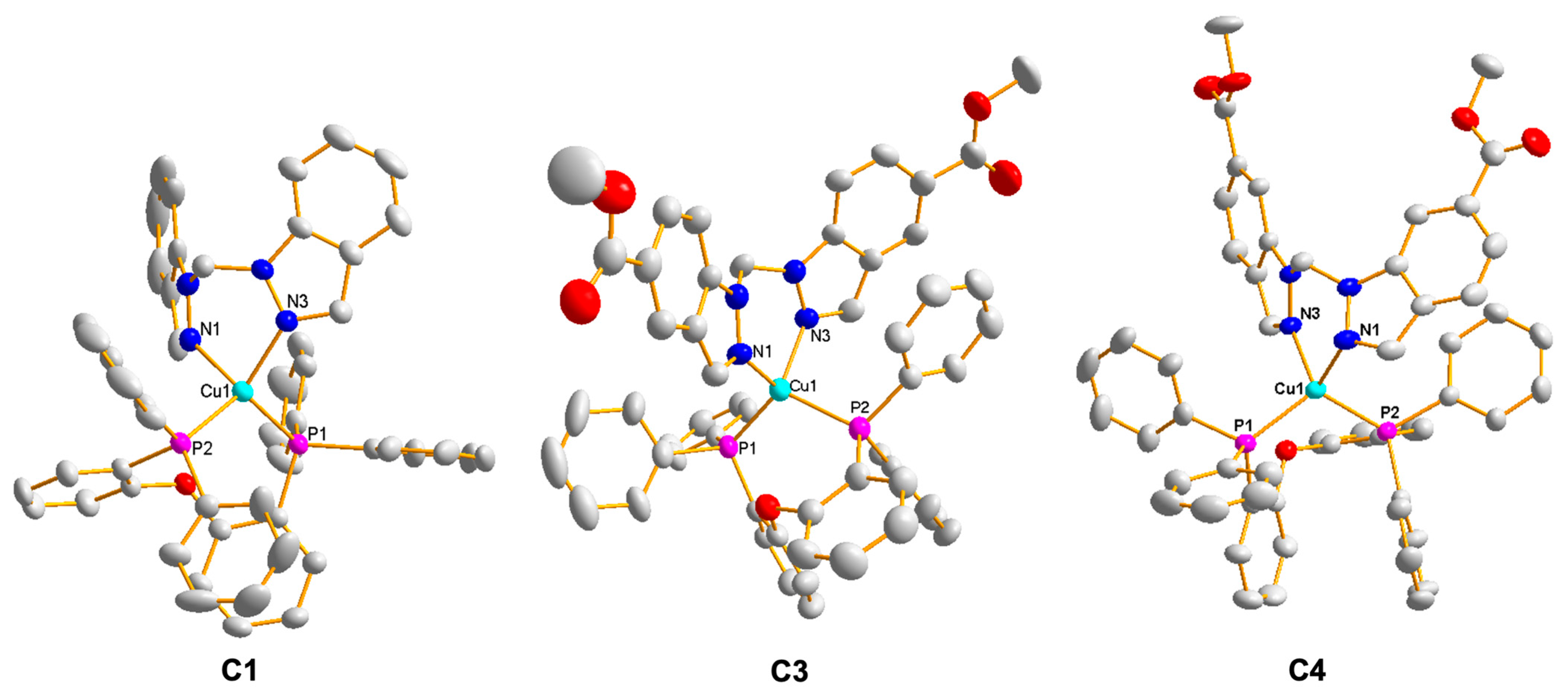
2.1.2. Synthesis and Characterisation of Heteroleptic Cu(I) Complexes with Bis(indazol-1-yl)methane Ligands
2.2. Cyclic Voltammetry
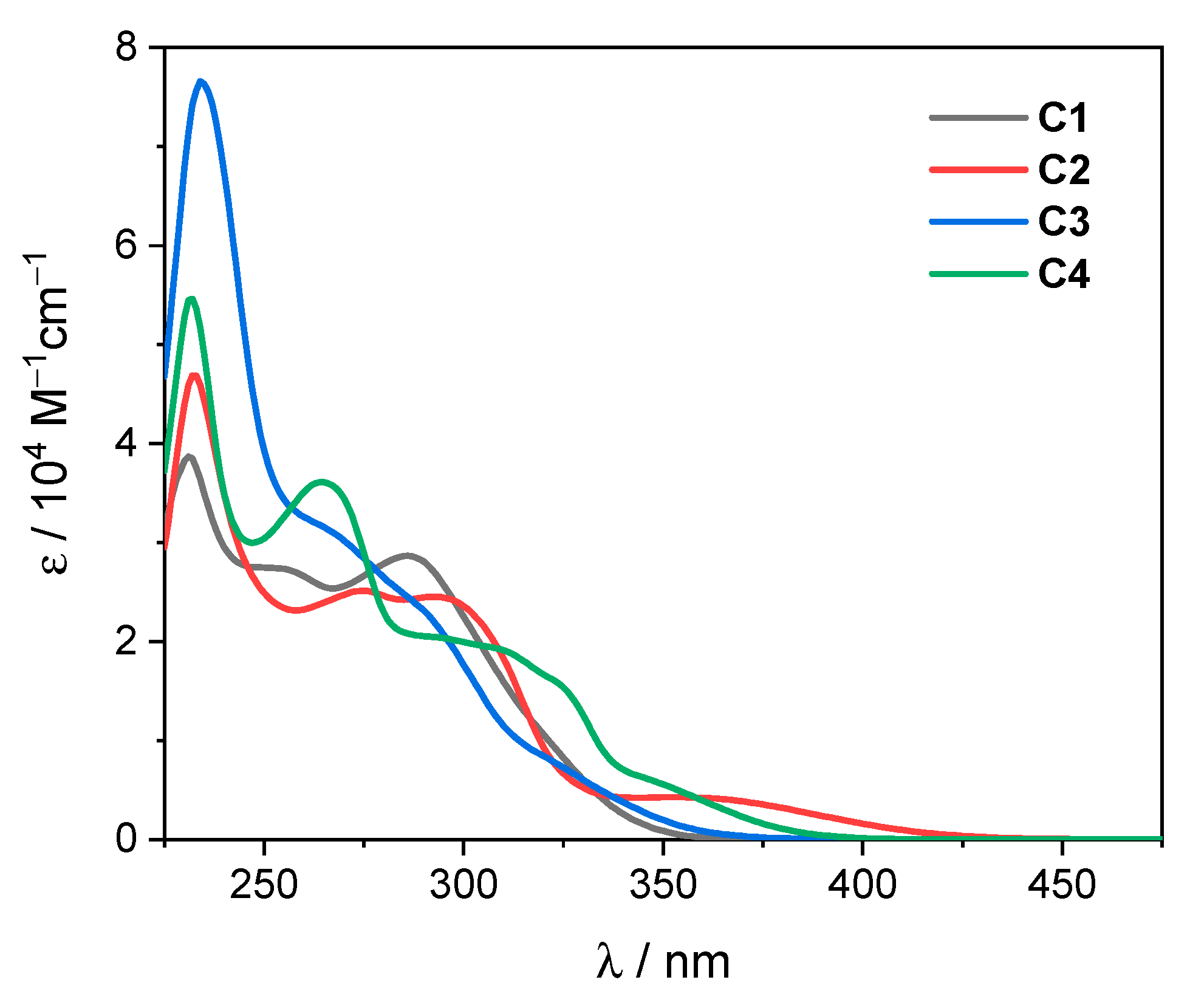
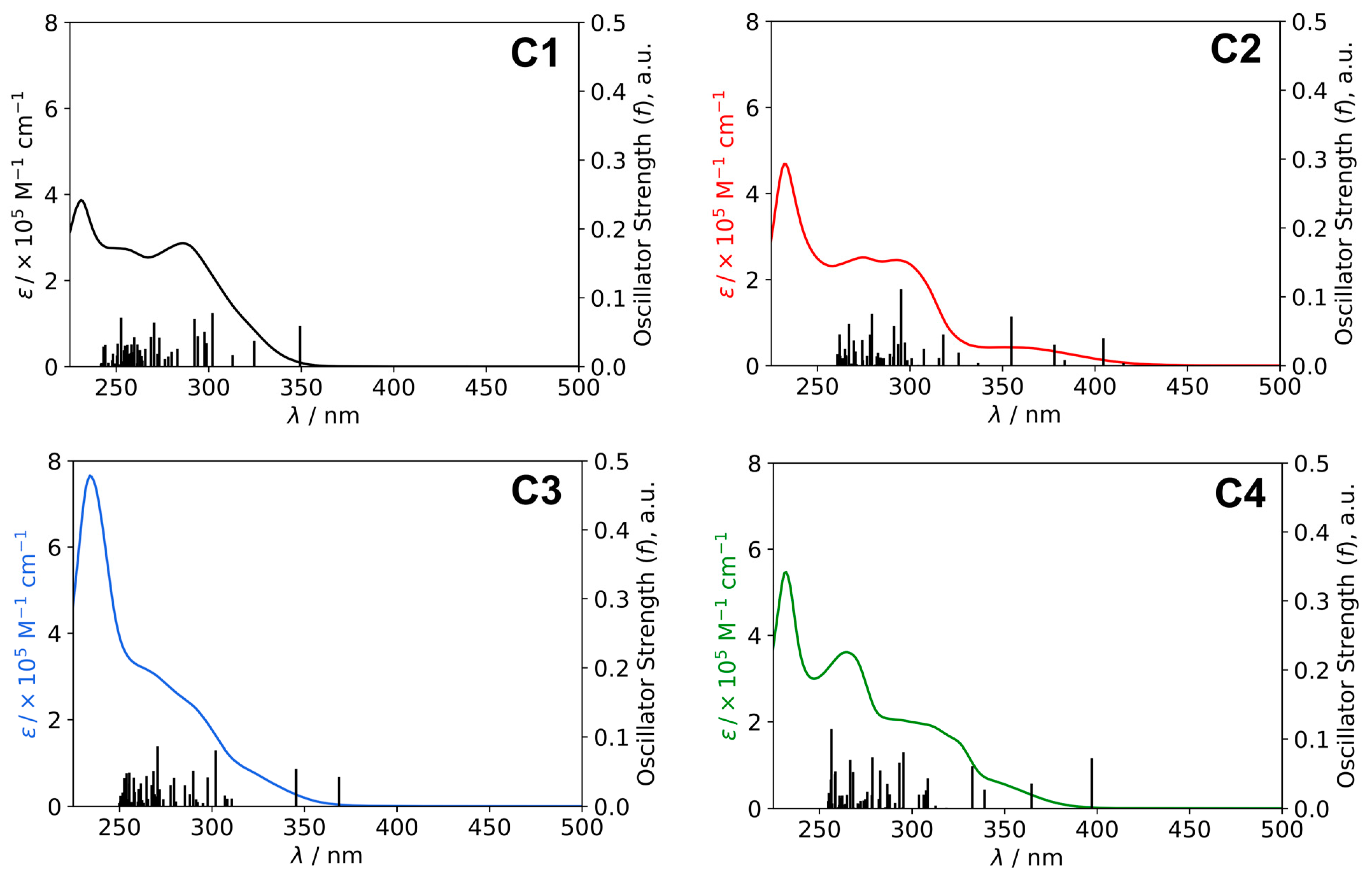
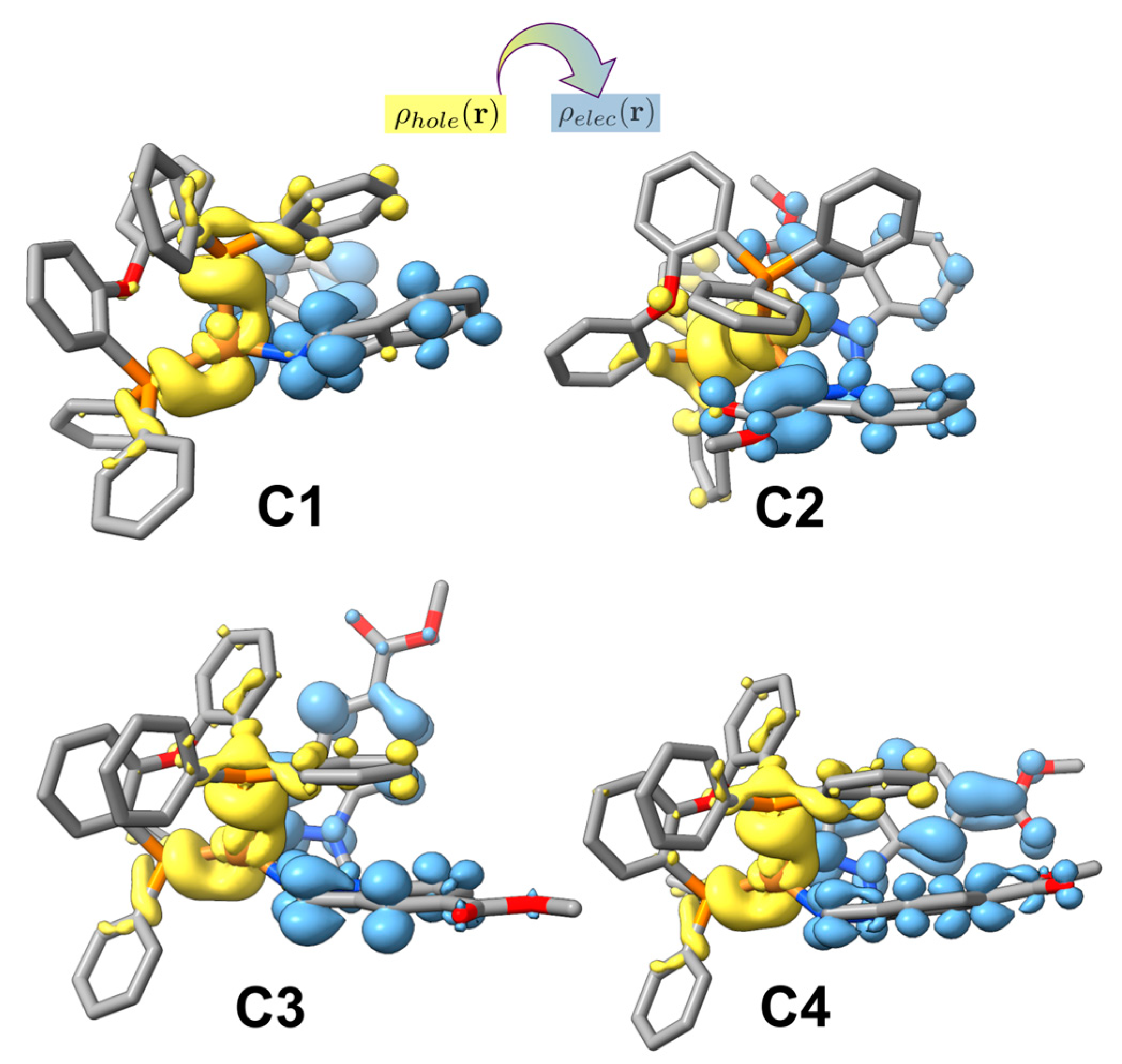
2.3. UV-Visible Absorption Studies
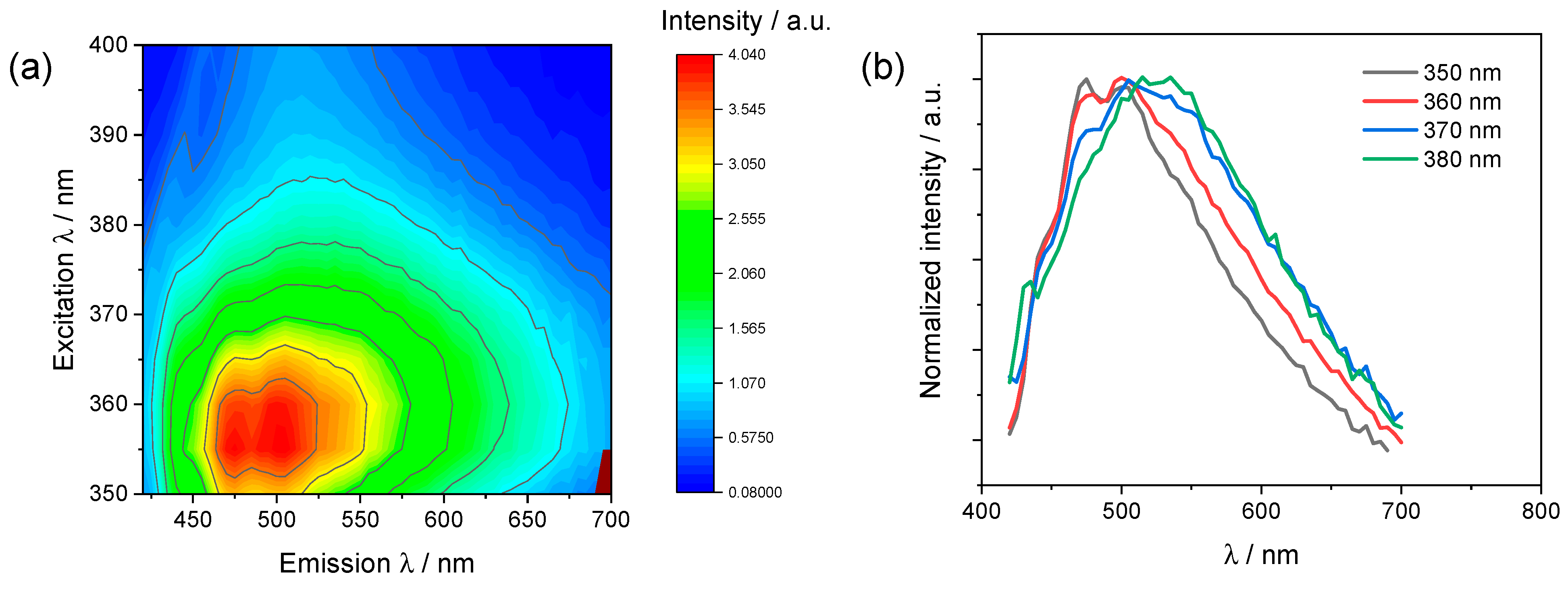
2.4. Emission Studies
3. Materials and Methods
3.1. General Information and Materials
3.2. General Synthetic Procedure for Bis(indazol-1-yl)methane Analogues L1-4
3.3. General Synthetic Procedure for Complexes C1-4
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wegeberg, C.; Wenger, O.S. Luminescent First-Row Transition Metal Complexes. JACS Au 2021, 1, 1860–1876. [Google Scholar] [CrossRef]
- Nicholls, T.P.; Bissember, A.C. Developments in visible-light-mediated copper photocatalysis. Tetrahedron Lett. 2019, 60, 150883. [Google Scholar] [CrossRef]
- Sinha, N.; Wenger, O.S. Photoactive Metal-to-Ligand Charge Transfer Excited States in 3d6 Complexes with Cr0, MnI, FeII, and CoIII. J. Am. Chem. Soc. 2023, 145, 4903–4920. [Google Scholar] [CrossRef]
- Beaudelot, J.; Oger, S.; Perusko, S.; Phan, T.A.; Teunens, T.; Moucheron, C.; Evano, G. Photoactive Copper Complexes: Properties and Applications. Chem. Rev. 2022, 122, 16365–16609. [Google Scholar] [CrossRef]
- Costa, R.D.; Orti, E.; Bolink, H.J.; Monti, F.; Accorsi, G.; Armaroli, N. Luminescent ionic transition-metal complexes for light-emitting electrochemical cells. Angew. Chem. Int. Ed. 2012, 51, 8178–8211. [Google Scholar] [CrossRef]
- Wang, L.D.; Fang, P.Y.; Zhao, Z.F.; Huang, Y.Y.; Liu, Z.W.; Bian, Z.Q. Rare Earth Complexes with 5d-4f Transition: New Emitters in Organic Light-Emitting Diodes. J. Phys. Chem. Lett. 2022, 13, 2686–2694. [Google Scholar] [CrossRef]
