
Synthetic Approaches to Piperazine-Containing Drugs Approved by FDA in the Period of 2011–2023 †

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Synthesis of Drugs Carrying a Piperazine Ring Decorated Only on N-Atoms
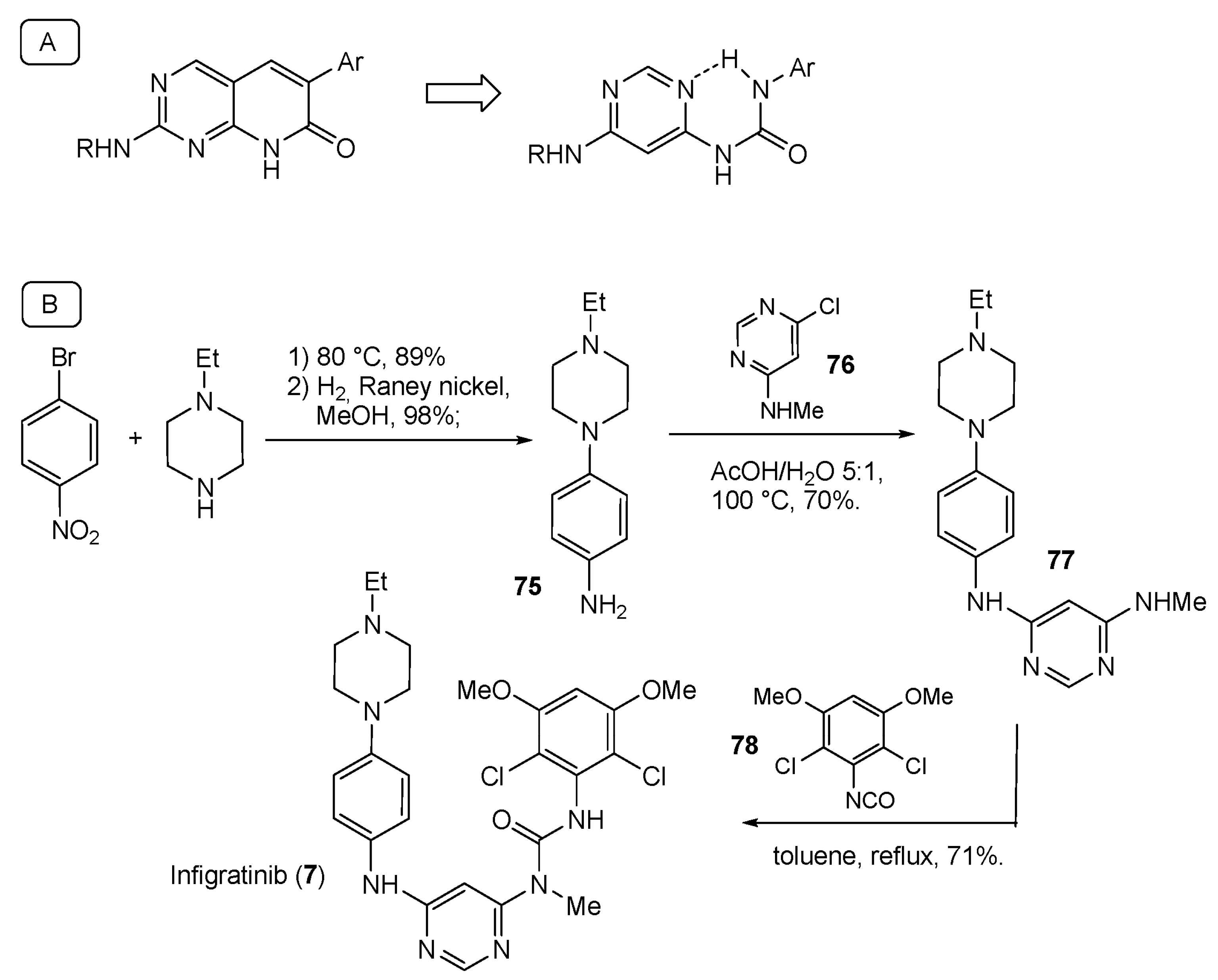
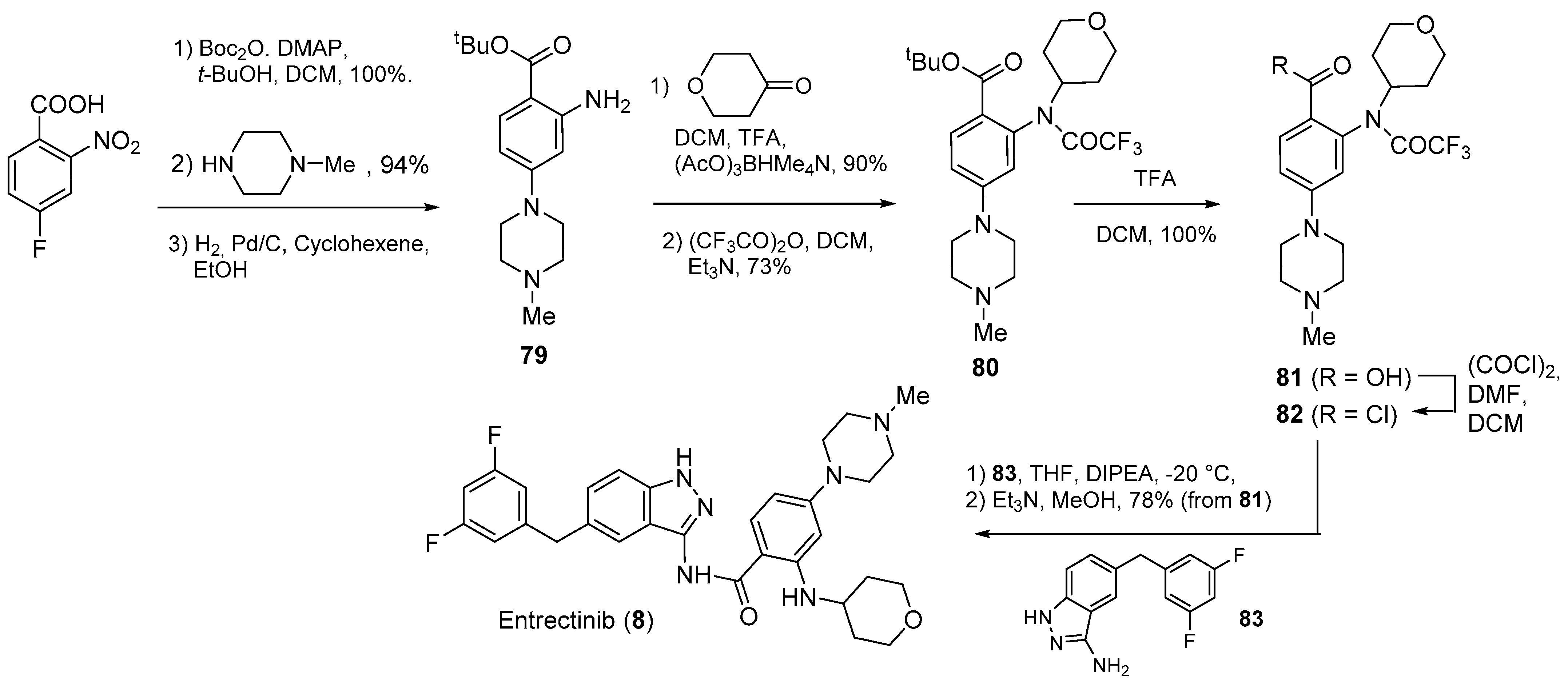
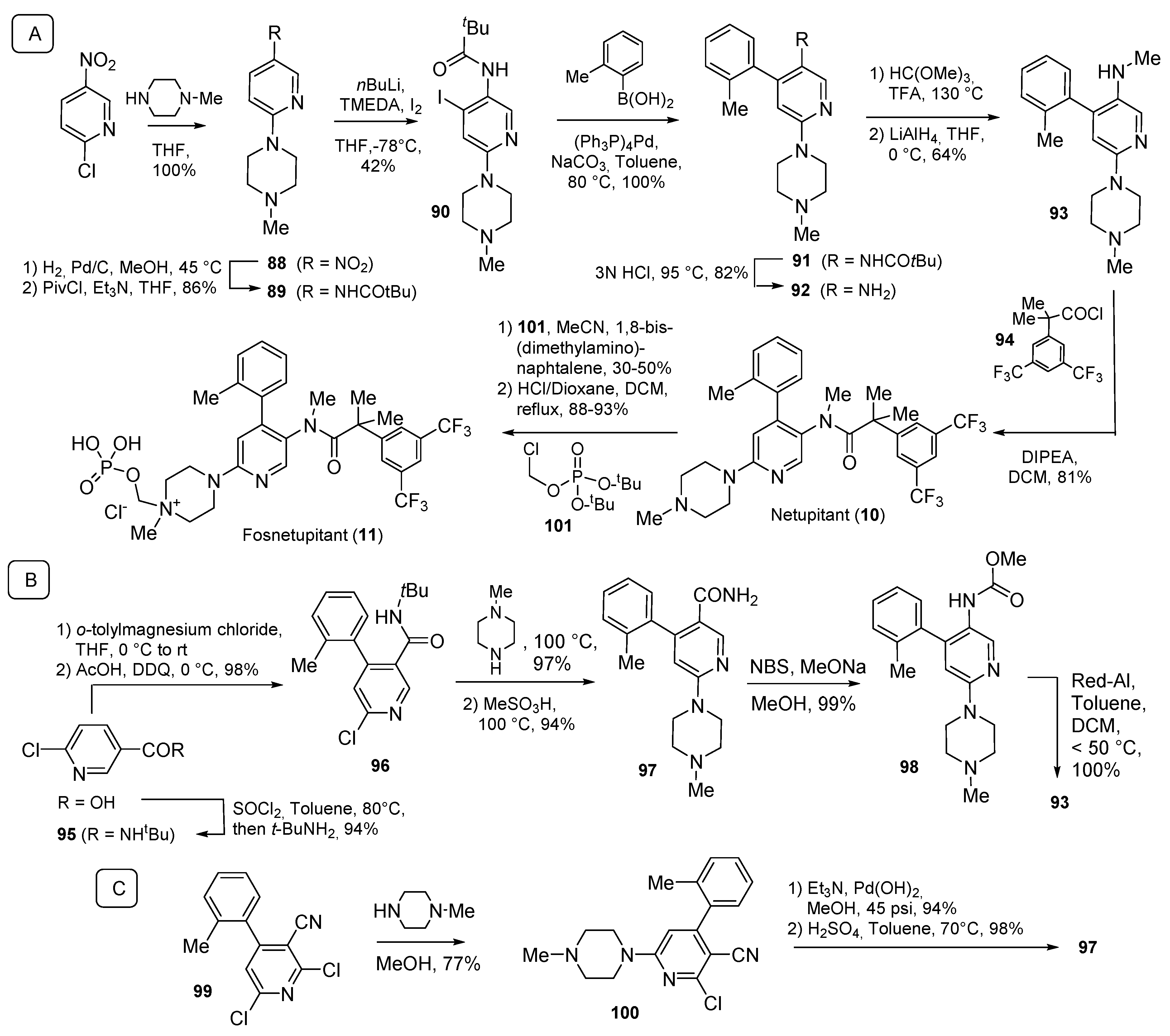
2.1. N-Aryl Derivatives
2.2. N-Alkyl Derivatives
2.3. N-Acyl Derivatives
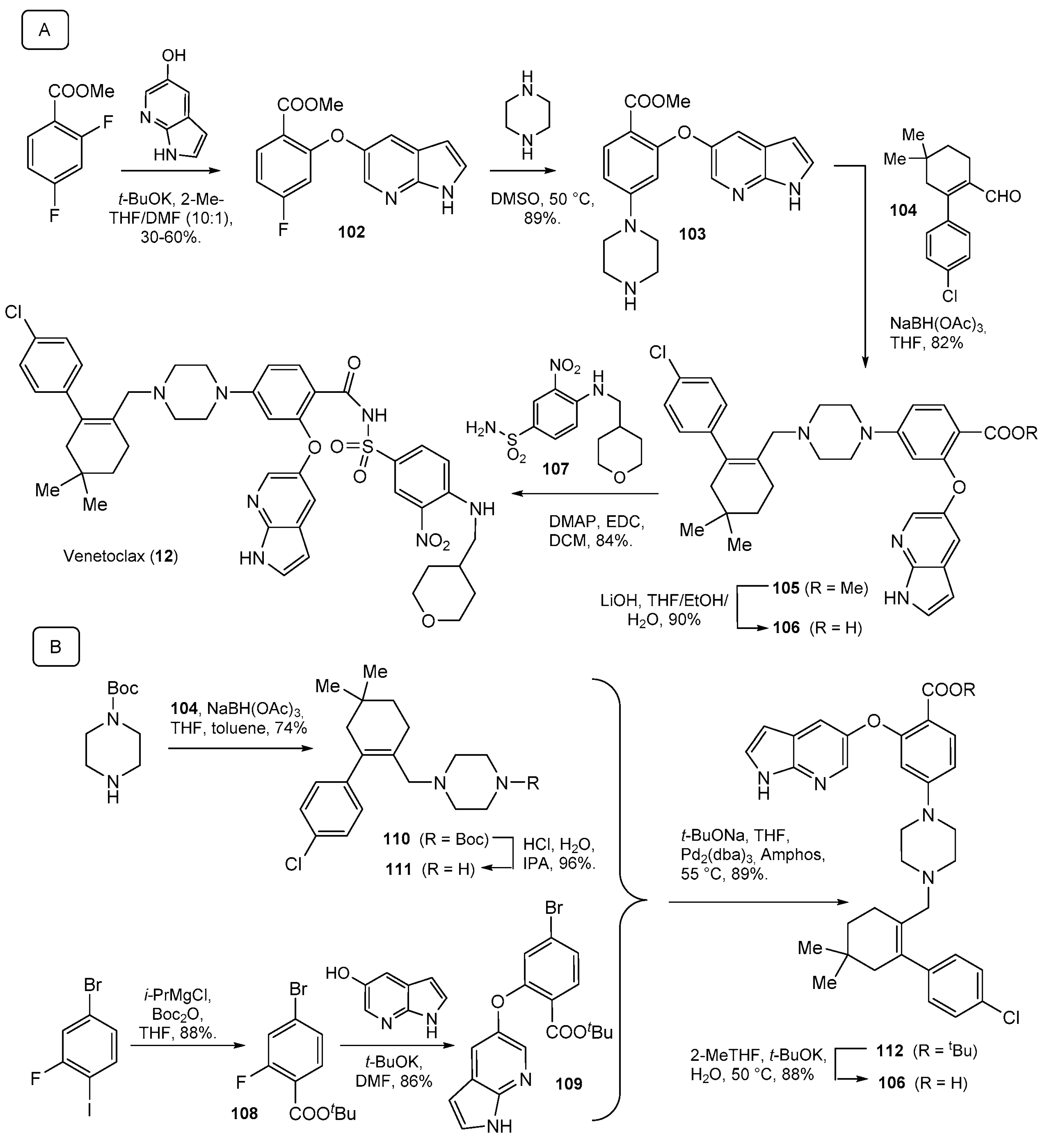
3. Synthesis of Drugs Carrying C-Substituents on the Piperazine Ring
4. Role of the Piperazine Moiety
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Taylor, R.D.; Maccoss, M.; Lawson, A.D.G. Rings in Drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A.; Loiseleur, O. Applications of Isosteres of Piperazine in the Design of Biologically Active Compounds: Part 1. J. Agric. Food Chem. 2022, 70, 10942–10971. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.N.; Manetti, D.; Braconi, L.; Dei, S.; Gabellini, A.; Teodori, E. The piperazine scaffold for novel drug discovery efforts: The evidence to date. Exp. Opin. Drug Discov. 2022, 17, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Dinsmore, C.J.; Beshore, D.C. Syntheses and transformations of piperazinone rings. A review. Org. Prep. Proced. Int. 2002, 34, 367–404. [Google Scholar] [CrossRef]
- Dömling, A.; Huang, Y. Piperazine Scaffolds via Isocyanide-Based Multicomponent Reactions. Synthesis 2010, 2010, 2859–2883. [Google Scholar] [CrossRef]
- Ye, Z.; Gettys, K.E.; Dai, M. Opportunities and challenges for direct C–H functionalization of piperazines. Beilstein J. Org. Chem. 2016, 12, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Gettys, K.E.; Ye, Z.; Dai, M. Recent Advances in Piperazine Synthesis. Synthesis 2017, 49, 2589–2604. [Google Scholar] [CrossRef]
- Seifinoferest, B.; Tanbakouchian, A.; Larijani, B.; Mahdavi, M. Ullmann-Goldberg and Buchwald-Hartwig C−N Cross Couplings: Synthetic Methods to Pharmaceutically Potential N-Heterocycles. Asian J. Org. Chem. 2021, 10, 1319–1344. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Zahler, R.E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.-A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Poratti, M.; Marzaro, G. Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; Vanderwel, S.N.; Zhou, H.; Keller, P.R.; Mcnamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Calienni, J.V.; Chen, G.-P.; Gong, B.; Kapa, P.K.; Saxena, V. Salt(S) of 7-Cyclopentyl-2-(5-Piperazin-1-Yl-Pyridin-2-Ylamino)-7h-Pyrrolo[2,3-d]Pyrimidine-6-Carboxylic Acid Dimethylamide and Processes of Making Thereof. U.S. Patent US2012115878A1, 10 May 2012. [Google Scholar]
- Duan, S.; Place, D.; Perfect, H.H.; Ide, N.D.; Maloney, M.; Sutherland, K.; Price Wiglesworth, K.E.; Wang, K.; Olivier, M.; Kong, F.; et al. Palbociclib Commercial Manufacturing Process Development. Part I: Control of Regioselectivity in a Grignard-Mediated SNAr Coupling. Org. Process Res. Dev. 2016, 20, 1191–1202. [Google Scholar] [CrossRef]
- Chekal, B.P.; Ewers, J.; Guinness, S.M.; Ide, N.D.; Leeman, K.R.; Post, R.J.; Rane, A.M.; Sutherland, K.; Wang, K.; Webster, M.; et al. Palbociclib Commercial Manufacturing Process Development. Part III. Deprotection Followed by Crystallization for API Particle Property Control. Org. Process Res. Dev. 2016, 20, 1217–1226. [Google Scholar] [CrossRef]
