

Chitosan Soft Matter Vesicles Loaded with Acetaminophen as Promising Systems for Modified Drug Release

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
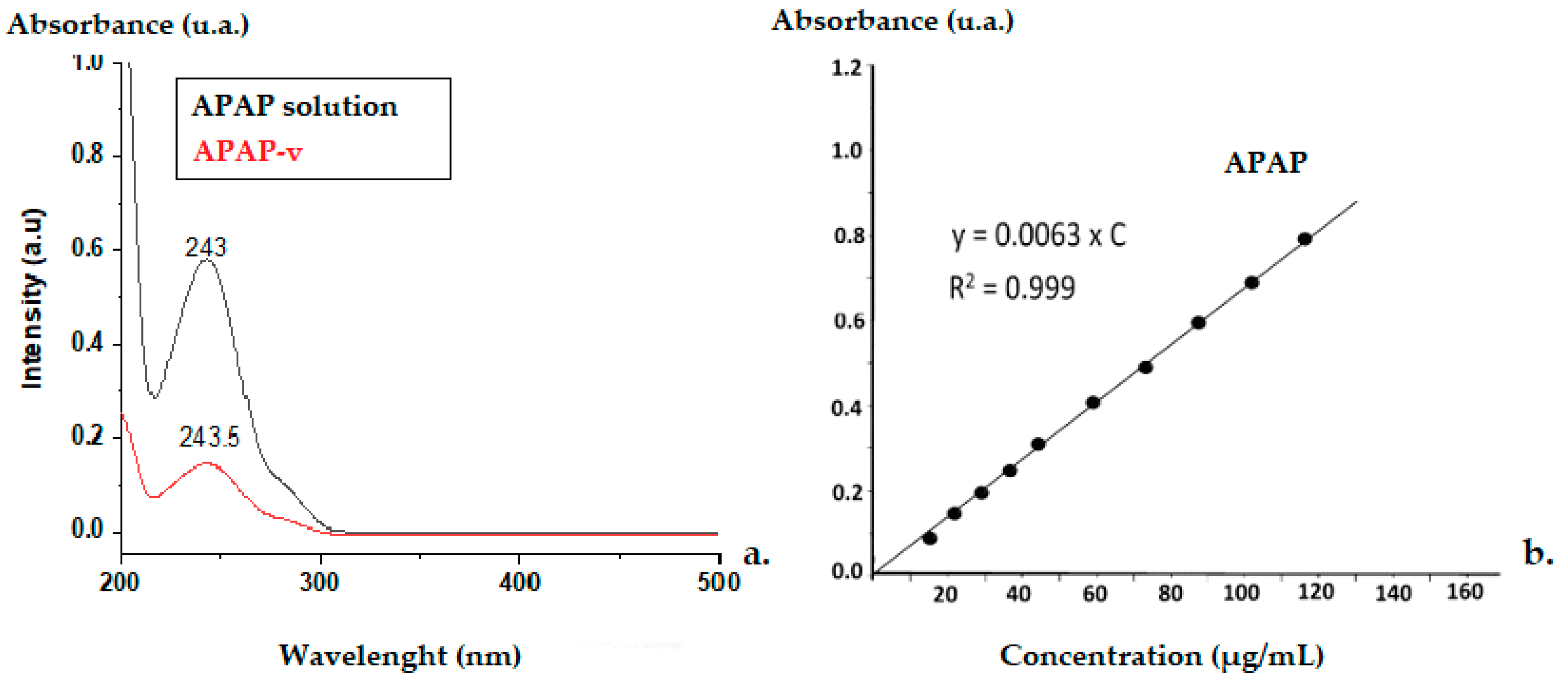
2.1. pH Value of Solutions with APAP
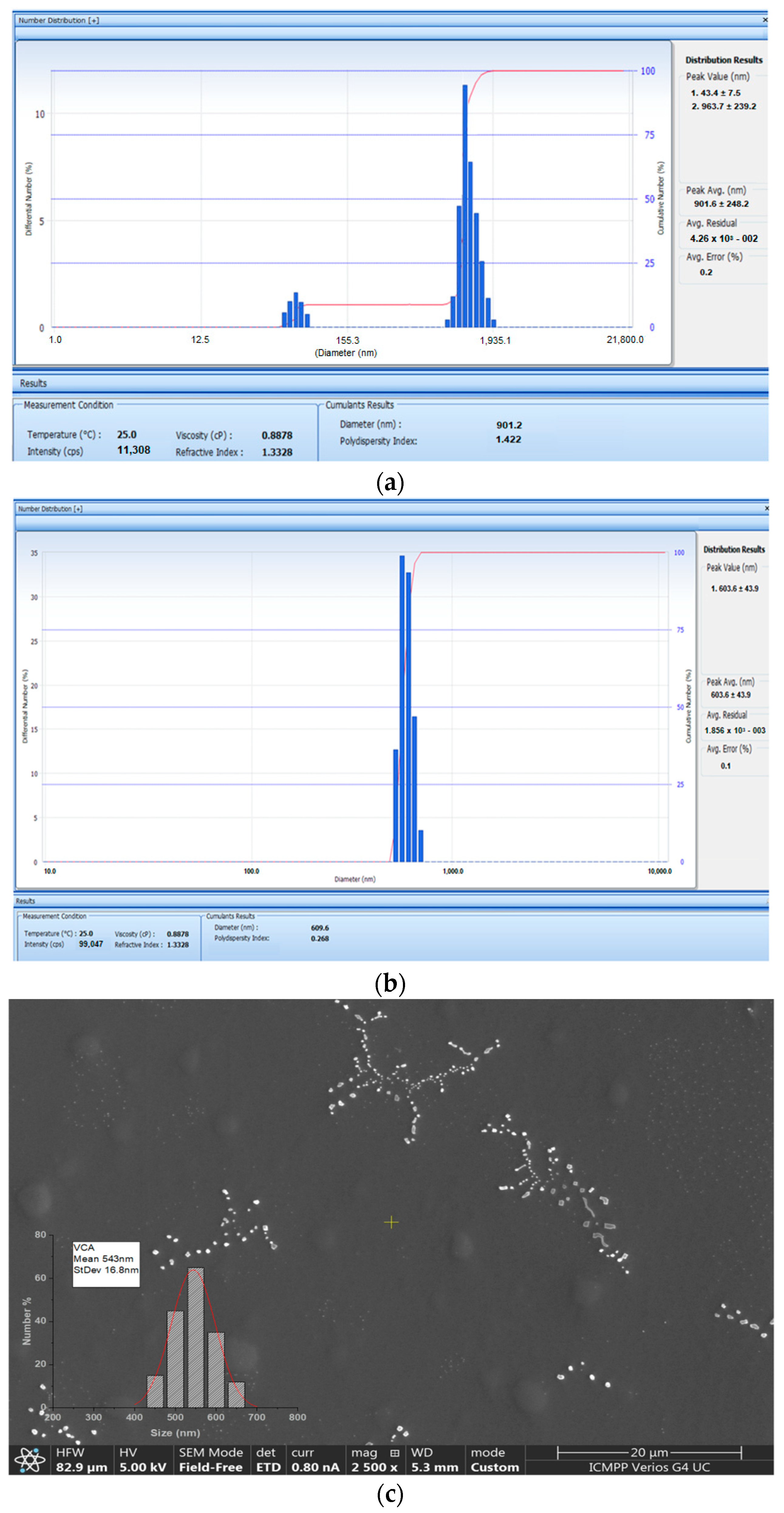