- Zhu, L.; Sha, C.W.; Lv, A.Q.; Xie, W.; Shen, K.; Chen, Y.M.; Xie, G.H.; Ma, H.L.; Li, H.B.; Hang, X.C. Tetradentate Pt(II) Complexes with Peripheral Hindrances for Highly Efficient Solution-Processed Blue Phosphorescent OLEDs. Inorg. Chem. 2022, 61, 10402–10409. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Chen, Z.; Zhu, L.Z.; Wu, Y.Q.; Xu, Y.Q.; Chen, S.M.; Wong, W.Y. High Performance NIR OLEDs with Emission Peak Beyond 760 nm and Maximum EQE of 6.39%. Adv. Opt. Mater. 2022, 10, 2200111. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photophysics, Photochemistry and Solar-Energy Conversion with Tris(Bipyridyl)Ruthenium(Ii) and Its Analogs. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Dumur, F. Recent Advances on Visible Light Metal-Based Photocatalysts for Polymerization under Low Light Intensity. Catalysts 2019, 9, 736. [Google Scholar] [CrossRef]
- Hockin, B.M.; Li, C.F.; Robertson, N.; Zysman-Colman, E. Photoredox catalysts based on earth-abundant metal complexes. Catal. Sci. Technol. 2019, 9, 889–915. [Google Scholar] [CrossRef]
- Wenger, O.S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. [Google Scholar] [CrossRef]
- Forster, C.; Heinze, K. Photophysics and photochemistry with Earth-abundant metals—Fundamentals and concepts. Chem. Soc. Rev. 2020, 49, 1057–1070. [Google Scholar] [CrossRef]
- Hossain, A.; Bhattacharyya, A.; Reiser, O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, eaav9713. [Google Scholar] [CrossRef]
- Liu, Y.R.; Yiu, S.C.; Ho, C.L.; Wong, W.Y. Recent advances in copper complexes for electrical/light energy conversion. Coord. Chem. Rev. 2018, 375, 514–557. [Google Scholar] [CrossRef]