- Besong, G.; Brain, C.T.; Brooks, C.A.; Congreve, M.S.; Dagostin, C.; He, G.; Hou, Y.; Howard, S.; Li, Y.; Lu, Y.; et al. Pyrrolopyrimidine Compounds as CDK Inhibitors. Patent WO2010020675A1, 25 February 2010. [Google Scholar]
- Pellegatti, L.; Hafner, A.; Sedelmeier, J. A Two-Step Continuous-Flow Procedure towards Ribociclib. J. Flow Chem. 2016, 6, 198–201. [Google Scholar] [CrossRef]
- Tavares, F.X.; Strum, J.C. CDK Inhibitors. U.S. Patent US2013237534A1, 12 September 2013. [Google Scholar]
- Smith, A.; White, H.S.; Tavares, F.X.; Krasutsky, S.; Chen, J.-X.; Dorrow, R.L.; Zhong, H. Synthesis of N-(Heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. Patent WO2018005865A1, 4 January 2018. [Google Scholar]
- Bang-Andersen, B.; Ruhland, T.; Jørgensen, M.; Smith, G.; Frederiksen, K.; Jensen, K.G.; Zhong, H.; Nielsen, S.M.; Hogg, S.; Mørk, A.; et al. Discovery of 1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A Novel Multimodal Compound for the Treatment of Major Depressive Disorder. J. Med. Chem. 2011, 54, 3206–3221. [Google Scholar] [CrossRef]
- Sanchez, C.; Asin, K.E.; Artigas, F. Vortioxetine, a novel antidepressant with multimodal activity: Review of preclinical and clinical data. Pharmacol. Ther. 2015, 145, 43–57. [Google Scholar] [CrossRef]
- Bishop, M.M.; Fixen, D.R.; Linnebur, S.A.; Pearson, S.M. Cognitive effects of vortioxetine in older adults: A systematic review. Ther. Adv. Psychopharmacol. 2021, 11, 20451253211026796. [Google Scholar] [CrossRef]
- Alcántara Montero, A.; Pacheco De Vasconcelos, S.R. Role of vortioxetine in the treatment of neuropathic pain. Rev. Esp. Anestesiol. Reanim. (Engl. Ed.) 2022, 69, 640–648. [Google Scholar] [CrossRef]
- Bang-Andersen, B.; Faldt, A.; Moerk, A.; Lopez De Diego, H.; Holm, R.; Stensboel, T.B.; Ringgaard, L.M.; Mealy, M.J.; Rock, M.H.; Brodersen, J.; et al. 1-[2-(2,4-Dimethylphenylsulfanyl)-phenyl] Piperazine as a Compound with Combined Serotonin Reuptake, 5-HT3 and 5-HT1a Activity for the Treatment of Cognitive Impairment. Patent WO2007144005A1, 21 December 2005. [Google Scholar]
- Zupancic, B. Synthesis of Vortioxetine via (2,4-Dimethylphenyl)(2-iodophenyl)sulfane Intermediate. Patent WO2015155153A1, 15 October 2015. [Google Scholar]
- Zupancic, B. New Process for the Synthesis of 1-(2-((2,4-Dimethylphenyl)thio)phenyl)piperazine. Patent WO2014161976A1, 9 October 2014. [Google Scholar]
- Zupancic, B.; Sterk, D.; Maras, N. Synthesis of Vortioxetine via (2-(Piperazine-1-yl)phenvl)lithium Intermediates. Patent WO2015079018A1, 6 April 2015. [Google Scholar]
- Zupancic, B. New Process for the Synthesis of 1-(2-((2,4-Dimethylphenyl)thio)phenyl)piperazine. Patent WO2014191548A1, 12 April 2014. [Google Scholar]
- Dhillon, S. Avapritinib: First Approval. Drugs 2020, 80, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Klug, L.R.; Kent, J.D.; Heinrich, M.C. Structural and clinical consequences of activation loop mutations in class III receptor tyrosine kinases. Pharmacol. Ther. 2018, 191, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.K.; Gardino, A.K.; Kim, J.L.; Hodous, B.L.; Shutes, A.; Davis, A.; Zhu, X.J.; Schmidt-Kittler, O.; Wilson, D.; Wilson, K.; et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 2017, 9, eaao1690. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-P.; Lusvarghi, S.; Wang, J.-C.; Hsiao, S.-H.; Huang, Y.-H.; Hung, T.-H.; Ambudkar, S.V. Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines. Mol. Pharm. 2019, 16, 3040–3052. [Google Scholar] [CrossRef]