2.2. Size Distribution of APAP-v
2.3. Zeta Potential of APAP-v
2.4. The Efficacy of APAP Entrapment in Vesicles
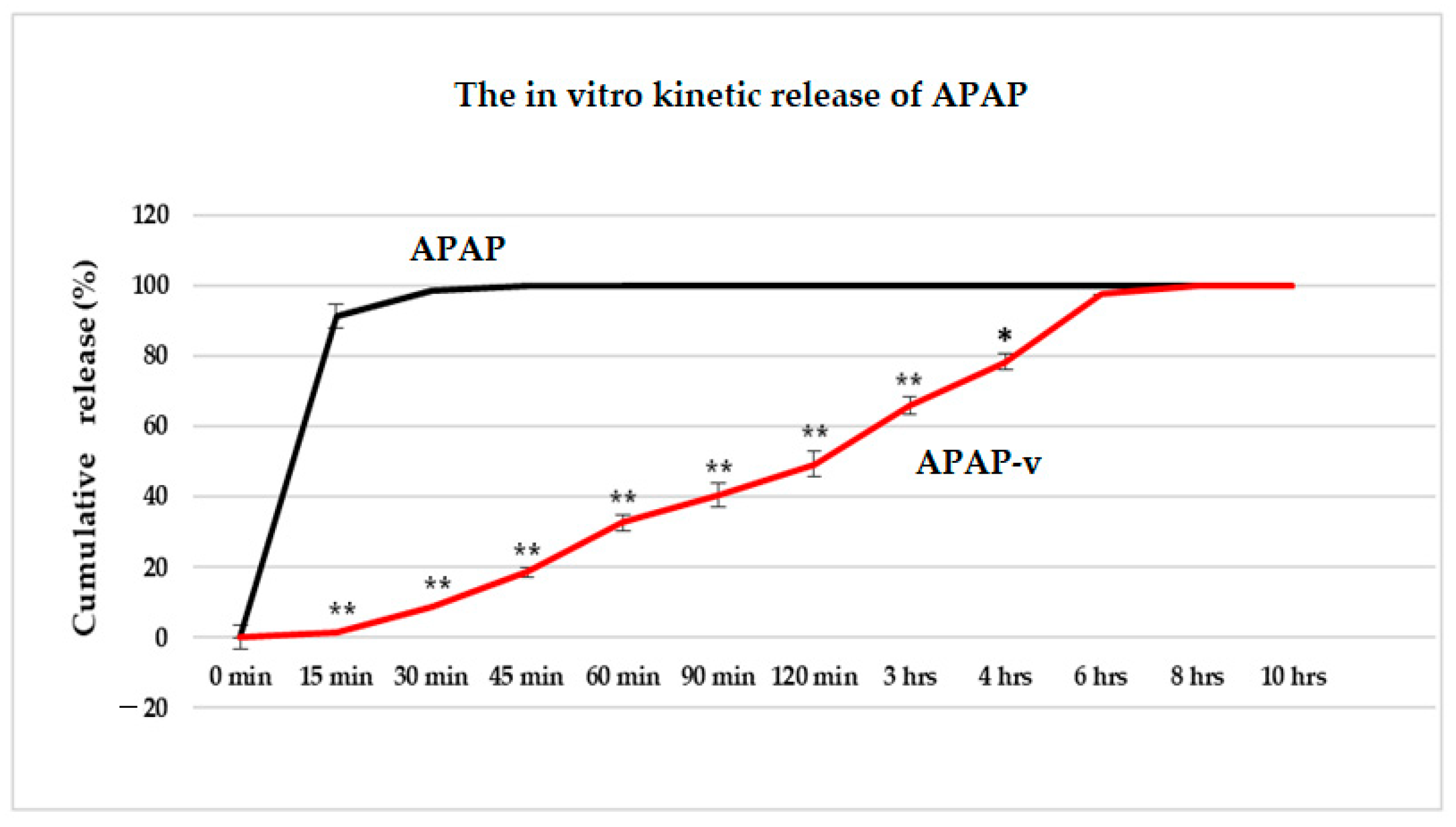
2.5. In Vitro Release Kinetics of APAP from APAP-v
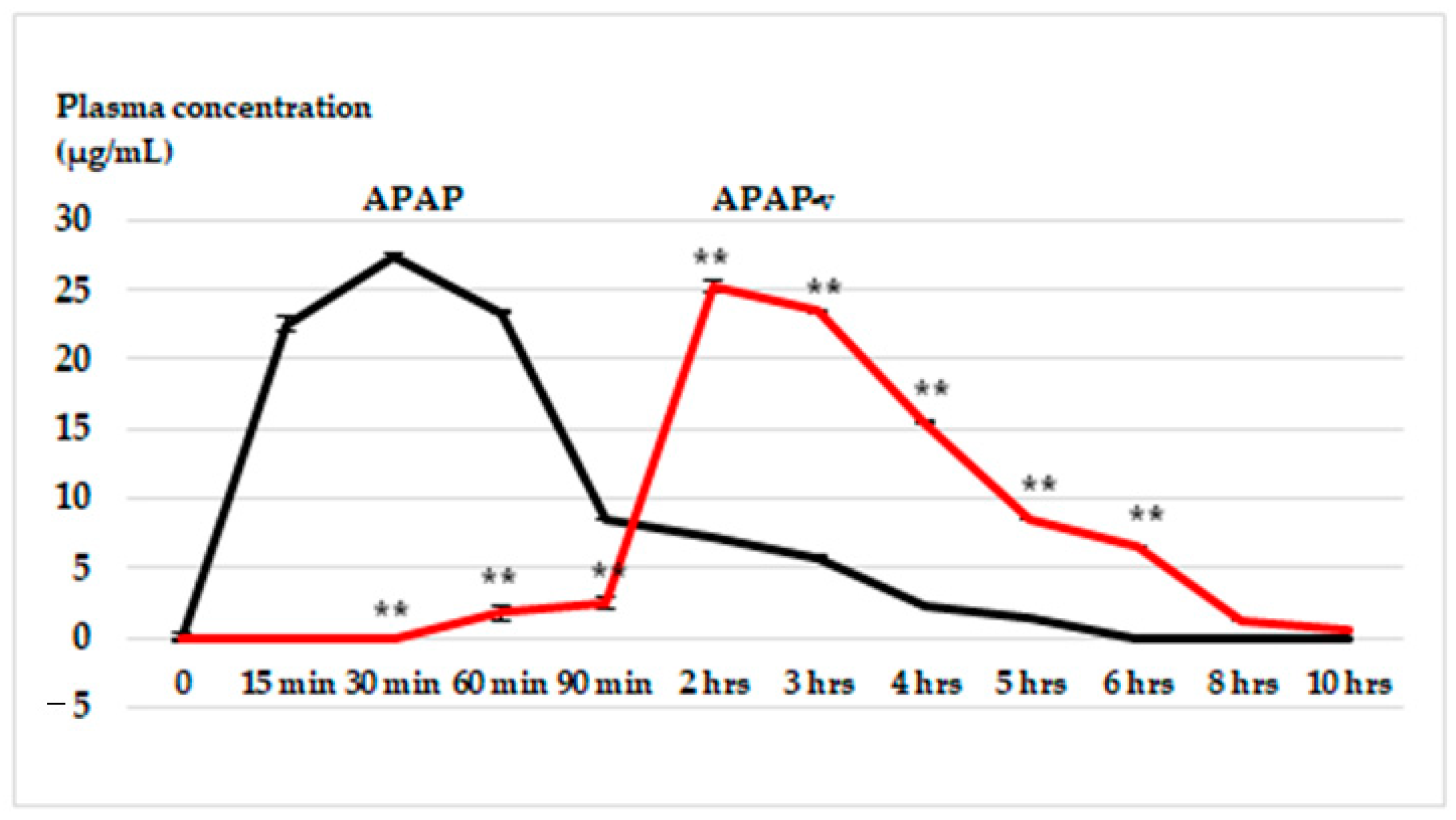
2.6. In Vivo Release Kinetics of APAP from APAP-v