- Zhong, M.B.; Pannecoucke, X.; Jubault, P.; Poisson, T. Recent advances in photocatalyzed reactions using well-defined copper(I) complexes. Beilstein J. Org. Chem. 2020, 16, 451–481. [Google Scholar] [CrossRef]
- Pirtsch, M.; Paria, S.; Matsuno, T.; Isobe, H.; Reiser, O. [Cu(dap)2Cl] As an Efficient Visible-Light-Driven Photoredox Catalyst in Carbon-Carbon Bond-Forming Reactions. Chem.-Eur. J. 2012, 18, 7336–7340. [Google Scholar] [CrossRef]
- Minozzi, C.; Caron, A.; Grenier-Petel, J.C.; Santandrea, J.; Collins, S.K. Heteroleptic Copper(I)-Based Complexes for Photocatalysis: Combinatorial Assembly, Discovery, and Optimization. Angew. Chem. Int. Ed. 2018, 57, 5477–5481. [Google Scholar] [CrossRef]
- Pettinari, C.; Marchetti, F.; Orbisaglia, S.; Pettinari, R.; Ngoune, J.; Gómez, M.; Santos, C.; Álvarez, E. Group 11 complexes with the bidentate di(1H-indazol-1-yl)methane and di(2H-indazol-2-yl)methane) ligands. CrystEngComm 2013, 15, 3892–3907. [Google Scholar] [CrossRef]
- Santos, C.; Gómez, M.; Álvarez, E.; Ngoune, J.; Marchetti, F.; Pettinari, R.; Pettinari, C. Group 9 and 10 complexes with the bidentate di(1H-indazol-1-yl)methane and di(2H-indazol-2-yl)methane ligands: Synthesis and structural characterization. New J. Chem. 2016, 40, 5695–5703. [Google Scholar] [CrossRef]
- Carrion, M.C.; Jalon, F.A.; Manzano, B.R.; Rodriguez, A.M.; Sepulveda, F.; Maestro, M. (Arene)ruthenium(II) complexes containing substituted bis(pyrazolyl)methane ligands–Catalytic behaviour in transfer hydrogenation of ketones. Eur. J. Inorg. Chem. 2007, 2007, 3961–3973. [Google Scholar] [CrossRef]
- Carrion, M.C.; Diaz, A.; Guerrero, A.; Jalon, F.A.; Manzano, B.R.; Rodriguez, A. New palladium and platinum polyfluorophenyl complexes with pyrazolyl N-donor ligands. Analysis of the restricted rotation of the poly fluorophenyl rings. New J. Chem. 2002, 26, 305–312. [Google Scholar] [CrossRef]