- Hodous, B.L.; Kim, J.L.; Wilson, K.J.; Wilson, D.; Zhang, Y. Compositions Useful for Treating Disorders Related to KIT. U.S. Patent US2015111887A1, 23 April 2015. [Google Scholar]
- Waetzig, J.; Mar, B.; Heinrich, B.; Wilkie, G.; Maceachern, L. Crystalline forms of (S)-1-(4-Fluorophenyl)-1-(2-(4-(6-(1-methyl-1h-pyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl)piperazinyl)-pyrimidin-5-yl)ethan-1-amine and Methods of Making. Patent WO2020210669A1, 15 October 2020. [Google Scholar]
- Porcs-Makkay, M.; Bertha, F.; Molnár, E.; Németh, G.; Horváth, S.; Szebelédi, I.; Bali, B.; Tellér, M.; Kátainé Fadgyas, K. Process for Obtaining Avapritinib and Its Intermediates. Patent WO2022180416A1, 9 January 2022. [Google Scholar]
- Kim, E.S. Letermovir: First Global Approval. Drugs 2018, 78, 147–152. [Google Scholar] [CrossRef]
- Lischka, P.; Hewlett, G.; Wunberg, T.; Baumeister, J.; Paulsen, D.; Goldner, T.; Ruebsamen-Schaeff, H.; Zimmermann, H. In Vitro and In Vivo Activities of the Novel Anticytomegalovirus Compound AIC246. Antimicrob. Agents Chemother. 2010, 54, 1290–1297. [Google Scholar] [CrossRef]
- Gentry, B.G.; Bogner, E.; Drach, J.C. Targeting the terminase: An important step forward in the treatment and prophylaxis of human cytomegalovirus infections. Antivir. Res. 2019, 161, 116–124. [Google Scholar] [CrossRef]
- Wunberg, T.; Baumeister, J.; Betz, U.; Jeske, M.; Lampe, T.; Nikolic, S.; Reefschlager, J.; Schohe-Loop, R.; Sussmeier, F.; Zimmermann, H.; et al. Substituted Dihydroquinazolines. U.S. Patent US2007191387A1, 16 August 2007. [Google Scholar]
- Paulus, K.; Schwab, W.; Grunder, D.; Van Hoogevest, P. Pharmaceutical Composition Containing an Antivirally Active Dihydroquinazoline Derivative. U.S. Patent US2015133461A1, 14 May 2015. [Google Scholar]
- Humphrey, G.R.; Dalby, S.M.; Andreani, T.; Xiang, B.; Luzung, M.R.; Song, Z.J.; Shevlin, M.; Christensen, M.; Belyk, K.M.; Tschaen, D.M. Asymmetric Synthesis of Letermovir Using a Novel Phase-Transfer-Catalyzed Aza-Michael Reaction. Org. Process Res. Dev. 2016, 20, 1097–1103. [Google Scholar] [CrossRef]
- Kang, C. Infigratinib: First Approval. Drugs 2021, 81, 1355–1360. [Google Scholar] [CrossRef]
- Furet, P.; Caravatti, G.; Guagnano, V.; Lang, M.; Meyer, T.; Schoepfer, J. Entry into a new class of protein kinase inhibitors by pseudo ring design. Bioorg. Med. Chem. Lett. 2008, 18, 897–900. [Google Scholar] [CrossRef]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, M.; Li, H.; Chen, L. Entrectinib, a new multi-target inhibitor for cancer therapy. Biomed. Pharmacother. 2022, 150, 112974. [Google Scholar] [CrossRef] [PubMed]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, T.; Lombardi, B.A.; Nesi, M.; Perrone, E.; Bossi, R.; Polucci, P. Indazole Derivatives as Kinase Inhibitors for the Treatment of Cancer. Patent WO2008074749A1, 26 June 2008. [Google Scholar]