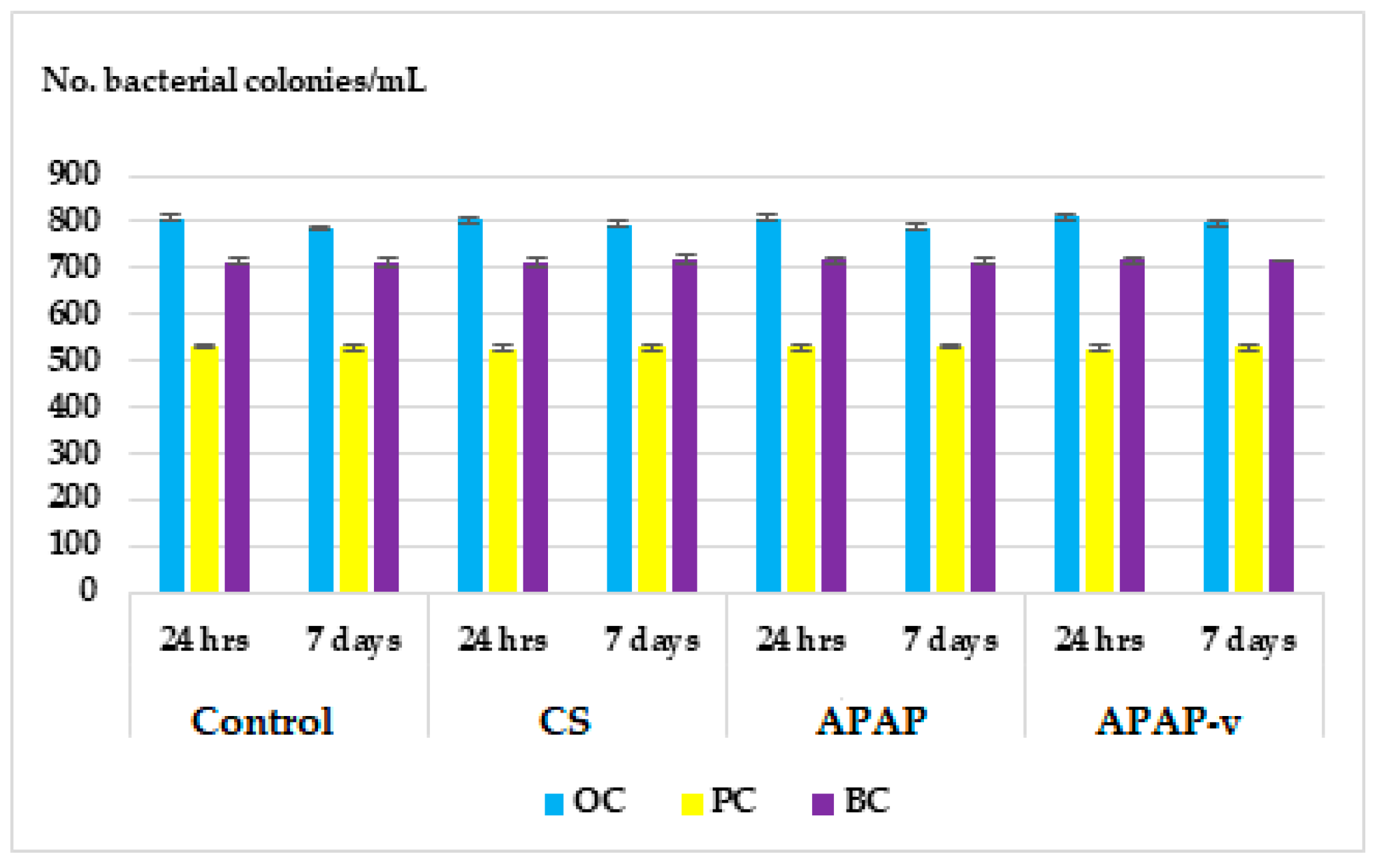
2.7. In Vitro Hemocompatibility
2.8. In Vivo Biocompatibility
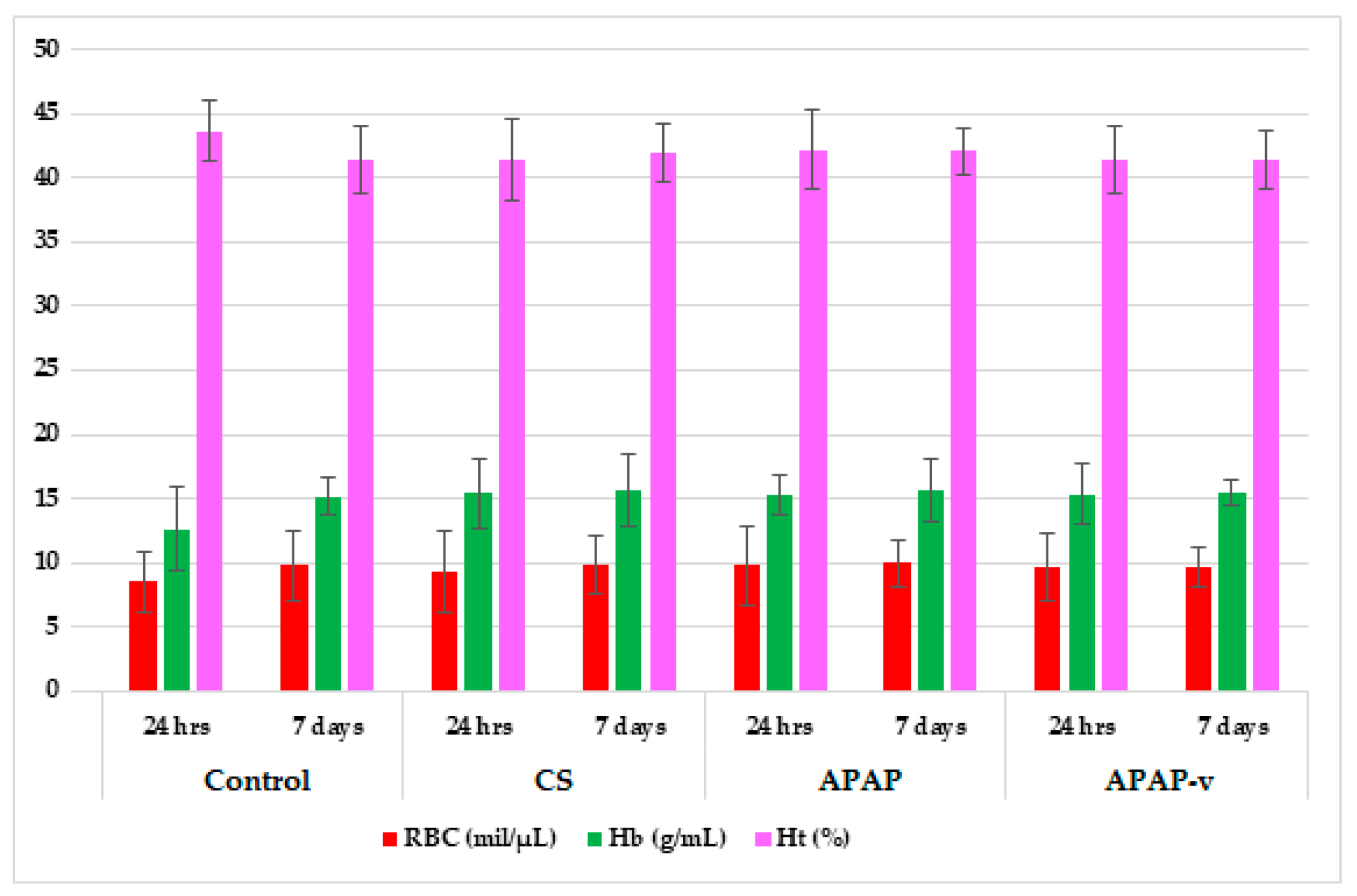
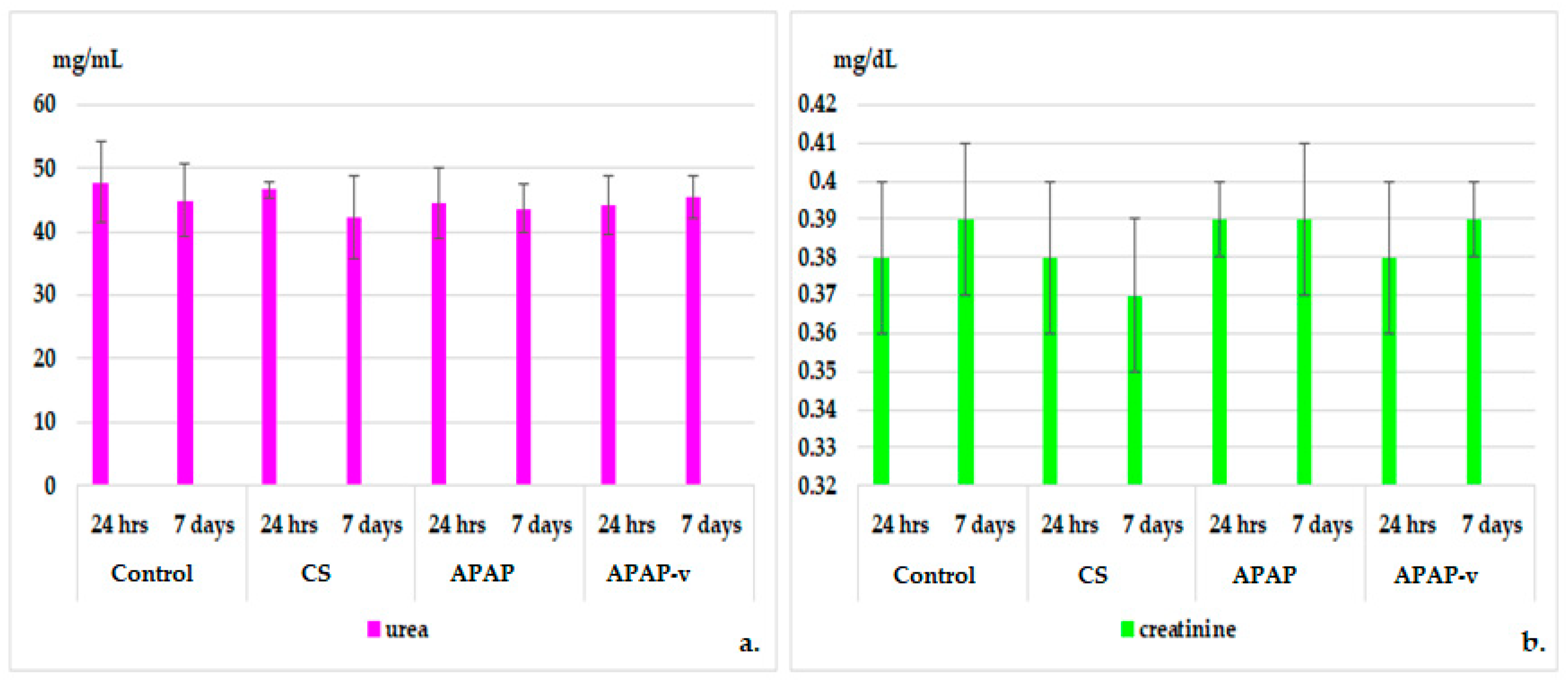
2.8.1. Hematological Tests
2.8.2. Histopathological Examination
3. Materials and Methods
3.1. Animals
3.2. Substances
3.3. The Design of APAP-v
- L-alpha-phosphatidylcholine (0.0075 g) was dissolved in 1 mL chloroform, and the solvent was evaporated using a Rotary evaporator RE-2000A (Ya Rong Biochemical Instrument Factory—Shanghai, China). This resulted in the formation of a dry lipid film.