- Carrion, M.C.; Sepulveda, F.; Jalon, F.A.; Manzano, B.R.; Rodriguez, A.M. Base-Free Transfer Hydrogenation of Ketones Using Arene Ruthenium(II) Complexes. Organometallics 2009, 28, 3822–3833. [Google Scholar] [CrossRef]
- Reger, D.L.; Foley, E.A.; Smith, M.D. Structural Impact of Multitopic Third-Generation Bis(1-pyrazolyl)methane Ligands: Double, Mononuclear Metallacyclic Silver(I) Complexes. Inorg. Chem. 2010, 49, 234–242. [Google Scholar] [CrossRef]
- Reger, D.L.; Foley, E.A.; Watson, R.P.; Pellechia, P.J.; Smith, M.D.; Grandjean, F.; Long, G.J. Monofluoride Bridged, Binuclear Metallacycles of First Row Transition Metals Supported by Third Generation Bis(1-pyrazolyl)methane Ligands: Unusual Magnetic Properties. Inorg. Chem. 2009, 48, 10658–10669. [Google Scholar] [CrossRef]
- Reger, D.L.; Foley, E.A.; Smith, M.D. Mononuclear Metallacyclic Silver(I) Complexes of Third Generation Bis(1-pyrazolyl)methane Ligands. Inorg. Chem. 2009, 48, 936–945. [Google Scholar] [CrossRef]
- Ballesteros, P.; Elguero, J.; Claramunt, R.M. Reactivity of Azoles Towards Benzaldehyde and Its Dimethylacetal—Synthesis of N,N′-Diazolylphenylmethanes. Tetrahedron 1985, 41, 5955–5963. [Google Scholar] [CrossRef]
- Gallegopreciado, M.C.L.; Ballesteros, P.; Claramunt, R.M.; Cano, M.; Heras, J.V.; Pinilla, E.; Monge, A. The Crystal and Molecular-Structure of Bis(Indazol-1-Yl)Pyridin-2′-Ylmethane (Bipm) and [Rh(Bipm)(Nbd)]Pf6. J. Organomet. Chem. 1993, 450, 237–244. [Google Scholar] [CrossRef]
- Lopez, M.C.; Jagerovic, N.; Ballesteros, P. Regioselective Synthesis of the Homochiral Ligand (4s,7r)-7,8,8-Trimethyl-4,5,6,7-Tetrahydro-4,7-Methanoindazol-2-Yl-Indazol-1′-Ylmethane. Tetrahedron-Asymmetry 1994, 5, 1887–1890. [Google Scholar] [CrossRef]