- Shirley, M. Avatrombopag: First Global Approval. Drugs 2018, 78, 1163–1168. [Google Scholar] [CrossRef]
- Zhang, J.; Alt, J.; Yang, M.; Baranwal, A.; Luby, T.M. AKR-501 Activates the Thrombopoietin Receptor through Interaction with the Transmembrane Domain. Blood 2008, 112, 5391. [Google Scholar] [CrossRef]
- Kuter, D.J. The structure, function, and clinical use of the thrombopoietin receptor agonist avatrombopag. Blood Rev. 2022, 53, 100909. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Watanuki, S.; Koga, Y.; Nagata, H.; Obitsu, K.; Wakayama, R.; Hirayama, F.; Suzuki, K.-I. 2-Acylaminothiazole Derivative or Salt Thereof. Patent EP1466912A1, 13 October 2004. [Google Scholar]
- Shirley, M. Netupitant/Palonosetron: A Review in Chemotherapy-Induced Nausea and Vomiting. Drugs 2021, 81, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Bös, M.; Stadler, H.; Schnider, P.; Hunkeler, W.; Godel, T.; Galley, G.; Ballard, T.M.; Higgins, G.A.; Poli, S.M.; et al. Design and synthesis of a novel, achiral class of highly potent and selective, orally active neurokinin-1 receptor antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 1362–1365. [Google Scholar] [CrossRef]
- Boes, M.; Branca, Q.; Galley, G.; Godel, T.; Hoffmann, T.; Hunkeler, W.; Schnider, P.; Stadler, H. 4-Phenyl-pyridine Derivatives. U.S. Patent US6297375B1, 2 October 2001. [Google Scholar]
- Hoffmann-Emery, F.; Hilpert, H.; Scalone, M.; Waldmeier, P. Efficient Synthesis of Novel NK1 Receptor Antagonists: Selective 1,4-Addition of Grignard Reagents to 6-Chloronicotinic Acid Derivatives. J. Org. Chem. 2006, 71, 2000–2008. [Google Scholar] [CrossRef]
- Harrington, P.J.; Johnston, D.; Moorlag, H.; Wong, J.-W.; Hodges, L.M.; Harris, L.; Mcewen, G.K.; Smallwood, B. Research and Development of an Efficient Process for the Construction of the 2,4,5-Substituted Pyridines of NK-1 Receptor Antagonists. Org. Process Res. Dev. 2006, 10, 1157–1166. [Google Scholar] [CrossRef]
- Fadini, L.; Manini, P.; Pietra, C.; Giuliano, C.; Lovati, E.; Cannella, R.; Venturini, A.; Stella, V.J. Substituted Piperaziniums for the Treatment of Emesis. U.S. Patent US9908907B2, 3 June 2018. [Google Scholar]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematology 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Chen, L.; Lanning, M.E.; Fletcher, S. Expanding the Cancer Arsenal with Targeted Therapies: Disarmament of the Antiapoptotic Bcl-2 Proteins by Small Molecules. J. Med. Chem. 2017, 60, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Ku, Y.-Y.; Chan, V.S.; Christesen, A.; Grieme, T.; Mulhern, M.; Pu, Y.-M.; Wendt, M.D. Development of a Convergent Large-Scale Synthesis for Venetoclax, a First-in-Class BCL-2 Selective Inhibitor. J. Org. Chem. 2019, 84, 4814–4829. [Google Scholar] [CrossRef] [PubMed]
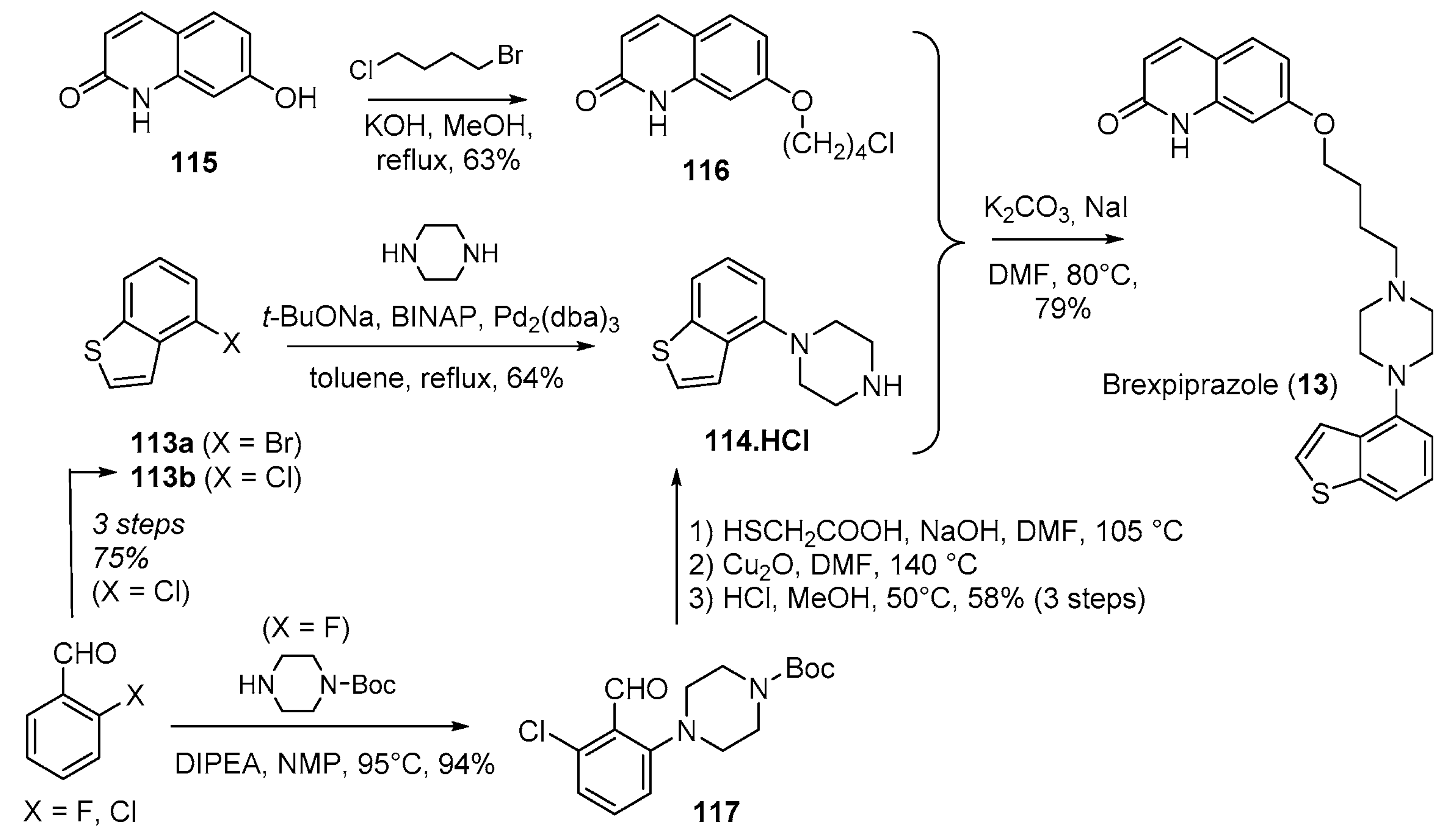
- Citrome, L. Brexpiprazole: A new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today 2015, 51, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Siwek, M.; Wojtasik-Bakalarz, K.; Krupa, A.J.; Chrobak, A.A. Brexpiprazole-Pharmacologic Properties and Use in Schizophrenia and Mood Disorders. Brain Sci. 2023, 13, 397. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Matsubara, J.; Oshima, K.; Kuroda, H.; Ito, N.; Miyamura, S.; Shimizu, S.; Tanaka, T.; Oshiro, Y.; Shimada, J.; et al. Piperazine-Substituted Benzothiophenes for Treatment of Mental Disorders. Patent WO2006112464A1, 26 October 2006. [Google Scholar]