- APAP (250 mg) was dissolved in 1 mL ethyl alcohol, and then diluted with double-distilled water up to 10 mL.
- The dry lipid film was rehydrated using the hydro-alcoholic APAP solution.
- The mixture was subjected to an ultrasonic field (25% amplitude) for 20 min at 29 °C, corresponding to an energy input of 20,000 kJ, using Bandelin 2450 SONOPULS ultrasonic homogenizers (Sigma-Aldrich- Steinheim, Germany). The lipid vesicles entrapping APAP were obtained.
- To coat the lipid vesicles with CS, 4 mL of a 1% CS solution was added to the dispersion of APAP-loaded vesicles. The mixture was magnetically stirred at 800 rpm for 10 min.
- The addition of acetic acid solution to the CS vesicle dispersion led to an acidic pH, enabling the protonation of the amino groups in the CS chain and facilitating its dissolution in water.
- The resulting dispersion was dialyzed for 2 h in double-distilled water, using tubular fiber membranes (type D6191-25EA) with a pore size of 12,000 Da MWCO (Sigma-Aldrich Chemical Co, Steinheim, Germany). This step aimed to achieve a pH value as close as possible to physiological levels, and the pH was monitored using a Sartorius Professional PP-50 pH meter from Sartorius Lab Instruments GmbH & Co. KG, Göttingen, Germany.
3.4. Characterization of Vesicles Entrapping APAP
3.5. In Vivo Release Kinetics of APAP from APAP-v
3.6. Assessing the In Vitro Hemocompatibility of Vesicles Entrapping APAP
3.7. Evaluation of the In Vivo Biocompatibility of Vesicles Entrapping APAP
3.8. Data Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mallet, C.; Desmeules, J.; Pegahi, R.; Eschalier, A. An updated review on the metabolite (AM404)-mediated central mechanism of action of paracetamol (acetaminophen): Experimental evidence and potential clinical impact. J. Pain Res. 2023, 16, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Przybyła, G.W.; Szychowski, K.A.; Gmiński, J. Paracetamol—An old drug with new mechanisms of action. Clin. Exp. Pharmacol. Physiol. 2020, 4891, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Esh, C.J.; Chrismas, B.C.R.; Mauger, A.R.; Taylor, L. Pharmacological hypotheses: Is acetaminophen selective in its cyclooxygenase inhibition? Pharmacol. Res. Perspect. 2021, 9, e00835. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.W.; Malonn, M.C. acetaminophen pharmacokinetic and toxicological aspects: A review. Rev. Científica Multidiscip. Núcleo Do Conhecimento 2021, 12, 18–27. [Google Scholar] [CrossRef]
- Graham, G.G.; Davies, M.J.; Day, R.O.; Mohamudally, A.; Scott, K.F. The modern pharmacology of paracetamol: Therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacology 2013, 21, 201–232. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Jaeschke, H. Metabolism and disposition of acetaminophen: Recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.D.; Stanescu, I.; Atkinson, H.C.; Anderson, B.J. Population pharmacokinetic modelling of acetaminophen and ibuprofen: The influence of body composition, formulation and feeding in healthy adult volunteers. Eur. J. Drug Metab. Pharmacokinet. 2022, 47, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef]
- Jafernik, K.; Ładniak, A.; Blicharska, E.; Czarnek, K.; Ekiert, H.; Wiącek, A.E.; Szopa, A. Chitosan-based nanoparticles as effective drug delivery systems—A review. Molecules 2023, 28, 1963. [Google Scholar] [CrossRef]
- Herdiana, Y.; Wathoni, N.; Shamsuddin, S.; Muchtaridi, M. Drug release study of the chitosan-based nanoparticles. Heliyon 2022, 8, e08674. [Google Scholar] [CrossRef]
- Kumar, R.; Sirvi, A.; Kaur, S.; Samal, S.K.; Roy, S.; Sangamwar, A.T. Polymeric micelles based on amphiphilic oleic acid modified carboxymethyl chitosan for oral drug delivery of bcs class iv compound: Intestinal permeability and pharmacokinetic evaluation. Eur. J. Pharm. Sci. 2020, 153, 105466. [Google Scholar] [CrossRef] [PubMed]
- Shoueir, K.R.; El-Desouky, N.; Rashad, M.M.; Ahmed, M.K.; Janowska, I.; El-Kemary, M. Chitosan based-nanoparticles and nanocapsules: Overview, physicochemical features, applications of a nanofibrous scaffold, and bioprinting. Int. J. Biol. Macromol. 2021, 167, 1176–1197. [Google Scholar] [CrossRef] [PubMed]
- Seidi, F.; Khodadadi Yazdi, M.; Jouyandeh, M.; Dominic, M.; Naeim, H.; Nezhad, M.N.; Bagheri, B.; Habibzadeh, S.; Zarrintaj, P.; Saeb, M.R.; et al. Chitosan-based blends for biomedical applications. Int. J. Biol. Macromol. 2021, 183, 1818–1850. [Google Scholar] [CrossRef] [PubMed]
- Sangnim, T.; Dheer, D.; Jangra, N.; Huanbutta, K.; Puri, V.; Sharma, A. Chitosan in oral drug delivery formulations: A review. Pharmaceutics 2023, 15, 2361. [Google Scholar] [CrossRef] [PubMed]