- Tabassum, S.; Parveen, S.; Arjmand, F. Synthesis and characterization of new synthetic oxygen carriers. A kinetic study of the reaction of the binuclear iron(III)-copper(II) complex with H2O2. Transit. Met. Chem. 2005, 30, 196–204. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, tau4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef]
- Femoni, C.; Muzzioli, S.; Palazzi, A.; Stagni, S.; Zacchini, S.; Monti, F.; Accorsi, G.; Bolognesi, M.; Armaroli, N.; Massi, M.; et al. New tetrazole-based Cu(I) homo- and heteroleptic complexes with various P boolean AND P ligands: Synthesis, characterization, redox and photophysical properties. Dalton Trans. 2013, 42, 997–1010. [Google Scholar] [CrossRef]
- Qin, L.; Zhang, Q.; Sun, W.; Wang, J.; Lu, C.; Cheng, Y.; Wang, L. Novel luminescent iminephosphine complex of copper(I) with high photochemical and electrochemical stability. Dalton Trans. 2009, 43, 9388–9391. [Google Scholar] [CrossRef]
- Henriquez, M.A.; Engl, S.; Jaque, P.; Gonzalez, I.A.; Natali, M.; Reiser, O.; Cabrera, A.R. Phosphine Evaluation on a New Series of Heteroleptic Copper(I) Photocatalysts with dpa Ligand [Cu(dpa)(P,P)]BF4. Eur. J. Inorg. Chem. 2021, 2021, 4020–4029. [Google Scholar] [CrossRef]
- Escobar, M.A.; Morales-Verdejo, C.; Arroyo, J.L.; Dreyse, P.; González, I.; Brito, I.; MacLeod-Carey, D.; Moreno da Costa, D.; Cabrera, A.R. Burning Rate Performance Study of Ammonium Perchlorate Catalyzed by Heteroleptic Copper(I) Complexes with Pyrazino[2,3-f][1,10]phenanthroline-Based Ligands. Eur. J. Inorg. Chem. 2021, 2021, 1632–1639. [Google Scholar] [CrossRef]