- Miyake, M.; Shimizu, M.; Tsuji, K.; Ikeda, K. Safe and Efficient Decarboxylation Process: A Practical Synthetic Route to 4-Chlorobenzo[b]thiophene. Org. Process Res. Dev. 2016, 20, 86–89. [Google Scholar] [CrossRef]
- Shinhama, K.; Utsumi, N.; Sota, M.; Fujieda, S.; Ogasawara, S. Method for Producing benzo[b]thiophene Compound. Patent WO2013015456A1, 31 January 2013. [Google Scholar]
- Kumar, A.S.; Kandanur, S.G.S.; Sen, S.; Oruganti, S. Delineating an alternate convergent synthesis of brexpiprazole: A novel use of commercial 6,7-dihydrobenzo[b]thiophen-4(5H)-one as precursor to an efficacious Buchwald–Hartwig amination step. J. Chem. Sci. 2018, 130, 72. [Google Scholar] [CrossRef]
- Wu, C.; Chen, W.; Jiang, D.; Jiang, X.; Shen, J. An Improved Synthesis of 4-(1-Piperazinyl)benzo[b]thiophene Dihydrochloride. Org. Process Res. Dev. 2015, 19, 555–558. [Google Scholar] [CrossRef]
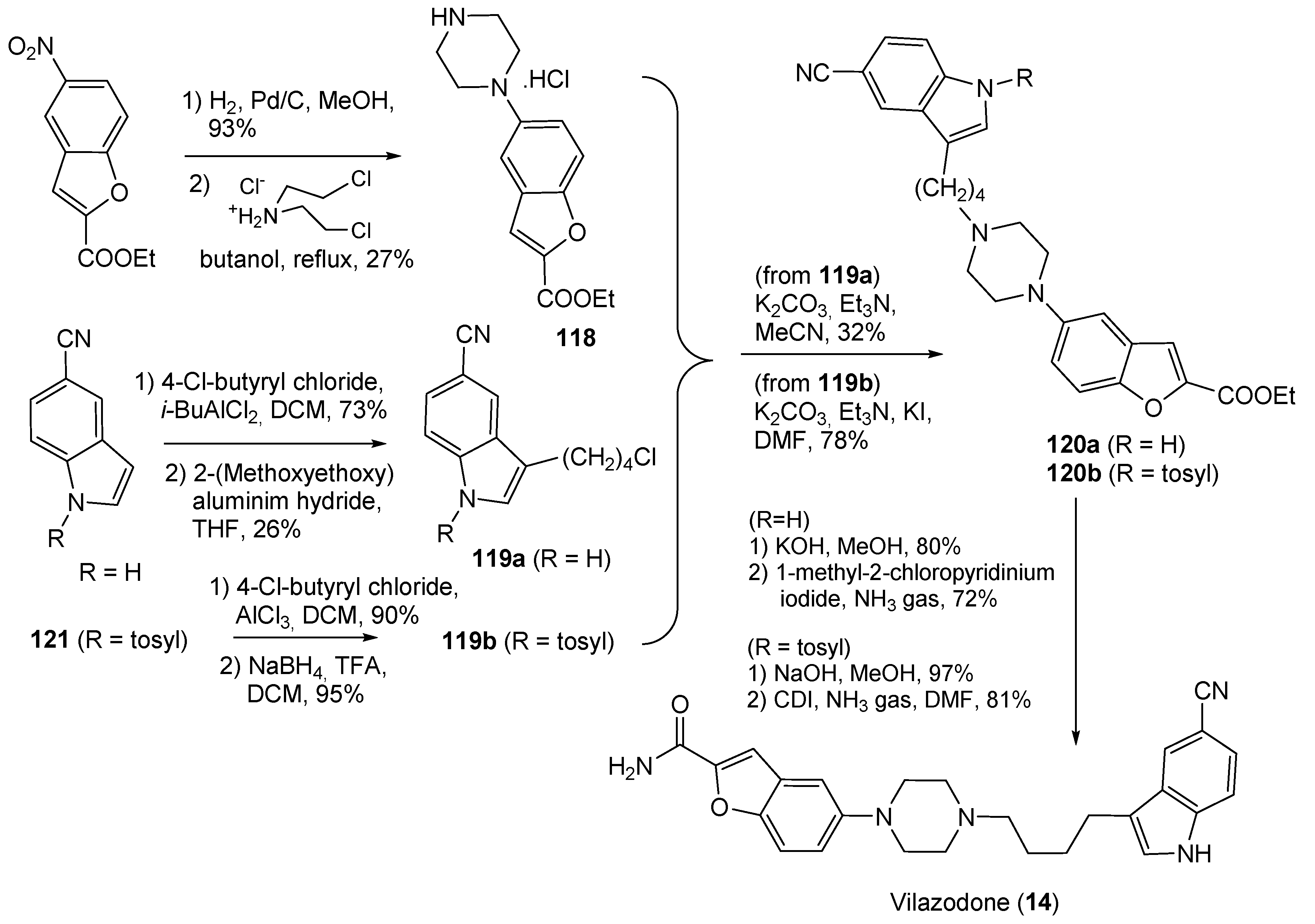
- Heinrich, T.; Böttcher, H.; Gericke, R.; Bartoszyk, G.D.; Anzali, S.; Seyfried, C.A.; Greiner, H.E.; Van Amsterdam, C. Synthesis and Structure−Activity Relationship in a Class of Indolebutylpiperazines as Dual 5-HT1A Receptor Agonists and Serotonin Reuptake Inhibitors. J. Med. Chem. 2004, 47, 4684–4692. [Google Scholar] [CrossRef]
- Hu, B.; Song, Q.; Xu, Y. Scale-Up Synthesis of Antidepressant Drug Vilazodone. Org. Process Res. Dev. 2012, 16, 1552–1557. [Google Scholar] [CrossRef]
- Borsini, F.; Evans, K.; Jason, K.; Rohde, F.; Alexander, B.; Pollentier, S. Pharmacology of Flibanserin. CNS Drug Rev. 2002, 8, 117–142. [Google Scholar] [CrossRef] [PubMed]
- Dooley, E.M.; Miller, M.K.; Clayton, A.H. Flibanserin: From Bench to Bedside. Sex. Med. Rev. 2017, 5, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignott, M. Benzimidazolone Derivatives. U.S. Patent US5576318A, 19 November 1996. [Google Scholar]
- Yang, F.; Wu, C.; Li, Z.; Tian, G.; Wu, J.; Zhu, F.; Zhang, J.; He, Y.; Shen, J. A Facile Route of Synthesis for Making Flibanserin. Org. Process Res. Dev. 2016, 20, 1576–1580. [Google Scholar] [CrossRef]
- Bugaenko, D.I.; Yurovskaya, M.A.; Karchava, A.V. N-Arylation of DABCO with Diaryliodonium Salts: General Synthesis of N-Aryl-DABCO Salts as Precursors for 1,4-Disubstituted Piperazines. Org. Lett. 2018, 20, 6389–6393. [Google Scholar] [CrossRef] [PubMed]
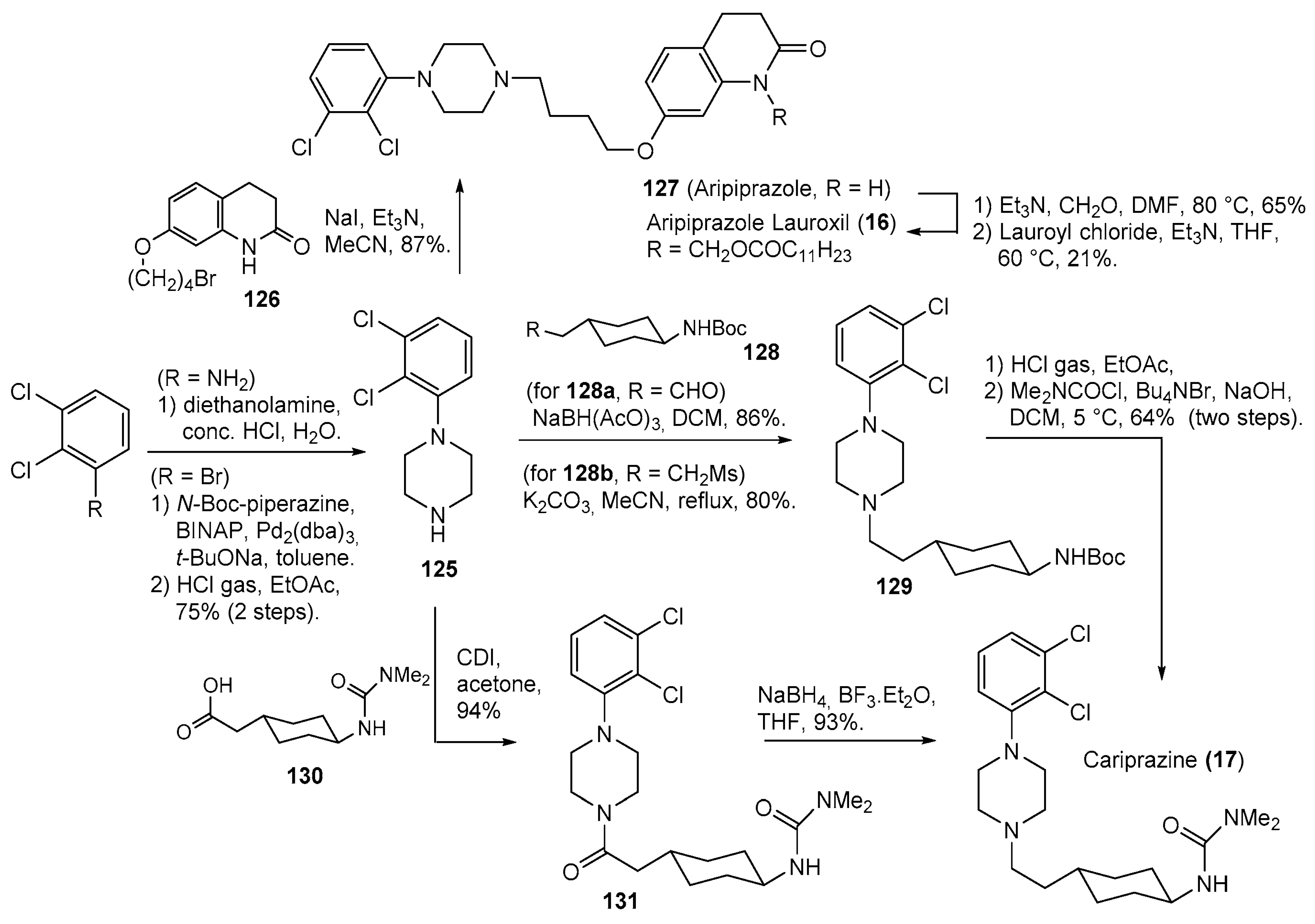
- Pahwa, M.; Sleem, A.; Elsayed, O.H.; Good, M.E.; El-Mallakh, R.S. New Antipsychotic Medications in the Last Decade. Curr. Psychiatry Rep. 2021, 23, 87. [Google Scholar] [CrossRef]
- Rohde, M.; M∅Rk, N.; Håkansson, A.E.; Jensen, K.G.; Pedersen, H.; Dige, T.; J∅Rgensen, E.B.; Holm, R. Biological conversion of aripiprazole lauroxil—An N-acyloxymethyl aripiprazole prodrug. Results Pharma Sci. 2014, 4, 19–25. [Google Scholar] [CrossRef]
- Ágai-Csongor, É.; Domány, G.; Nógrádi, K.; Galambos, J.; Vágó, I.; Keserű, G.M.; Greiner, I.; Laszlovszky, I.; Gere, A.; Schmidt, É.; et al. Discovery of cariprazine (RGH-188): A novel antipsychotic acting on dopamine D3/D2 receptors. Bioorg. Med. Chem. Lett. 2012, 22, 3437–3440. [Google Scholar] [CrossRef]