- Barange, S.; Singh, S.; Ahmad, S.; Ahmad, S.W.; Manjhi, S.K.; Thakur, A.S.; Rathi, J.C.; Sharma, R. Preparation and optimization of paracetamol loaded niosomes. JETIR 2022, 9, d452–d464, eISSN: 2349-5162. [Google Scholar]
- Shariare, M.H.; Masum, A.-A.; Alshehri, S.; Alanazi, F.K.; Uddin, J.; Kazi, M. Preparation and optimization of pegylated nano graphene oxide-based delivery system for drugs with different molecular structures using design of experiment (DoE). Molecules 2021, 26, 1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, S.; Sun, L.; Huang, C. Controlled delivery of paracetamol and protein at different stages from core–shell biodegradable microspheres. Carbohydr. Polym. 2010, 79, 437–444. [Google Scholar] [CrossRef]
- Al-Nemrawi, N.K.; Dave, R.H. Formulation and characterization of acetaminophen nanoparticles in orally disintegrating films. Drug Deliv. 2016, 23, 540–549. [Google Scholar] [CrossRef]
- Hallaj-Nezhadi, S.; Hassan, M. Nanoliposome-based antibacterial drug delivery. Drug Deliv. 2015, 22, 581–589. [Google Scholar] [CrossRef]
- Gomez, G.A.; Hosseinidoust, Z. Liposomes for antibiotic encapsulation and delivery. ACS Infect. Dis. 2020, 6, 896–908. [Google Scholar] [CrossRef]
- Yusuf, A.; Almotairy, A.R.Z.; Henidi, H.; Alshehri, O.Y.; Aldughaim, M.S. Nanoparticles as drug delivery systems: A review of the implication of nanoparticles’ physicochemical properties on responses in biological systems. Polymers 2023, 15, 1596. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, G.; Zhang, J. A review of liposomes as a drug delivery system: Current status of approved products, regulatory environments, and future perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.-C. Immunological and toxicological considerations for the design of liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef]
- Barba, A.A.; Bochicchio, S.; Bertoncin, P.; Lamberti, G.; Dalmoro, A. Coating of nanolipid structures by a novel simil-microfluidic technique: Experimental and theoretical approaches. Coatings 2019, 9, 491. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Prajapati, B.; Dutta, A.; Paliwal, H. Medical application of liposomes. J. Explor. Res. Pharmacol. 2023, 8, 1–10. [Google Scholar] [CrossRef]
- Garcia-Fuentes, M.; Prego, C.; Torres, D.; Alonso, M.J. A comparative study of the potential of solid triglyceride nanostructures coated with chitosan or poly(ethylene glycol) as carriers for oral calcitonin delivery. Eur. J. Pharm. Sci. 2005, 25, 133–143. [Google Scholar] [CrossRef]
- Mohite, P.; Singh, S.; Pawar, A.; Sangale, A.; Prajapati, B.G. Lipid-based oral formulation in capsules to improve the delivery of poorly water-soluble drugs. Front. Drug Deliv. 2023, 3, 1232012. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Quispe, C.; Butnariu, M.; Rotariu, L.S.; Sytar, O.; Sestito, S.; Rapposelli, S.; Akram, M.; Iqbal, M.; Krishna, A.; et al. Chitosan nanoparticles as a promising tool in nanomedicine with particular emphasis on oncological treatment. Cancer Cell Int. 2021, 21, 318. [Google Scholar] [CrossRef]
- Kukushkina, E.A.; Hossain, S.I.; Sportelli, M.C.; Ditaranto, N.; Picca, R.A.; Cioffi, N. Ag-based synergistic antimicrobial composites. A critical review. Nanomaterials 2021, 11, 1687. [Google Scholar] [CrossRef]
- Pathak, K.; Misra, S.K.; Sehgal, A.; Singh, S.; Bungau, S.; Najda, A.; Gruszecki, R.; Behl, T. Biomedical applications of quaternized chitosan. Polymers 2021, 13, 2514. [Google Scholar] [CrossRef]
- Confederat, L.G.; Tuchilus, C.G.; Dragan, M.; Sha’at, M.; Dragostin, O.M. Preparation and antimicrobial activity of chitsan and its derivatives: A concise review. Molecules 2021, 26, 3694. [Google Scholar] [CrossRef]
- Boroumand, H.; Badie, F.; Mazaheri, S.; Seyedi, Z.S.; Nahand, J.S.; Nejati, M.; Baghi, H.B.; Abbasi-Kolli, M.; Badehnoosh, B.; Ghandali, M.; et al. Chitosan-based nanoparticles against viral infections. Front. Cell. Infect. Microbiol. 2021, 11, 643953. [Google Scholar] [CrossRef]
- Herdiana, Y.; Wathoni, N.; Shamsuddin, S.; Joni, I.M.; Muchtaridi, M. Chitosan-based nanoparticles of targeted drug delivery system in breast cancer treatment. Polymers 2021, 13, 1717. [Google Scholar] [CrossRef]
- Abd-Elhakeem, A.; Radwan, M.S.; Rady, A.H. Chitosan nanoparticles as hepato-protective agent against alcohol and fatty diet stress in rats. J. Biochem. Int. 2017, 4, 5–10. Available online: https://www.ikprress.org/index.php/JOBI/article/view/3368 (accessed on 23 October 2023).