- Gonzalez, I.A.; Henriquez, M.A.; Cortes-Arriagada, D.; Natali, M.; Daniliuc, C.G.; Dreyse, P.; Maze, J.; Rojas, R.S.; Salas, C.O.; Cabrera, A.R. Heteroleptic Cu(I) complexes bearing methoxycarbonyl-imidoylindazole and POP ligands—An experimental and theoretical study of their photophysical properties. New J. Chem. 2018, 42, 12576–12586. [Google Scholar] [CrossRef]
- Cabrera, A.R.; Gonzalez, I.A.; Cortes-Arriagada, D.; Natali, M.; Berke, H.; Daniliuc, C.G.; Camarada, M.B.; Toro-Labbe, A.; Rojas, R.S.; Salas, C.O. Synthesis of new phosphorescent imidoyl-indazol and phosphine mixed ligand Cu(I) complexes structural characterization and photophysical properties. Rsc. Adv. 2016, 6, 5141–5153. [Google Scholar] [CrossRef]
- Catalan, J. On the solvatochromism, dimerization and tautomerism of indazole. Arkivoc 2014, 2, 57–70. [Google Scholar] [CrossRef]
- Gonzalez, I.; Natali, M.; Cabrera, A.R.; Loeb, B.; Maze, J.; Dreyse, P. Substituent influence in phenanthroline-derived ancillary ligands on the excited state nature of novel cationic Ir(III) complexes. New J. Chem. 2018, 42, 6644–6654. [Google Scholar] [CrossRef]
- Indelli, M.T.; Ghirotti, M.; Prodi, A.; Chiorboli, C.; Scandola, F.; McClenaghan, N.D.; Puntoriero, F.; Campagna, S. Solvent switching of intramolecular energy transfer in bichromophoric systems: Photophysics of (2,2′-bipyridine)tetracyanoruthenate(II)/pyrenyl complexes. Inorg. Chem. 2003, 42, 5489–5497. [Google Scholar] [CrossRef]
- Zhang, Y.; Schulz, M.; Wächtler, M.; Karnahl, M.D.B. Heteroleptic diimine–diphosphine Cu(I) complexes as an alternative towards noble-metal based photosensitizers: Design strategies, photophysical properties and perspective applications. Coord. Chem. Rev. 2018, 356, 127–146. [Google Scholar] [CrossRef]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. A Found. Adv. 1995, 51 Pt 1, 33–38. [Google Scholar] [CrossRef]