- Stahl, S.M. Mechanism of action of cariprazine. CNS Spectr. 2016, 21, 123–127. [Google Scholar] [CrossRef]
- Oshiro, Y.; Sato, S.; Kurahashi, N.; Tanaka, T.; Kikuchi, T.; Tottori, K.; Uwahodo, Y.; Nishi, T. Novel Antipsychotic Agents with Dopamine Autoreceptor Agonist Properties: Synthesis and Pharmacology of 7-[4-(4-Phenyl-1-piperazinyl)butoxy]-3,4-dihydro-2(1H)-quinolinone Derivatives. J. Med. Chem. 1998, 41, 658–667. [Google Scholar] [CrossRef]
- Pollard, C.B.; Wicker, T.H., Jr. Derivatives of Piperazine. XXIV. Synthesis of 1-Arylpiperazines and Amino Alcohol Derivatives. J. Am. Chem. Soc. 1954, 76, 1853–1855. [Google Scholar] [CrossRef]
- Perry, J.M.; Hickey, M.B.; Remenar, J.F.; Vandiver, J. Pharmaaceutical Compositions Comprising fatty Acid Esters. Patent WO2013142198A1, 26 September 2013. [Google Scholar]
- Againe Csongor, E.; Galambos, J.; Nogradi, K.; Vago, I.; Gyertyan, I.; Kiss, B.; Laszlovszky, I.; Laszy, J.; Saghy, K. (Thio)carbamoyl-cyclohexane Derivatives as D3/D2 Receptor Antagonists. Patent WO2005012266A1, 10 February 2005. [Google Scholar]
- Bhosle, S.D.; Itage, S.V.; Gangapuram, B.; Eppa, G.; Bhosale, R.S.; Yadav, J.S. Review of Synthetic Approaches toward the Synthesis of Cariprazine, an Antipsychotic Drug. Org. Process Res. Dev. 2022, 26, 493–507. [Google Scholar] [CrossRef]
- Againe Csongor, E.; Czibule, L.; Seboek, F.; Juhasz, B.; Galambos, J.; Nogradi, K. Process for the Preparation of Piperazine Derivatives. Patent WO2010070371A1, 24 June 2010. [Google Scholar]
- Czibula, L.; Againe Csongor, E.; Nogradi, K.; Juhasz, B.; Sebok, F.; Galambos, J.; Vago, I. Piperazine Salt and a Process for the Preparation Thereof. U.S. Patent US2011275816A1, 10 November 2011. [Google Scholar]
- Neu, J.; Garadnay, S.; Szabó, T. Industrial Process for the Preparation of Cariprazine. Patent WO2018007986A1, 11 January 2018. [Google Scholar]
- Afanasyev, O.I.; Kuchuk, E.; Usanov, D.L.; Chusov, D. Reductive Amination in the Synthesis of Pharmaceuticals. Chem. Rev. 2019, 119, 11857–11911. [Google Scholar] [CrossRef] [PubMed]
- Magano, J.; Dunetz, J.R. Large-Scale Carbonyl Reductions in the Pharmaceutical Industry. Org. Process Res. Dev. 2012, 16, 1156–1184. [Google Scholar] [CrossRef]
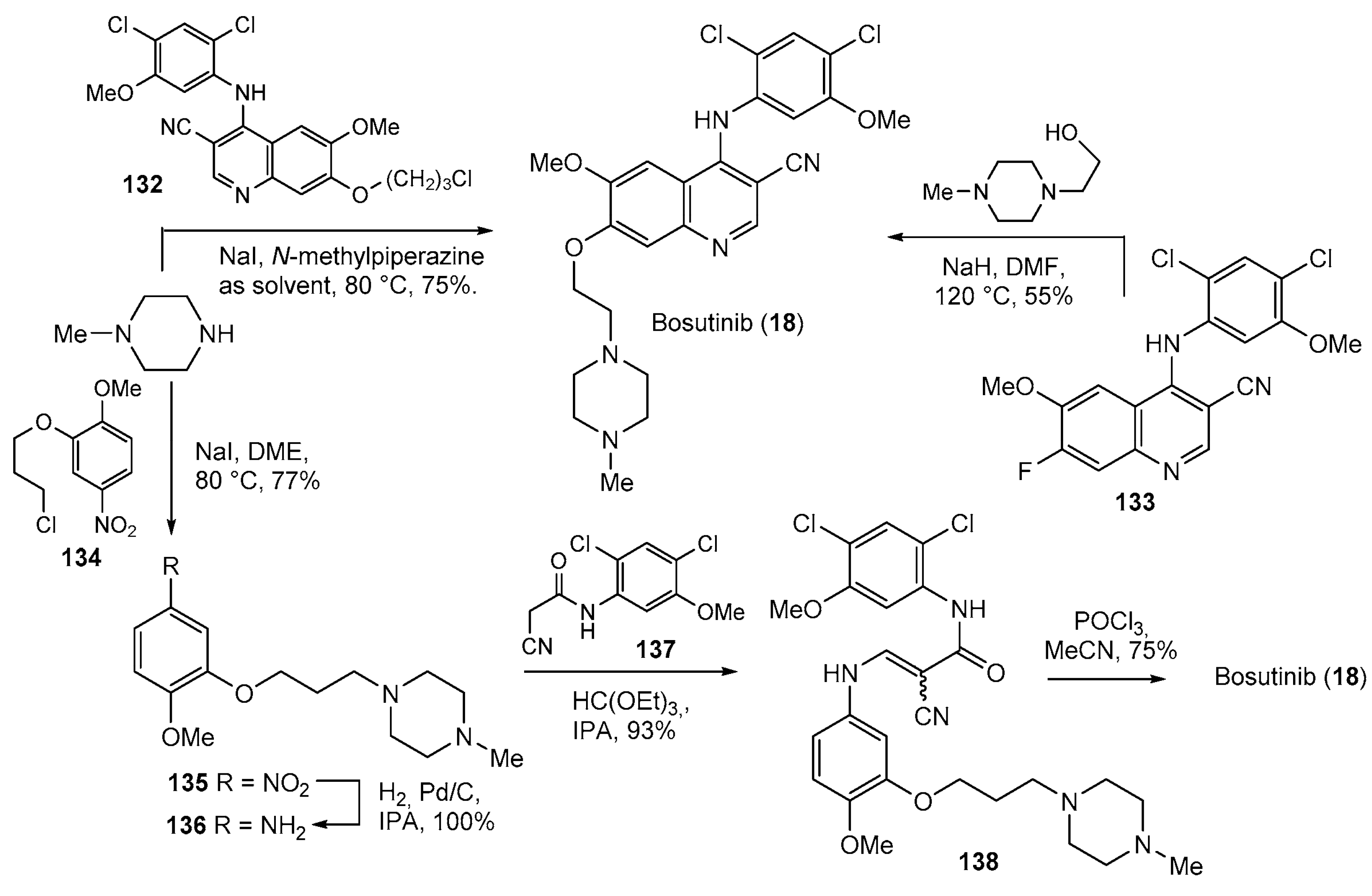
- Roskoski, R. Targeting BCR-Abl in the treatment of Philadelphia-chromosome positive chronic myelogenous leukemia. Pharmacol. Res. 2022, 178, 106156. [Google Scholar] [CrossRef] [PubMed]
- Yumura, M.; Nagano, T.; Nishimura, Y. Novel Multitarget Therapies for Lung Cancer and Respiratory Disease. Molecules 2020, 25, 3987. [Google Scholar] [CrossRef] [PubMed]
- Boschelli, D.H.; Ye, F.; Wang, Y.D.; Dutia, M.; Johnson, S.L.; Wu, B.; Miller, K.; Powell, D.W.; Yaczko, D.; Young, M.; et al. Optimization of 4-Phenylamino-3-quinolinecarbonitriles as Potent Inhibitors of Src Kinase Activity. J. Med. Chem. 2001, 44, 3965–3977. [Google Scholar] [CrossRef] [PubMed]
- Boschelli, D.H.; Wang, Y.D.; Johnson, S.; Wu, B.; Ye, F.; Barrios Sosa, A.C.; Golas, J.M.; Boschelli, F. 7-Alkoxy-4-phenylamino-3-quinolinecar-bonitriles as Dual Inhibitors of Src and Abl Kinases. J. Med. Chem. 2004, 47, 1599–1601. [Google Scholar] [CrossRef]