- Midekessa, G.; Godakumara, K.; Ord, J.; Viil, J.; Lättekivi, F.; Dissanayake, K.; Kopanchuk, S.; Rinken, A.; Andronowska, A.; Bhattacharjee, S.; et al. Zeta potential of extracellular vesicles: Toward understanding the attributes that determine colloidal stability. ACS Omega 2020, 5, 16701–16710. [Google Scholar] [CrossRef]
- Sebaaly, C.; Trifan, A.; Sieniawska, E.; Greige-Gerges, H. Chitosan-coating effect on the characteristics of liposomes: A focus on bioactive compounds and essential oils: A review. Processes 2021, 9, 445. [Google Scholar] [CrossRef]
- Ladiè, R.; Cosentino, C.; Tagliaro, I.; Antonini, C.; Bianchini, G.; Bertini, S. Supramolecular structuring of hyaluronan-lactose-modified Chitosan matrix: Towards high-performance biopolymers with excellent biodegradation. Biomolecules 2021, 11, 389. [Google Scholar] [CrossRef]
- Harugade, A.; Sherje, A.P.; Pethe, A. Chitosan: A review on properties, biological activities and recent progress in biomedical applications. React. Funct. Polym. 2023, 191, 105634. [Google Scholar] [CrossRef]
- Yan, L.; Crayton, S.H.; Thawani, J.P.; Amirshaghaghi, A.; Tsourkas, A.; Cheng, Z. A pH-responsive drug-delivery platform based on glycol chitosan-coated liposomes. Small 2015, 11, 4870–4874. [Google Scholar] [CrossRef] [PubMed]
- Rusu, A.G.; Chiriac, A.P.; Nita, L.E.; Rosca, I.; Pinteala, M.; Mititelu-Tartau, L. Chitosan derivatives in macromolecular co-assembly nanogels with potential for biomedical applications. Biomacromolecules 2020, 21, 4231–4243. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, C.N.; Gomes, A.J.; Rocha, F.S.; De Tommaso, J.; Patience, G.S. Experimental methods in chemical engineering: Zeta potential. Can. J. Chem. Eng. 2021, 99, 627–639. [Google Scholar] [CrossRef]
- Taqvi, S.; Bassioni, G. Understanding wettability through zeta potential measurements [Internet]. In Wettability and Interfacial Phenomena—Implications for Material Processing; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Carrasco-Sandoval, J.; Aranda, M.; Henríquez-Aedo, K.; Fernández, M.; López-Rubio, A.; Fabra, M.J. Impact of molecular weight and deacetylation degree of chitosan on the bioaccessibility of quercetin encapsulated in alginate/chitosan-coated zein nanoparticles. Int. J. Biol. Macromol. 2023, 242 Pt 2, 124876. [Google Scholar] [CrossRef] [PubMed]
- Silva, H.D.; Beldíkova, E.; Poejo, J.; Abrunhosa, L.; Serra, A.T.; Duarte, C.M.M.; Br’anyik, T.; Cerqueira, M.A.; Pinheiro, A.C.; Vicente, A.A. Evaluating the effect of chitosan layer on bioaccessibility and cellular uptake of curcumin nanoemulsions. J. Food Eng. 2019, 243, 89–100. [Google Scholar] [CrossRef]
- Nguyen, T.X.; Huang, L.; Liu, L.; Elamin Abdalla, A.M.; Gauthier, M.; Yang, G. Chitosan-coated nano-liposomes for the oral delivery of berberine hydrochloride. J. Mater. Chem. B 2014, 2, 7149–7159. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.X.; Li, B.K.; Yin, D.K.; Liang, J.; Li, S.S.; Peng, D.Y. Layer-by-layer assembly of chitosan stabilized multilayered liposomes for paclitaxel delivery. Carbohydr. Polym. 2014, 111, 298–304. [Google Scholar] [CrossRef]
- Alshraim, M.O.; Sangi, S.; Harisa, G.I.; Alomrani, A.H.; Yusuf, O.; Badran, M.M. Chitosan-Coated Flexible Liposomes Magnify the Anticancer Activity and Bioavailability of Docetaxel: Impact on Composition. Molecules 2019, 24, 250. [Google Scholar] [CrossRef]
- Goyal, T.; Schmotzer, C.L. Validation of hemolysis index thresholds optimizes detection of clinically significant hemolysis. Am. J. Clin. Pathol. 2015, 143, 579–583. [Google Scholar] [CrossRef]
- de Lima, J.M.; Sarmento, R.R.; de Souza, J.R.; Brayner, F.A.; Feitosa, A.P.; Padilha, R.; Alves, L.C.; Porto, I.J.; Batista, R.F.; de Oliveira, J.E.; et al. Evaluation of hemagglutination activity of chitosan nanoparticles using human erythrocytes. BioMed Res. Int. 2015, 2015, 247965. [Google Scholar] [CrossRef]
- Jesus, S.; Marques, A.P.; Duarte, A.; Soares, E.; Costa, J.P.; Colaço, M.; Schmutz, M.; Som, C.; Borchard, G.; Wick, P.; et al. Chitosan nanoparticles: Shedding light on immunotoxicity and hemocompatibility. Front. Bioeng. Biotechnol. 2020, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sun, T.; Zhong, R.; Ma, L.; You, C.; Tian, M.; Li, H.; Wang, C. Effects of chitosan oligosaccharides on human blood components. Front. Pharmacol. 2018, 9, 1412. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Zeng, Y.; Zaldivar-Silva, D.; Agüero, L.; Wang, S. Chitosan-based hemostatic hydrogels: The concept, mechanism, application, and prospects. Molecules 2023, 28, 1473. [Google Scholar] [CrossRef] [PubMed]
- Lala, V.; Zubair, M.; Minter, D.A. Liver function tests. [Updated 2023 Jul 30]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482489/ (accessed on 17 October 2023).
- Gounden, V.; Bhatt, H.; Jialal, I. Renal function tests. [Updated 2023 Jul 17]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507821/ (accessed on 25 October 2023).
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14 (Suppl. S2), 49. [Google Scholar] [CrossRef] [PubMed]
- Legea nr. 206/27 mai 2004. Available online: http://legislatie.just.ro/Public/DetaliiDocument/52457 (accessed on 29 October 2023).
- European Union. DIRECTIVE 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes; European Union: Brussels, Belgium, 2010; Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:en:PDF (accessed on 23 October 2023).