- Bruker. SAINT; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Weber, M.D.; Viciano-Chumillas, M.; Armentano, D.; Cano, J.; Costa, R.D. Sigma-Hammett parameter: A strategy to enhance both photo- and electro-luminescence features of heteroleptic copper(i) complexes. Dalton Trans. 2017, 46, 6312–6323. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Eox/V vs. Fc+/Fc b | Ered/V vs. Fc+/Fc b | ΔEox/red/V |
|---|---|---|---|
| C1 | 0.68 | −1.41 | 2.09 |
| C2 | 0.65 | −1.32 | 1.97 |
| C3 | 0.64 | −1.30 | 1.94 |
| C4 | 0.68 | −1.22 | 1.90 |
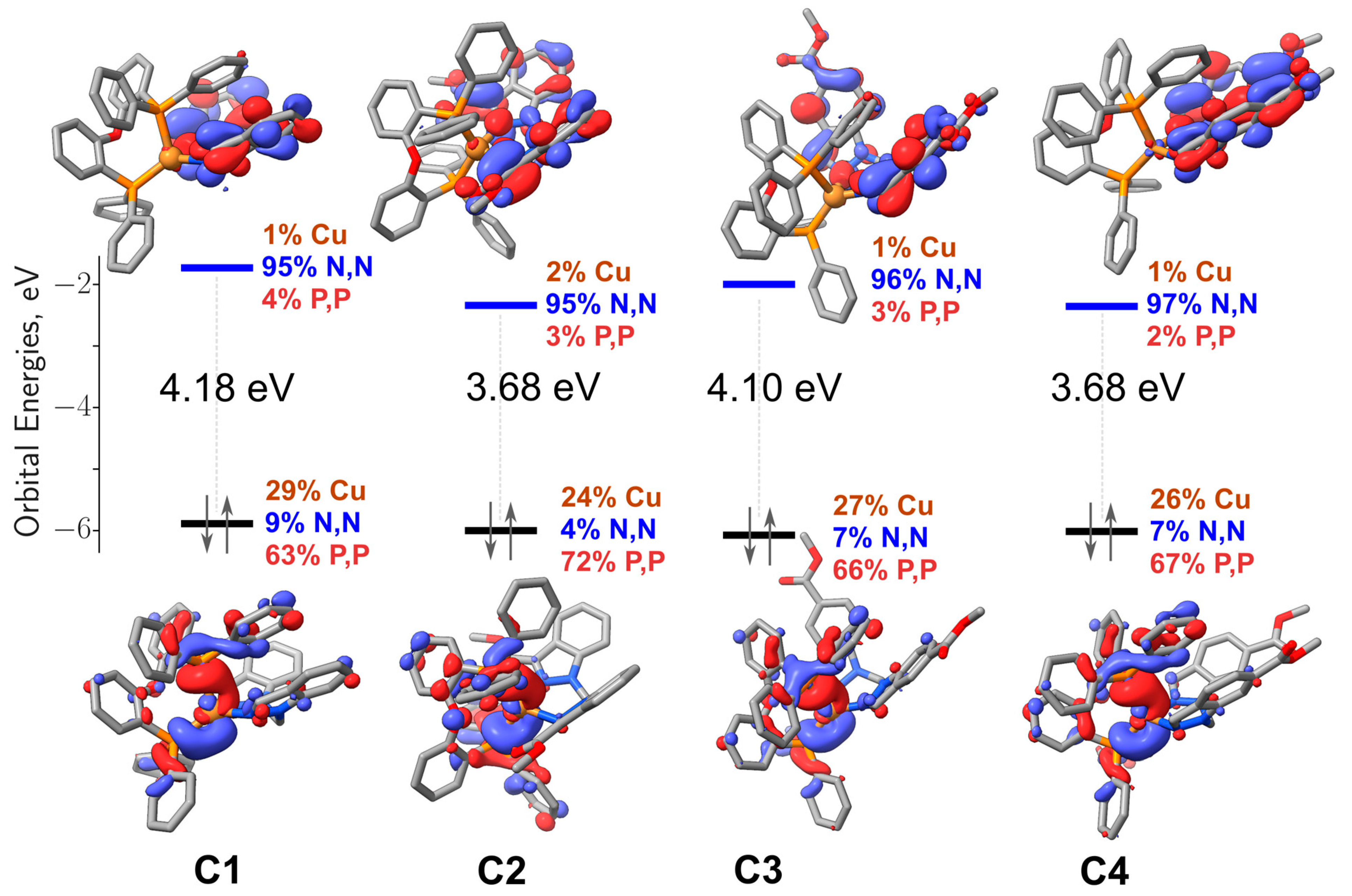
| State | Absorption λ [nm] (E [eV]) | f | Monoexcitations (CI Coef. and% Contribution) | Description of the Electronic Transition | |
|---|---|---|---|---|---|
| C1 | S1 | 349 (3.55) | 0.059 | H→L (0.70; 98%) | Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT |
| C2 | S1 | 415 (2.99) | 0.004 | H–1→L (−0.37; 27%) H→L (0.59; 71%) | Cu(d) + P,P(π) + N,N(π) → N,N(π*); 1MLCT/1LLCT/1IL Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT |
| S2 | 404 (3.06) | 0.040 | H–1→L (0.59; 70%) H→L (0.37; 27%) | Cu(d) + P,P(π) + N,N(π) → N,N(π*); 1MLCT/1LLCT/1IL Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT | |
| S3 | 384 (3.23) | 0.008 | H–1→L + 1 (−0.16, 5%) H→L + 1 (0.67, 91%) | Cu(d) + P,P(π) + N,N(π) → N,N(π*); 1MLCT/1LLCT/1IL Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT | |
| S4 | 378 (3.28) | 0.030 | H–1→L + 1 (0.68; 91%) H→L + 1 (0.16; 5%) | Cu(d) + P,P(π) + N,N(π) → N,N(π*); 1MLCT/1LLCT/1IL Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT | |
| C3 | S1 | 369 (3.36) | 0.043 | H→L (0.69; 96%) | Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT |
| S2 | 345 (3.59) | 0.054 | H→L + 1 (0.69; 96%) | Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT | |
| C4 | S1 | 397 (3.12) | 0.073 | H→L (0.70; 98%) | Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT |
| S2 | 365 (3.40) | 0.036 | H→L + 1 (0.70; 98%) | Cu(d) + P,P(π) → N,N(π*); 1MLCT/1LLCT |
| Complex | CH2Cl2 at Room Temperature | 77 K | ||
|---|---|---|---|---|
| λem [nm] | Φ/% | τ [μs] d | λem [nm] | |
| C1 | 440, 466, 495 | 2.2 a | 4.2 | 431, 489, 490, 525 |
| C2 | 545 | <0.1 b | 1.3, 10 | 449, 477, 514 |
| C3 | 473, 533 | 0.3 b,c | 1.8, 7.2 | 435, 464, 495, 531 |
| C4 | 574 | 2.5 b | 2.9, 7.7 | 455, 489, 522 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-da Costa, D.; Zúñiga-Loyola, C.; Droghetti, F.; Robles, S.; Villegas-Menares, A.; Villegas-Escobar, N.; Gonzalez-Pavez, I.; Molins, E.; Natali, M.; Cabrera, A.R. Air- and Water-Stable Heteroleptic Copper (I) Complexes Bearing Bis(indazol-1-yl)methane Ligands: Synthesis, Characterisation, and Computational Studies. Molecules 2024, 29, 47. https://doi.org/10.3390/molecules29010047
Moreno-da Costa D, Zúñiga-Loyola C, Droghetti F, Robles S, Villegas-Menares A, Villegas-Escobar N, Gonzalez-Pavez I, Molins E, Natali M, Cabrera AR. Air- and Water-Stable Heteroleptic Copper (I) Complexes Bearing Bis(indazol-1-yl)methane Ligands: Synthesis, Characterisation, and Computational Studies. Molecules. 2024; 29(1):47. https://doi.org/10.3390/molecules29010047
Chicago/Turabian StyleMoreno-da Costa, David, César Zúñiga-Loyola, Federico Droghetti, Stephania Robles, Alondra Villegas-Menares, Nery Villegas-Escobar, Ivan Gonzalez-Pavez, Elies Molins, Mirco Natali, and Alan R. Cabrera. 2024. "Air- and Water-Stable Heteroleptic Copper (I) Complexes Bearing Bis(indazol-1-yl)methane Ligands: Synthesis, Characterisation, and Computational Studies" Molecules 29, no. 1: 47. https://doi.org/10.3390/molecules29010047