- Sutherland, K.; Feigelson, G.B.; Boschelli, D.H.; Blum, D.M.; Strong, H.L. Process for Preparation of 4-amino-3-quinolinecarbonitriles. Patent US2005043537A1, 24 February 2005. [Google Scholar]
- Huang, W.-S.; Metcalf, C.A.; Sundaramoorthi, R.; Wang, Y.; Zou, D.; Thomas, R.M.; Zhu, X.; Cai, L.; Wen, D.; Liu, S.; et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a Potent, Orally Active Pan-Inhibitor of Breakpoint Cluster Region-Abelson (BCR-ABL) Kinase Including the T315I Gatekeeper Mutant. J. Med. Chem. 2010, 53, 4701–4719. [Google Scholar] [CrossRef]
- Roth, G.J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J. Med. Chem. 2009, 52, 4466–4480. [Google Scholar] [CrossRef]
- Heckel, A.; Roth, G.J.; Walter, R.; Van Meel, J.; Redemann, N.; Tontsch-Grunt, U.; Spevak, W.; Hilberg, F. 6-Position Substituted Indoline, Production and Use Thereof as a Medicament. Patent WO0127081A1, 19 April 2001. [Google Scholar]
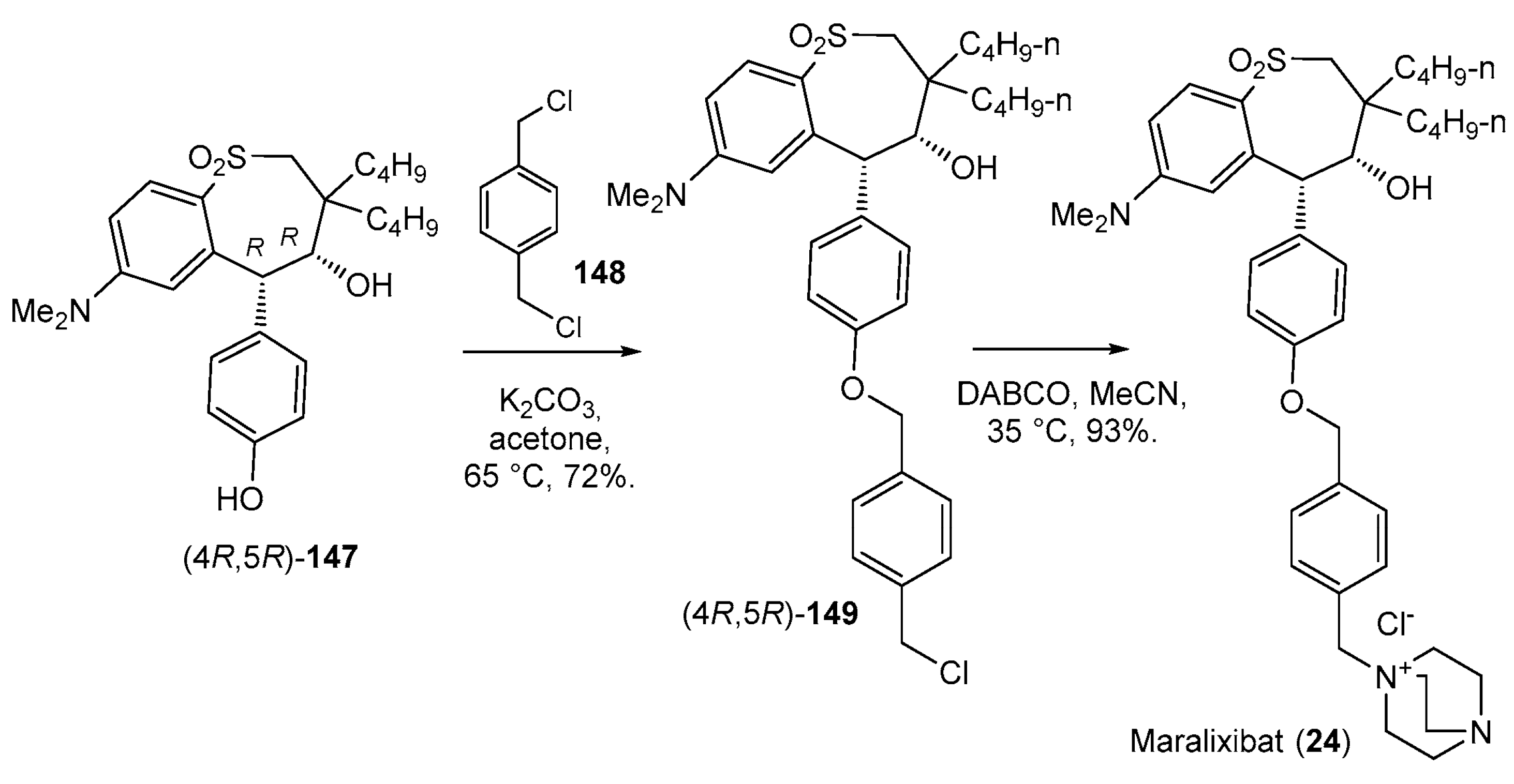
- Shirley, M. Maralixibat: First Approval. Drugs 2021, 82, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.C.; Huang, H.-C.; Li, J.J.; Miller, R.E.; Reitz, D.B.; Tremont, S.J. Substituted 5-aryl-benzothiepines Having Activity as Inhibitors of Ileal Bile Acid Transport and Taurocholate Uptake. U.S. Patent US6107494A, 22 August 2000. [Google Scholar]
- Mudipalli, P.S.; Pozzo, M.J.; Park, J.M. Method for the Preparation of Crystalline Tetrahydrobenzothiepines. Patent WO03022804A2, 20 March 2003. [Google Scholar]
- Coates, D.A.; Gelbert, L.M.; Knobeloch, J.M.; De Dios Magana, A.; De Prado Gonzalez, A.; Filadelfa Del Prado, C.M.; Garcia Paredes, M.C.; Martin De La Nava, E.M.; Martin Ortega Finger, M.D.; Martinez Perez, J.A.; et al. Protein Kinase Inhibitors. U.S. Patent US7855211B2, 21 December 2010. [Google Scholar]
- Chan, E.M. Combination Therapy for Cancer. Patent WO2015130540A1, 3 September 2015. [Google Scholar]
- Frederick, M.O.; Kjell, D.P. A synthesis of abemaciclib utilizing a Leuckart–Wallach reaction. Tetrahedron Lett. 2015, 56, 949–951. [Google Scholar] [CrossRef]
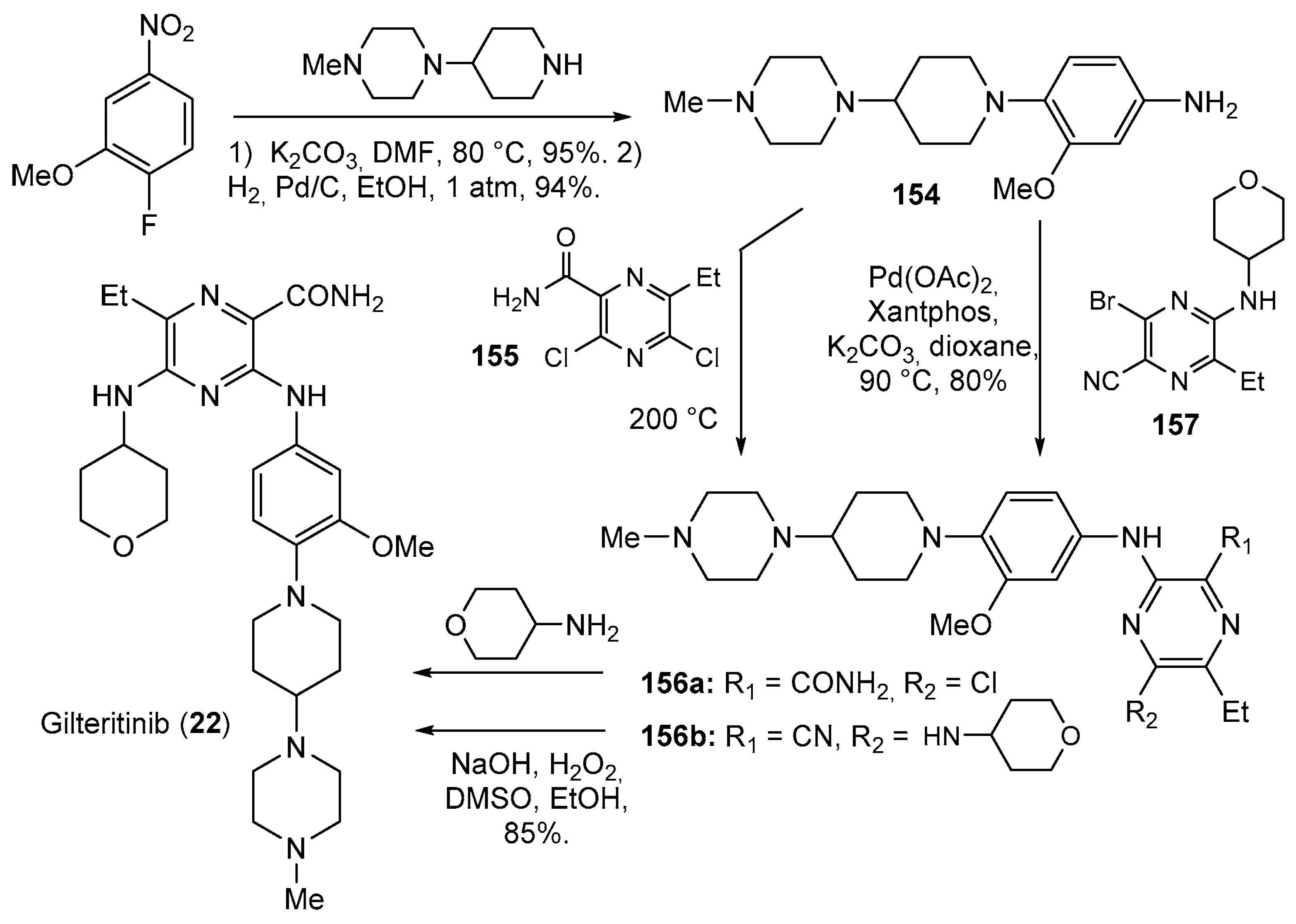
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. New Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef] [PubMed]
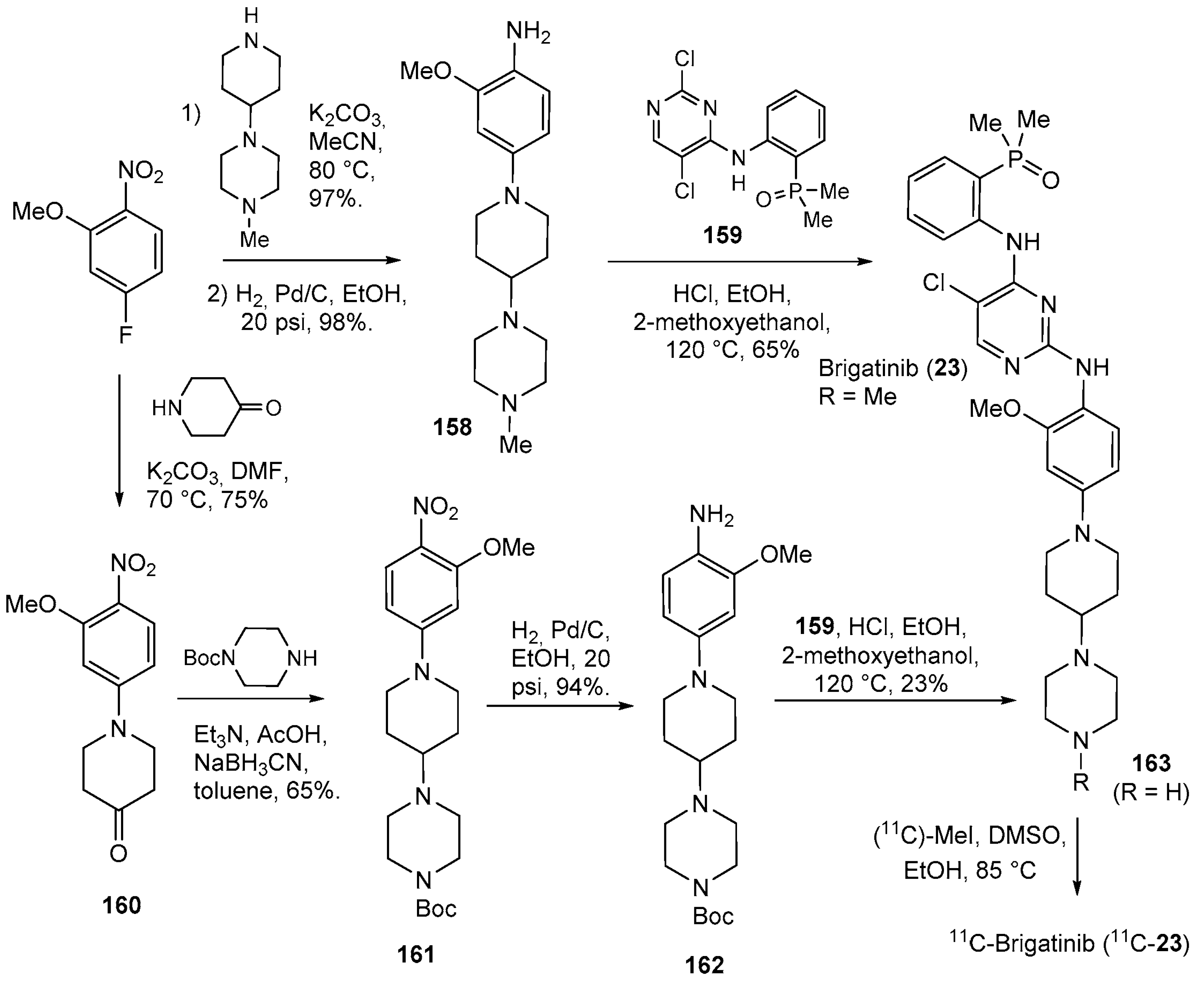
- Huang, W.-S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef]
- Shimada, I.; Kurosawa, K.; Matsuya, T.; Iikubo, K.; Kondoh, Y.; Kamikawa, A.; Tomiyama, H.; Iwai, Y. Diamino Heterocyclic Carboxamide Compound. U.S. Patent US8969336B2, 3 March 2010. [Google Scholar]
- Yue, Q.; Zhou, Z.; Gao, Q.; Baofu, Z. Synthesis Method of 3,5-Disubstituted-pyrazine-2-formamide Compound. Patent CN106083821A, 9 November 2016. [Google Scholar]
- Iikubo, K.; Kondoh, Y.; Shimada, I.; Matsuya, T.; Mori, K.; Ueno, Y.; Okada, M. Discovery of N-{2-Methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl}-N′-[2-(propane-2-sulfonyl)phenyl]-1,3,5-triazine-2,4-diamine (ASP3026), a Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor. Chem. Pharm. Bull. 2018, 66, 251–262. [Google Scholar] [CrossRef]