- Träger, J.; Meister, A.; Hause, G.; Harauz, G.; Hinderberger, D. Shaping membrane interfaces in lipid vesicles mimicking the cytoplasmic leaflet of myelin through variation of cholesterol and myelin basic protein contents. Biochim. Biophys. Acta Biomembr. 2023, 1865, 184179. [Google Scholar] [CrossRef]
- Disalvo, A.; Frias, M.A. Surface characterization of lipid biomimetic systems. Membranes 2021, 11, 821. [Google Scholar] [CrossRef]
- Nsairat, H.; Khater, D.; Sayed, U.; Odeh, F.; Al Bawab, A.; Alshaer, W. Liposomes: Structure, composition, types, and clinical applications. Heliyon 2022, 8, e09394. [Google Scholar] [CrossRef]
- Guillot, A.J.; Martínez-Navarrete, M.; Garrigues, T.M.; Melero, A. Skin drug delivery using lipid vesicles: A starting guideline for their development. J. Control. Release 2023, 355, 624–654. [Google Scholar] [CrossRef]
- Le, Q.V.; Lee, J.; Lee, H.; Shim, G.; Oh, Y.K. Cell membrane-derived vesicles for delivery of therapeutic agents. Acta Pharm. Sin. B. 2021, 11, 2096–2113. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Kiselev, M.A. Methods of liposomes preparation: Formation and control factors of versatile nanocarriers for biomedical and nanomedicine application. Pharmaceutics 2022, 14, 543. [Google Scholar] [CrossRef] [PubMed]
- Tartau, L.; Cazacu, A.; Melnig, V. Ketoprofen-liposomes formulation for clinical therapy. J. Mater. Sci. Mater. Med. 2012, 23, 2499–2507. [Google Scholar] [CrossRef] [PubMed]
- Pauna, A.R.; Mititelu-Tartau, L.; Bogdan, M.; Meca, A.D.; Popa, G.E.; Pelin, A.M.; Drochioi, C.I.; Pricop, D.A.; Pavel, L.L. Synthesis, characterization and biocompatibility evaluation of novel chitosan lipid micro-systems for modified release of diclofenac sodium. Biomedicines 2023, 11, 453. [Google Scholar] [CrossRef] [PubMed]
- Sæbø, I.P.; Bjørås, M.; Franzyk, H.; Helgesen, E.; Booth, J.A. Optimization of the hemolysis assay for the assessment of cytotoxicity. Int. J. Mol. Sci. 2023, 24, 2914. [Google Scholar] [CrossRef]
- Parasuraman, S.; Raveendran, R.; Kesavan, R. Blood sample collection in small laboratory animals. J. Pharmacol. Pharmacother. 2010, 1, 87–93. [Google Scholar] [CrossRef]
- Marin, N.; Moragon, A.; Gil, D.; Garcia-Garcia, F.; Bisbal, V. Acclimation and blood sampling: Effects on stress markers in C57Bl/6J mice. Animals 2023, 13, 2816. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | pH |
|---|---|
| APAP solution (APAP) | 6.00 |
| APAP vesicles with CS (prior to the dialysis process) (APAP-v nondialyzed) | 4.65 |
| Dialyzed APAP vesicles with CS (APAP-v) | 6.63 |
| Group | Triton X-100 | Negative Control | CS | APAP | APAP-v |
|---|---|---|---|---|---|
| Hemolysis % | 88.45 ± 4.17 ** | 0.07 ± 0.01 | 1.64 ± 0.15 | 2.38 ± 0.25 | 2.52 ± 0.21 |
| Groups. | Time Elapsed | White Blood Count | ||||
|---|---|---|---|---|---|---|
| % | ||||||
| PMN | Ly | E | M | B | ||
| Control | 24 h | 19.57 ± 3.23 | 78.20 ± 4.59 | 0.35 ± 0.17 | 1.67 ± 1.13 | 0.21 ± 0.03 |
| 7 days | 19.96 ± 2.59 | 77.31 ± 3.72 | 0.41 ± 0.26 | 2.11 ± 1.42 | 0.21 ± 0.07 | |
| CS | 24 h | 18.35 ± 2.57 | 79.45 ± 4.42 | 0.45 ± 0.35 | 1.55 ± 1.59 | 0.20 ± 0.05 |
| 7 days | 19.17 ± 1.71 | 78.41 ± 1.82 | 0.53 ± 0.42 | 1.68 ± 1.67 | 0.21 ± 0.07 | |
| APAP | 24 h | 18.70 ± 3.03 | 78.47 ± 2.87 | 0.55 ± 0.23 | 2.07 ± 1.03 | 0.21 ± 0.11 |
| 7 days | 19.67 ± 2.01 | 77.43 ± 2.23 | 0.47 ± 0.59 | 2.15 ± 1.26 | 0.22 ± 0.13 | |
| APAP-v | 24 h | 20.84 ± 3.81 | 75.96 ± 3.71 | 0.58 ± 0.87 | 2.41 ± 0.42 | 0.21 ± 0.07 |
| 7 days | 21.42 ± 4.07 | 76.10 ± 3.23 | 0.57 ± 0.76 | 2.35 ± 0.70 | 0.20 ± 0.18 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilițanu, L.N.; Mititelu-Tarțău, L.; Popa, E.G.; Bucă, B.R.; Gurzu, I.L.; Fotache, P.A.; Pelin, A.-M.; Pricop, D.A.; Pavel, L.L. Chitosan Soft Matter Vesicles Loaded with Acetaminophen as Promising Systems for Modified Drug Release. Molecules 2024, 29, 57. https://doi.org/10.3390/molecules29010057
Hilițanu LN, Mititelu-Tarțău L, Popa EG, Bucă BR, Gurzu IL, Fotache PA, Pelin A-M, Pricop DA, Pavel LL. Chitosan Soft Matter Vesicles Loaded with Acetaminophen as Promising Systems for Modified Drug Release. Molecules. 2024; 29(1):57. https://doi.org/10.3390/molecules29010057
Chicago/Turabian StyleHilițanu, Loredana Nicoleta, Liliana Mititelu-Tarțău, Eliza Grațiela Popa, Beatrice Rozalina Bucă, Irina Luciana Gurzu, Paula Alina Fotache, Ana-Maria Pelin, Daniela Angelica Pricop, and Liliana Lăcrămioara Pavel. 2024. "Chitosan Soft Matter Vesicles Loaded with Acetaminophen as Promising Systems for Modified Drug Release" Molecules 29, no. 1: 57. https://doi.org/10.3390/molecules29010057
APA StyleHilițanu, L. N., Mititelu-Tarțău, L., Popa, E. G., Bucă, B. R., Gurzu, I. L., Fotache, P. A., Pelin, A.-M., Pricop, D. A., & Pavel, L. L. (2024). Chitosan Soft Matter Vesicles Loaded with Acetaminophen as Promising Systems for Modified Drug Release. Molecules, 29(1), 57. https://doi.org/10.3390/molecules29010057