- Högnäsbacka, A.A.; Poot, A.J.; Kooijman, E.; Schuit, R.C.; Schreurs, M.; Verlaan, M.; Beaino, W.; Van Dongen, G.a.M.S.; Vugts, D.J.; Windhorst, A.D. Synthesis and Preclinical Evaluation of [Methylpiperazine-11C]brigatinib as a PET Tracer Targeting Both Mutated Epidermal Growth Factor Receptor and Anaplastic Lymphoma Kinase. J. Med. Chem. 2023, 66, 12130–12140. [Google Scholar] [CrossRef]
- Kung, C.; Hixon, J.; Choe, S.; Marks, K.; Gross, S.; Murphy, E.; Delabarre, B.; Cianchetta, G.; Sethumadhavan, S.; Wang, X.; et al. Small Molecule Activation of PKM2 in Cancer Cells Induces Serine Auxotrophy. Chem. Biol. 2012, 19, 1187–1198. [Google Scholar] [CrossRef]
- Kung, C.; Hixon, J.; Kosinski, P.A.; Cianchetta, G.; Histen, G.; Chen, Y.; Hill, C.; Gross, S.; Si, Y.; Johnson, K.; et al. AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood 2017, 130, 1347–1356. [Google Scholar] [CrossRef]
- Rab, M.a.E.; Van Oirschot, B.A.; Kosinski, P.A.; Hixon, J.; Johnson, K.; Chubukov, V.; Dang, L.; Pasterkamp, G.; Van Straaten, S.; Van Solinge, W.W.; et al. AG-348 (Mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes. Haematologica 2020, 106, 238–249. [Google Scholar] [CrossRef]
- Salituro, F.G.; Saunders, J.O.; Yan, S. Therapeutic Compounds and Compositions. U.S. Patent US2010331307A1, 30 December 2010. [Google Scholar]
- Sizemore, J.; Guo, L.; Mirmehrabi, M.; Su, Y. Crystalline Forms of n-(4-(4-(cyclopropylmethyl) piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide. Patent WO2019104134A1, 31 May 2019. [Google Scholar]
- Dhillon, S. Zavegepant: First Approval. Drugs 2023, 83, 825–831. [Google Scholar] [CrossRef]
- Cann, R.O.; Chen, C.-P.H.; Gao, Q.; Hanson, R.L.; Hsieh, D.; Li, J.; Lin, D.; Parsons, R.L.; Pendri, Y.; Nielsen, R.B.; et al. Selection of an Enantioselective Process for the Preparation of a CGRP Receptor Inhibitor. Org. Process Res. Dev. 2012, 16, 1953–1966. [Google Scholar] [CrossRef]
- Chaturvedula, P.V.; Mercer, S.E.; Pin, S.S.; Thalody, G.; Xu, C.; Conway, C.M.; Keavy, D.; Signor, L.; Cantor, G.H.; Mathias, N.; et al. Discovery of (R)-N-(3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl)-1-oxopropan-2-yl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)piperidine-1-carboxamide (BMS-742413): A potent human CGRP antagonist with superior safety profile for the treatment of migraine through intranasal delivery. Bioorg. Med. Chem. Lett. 2013, 23, 3157–3161. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Bolaňos, B.; Smith, M.; Palde, P.B.; Cuenca, P.D.; Vanarsdale, T.L.; Niessen, S.; Zhang, L.; Behenna, D.; Ornelas, M.A.; et al. Dissecting the molecular determinants of clinical PARP1 inhibitor selectivity for tankyrase1. J. Biol. Chem. 2021, 296, 100251. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.V. Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.-L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A Novel Bioavailable Inhibitor of Poly(ADP-ribose) Polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Menear, K.A.; Ottridge, A.P.; Londesbrough, D.J.; Hallett, M.R.; Mullholland, K.R.; Pittam, J.D.; Laffan, D.D.P.; Ashworth, I.W.; Jones, M.F.; Cherryman, J.H. Phthalazinone Derivative. Patent WO2008047082A2, 24 April 2008. [Google Scholar]
- Zmuda, F.; Malviya, G.; Blair, A.; Boyd, M.; Chalmers, A.J.; Sutherland, A.; Pimlott, S.L. Synthesis and Evaluation of a Radioiodinated Tracer with Specificity for Poly(ADP-ribose) Polymerase-1 (PARP-1) in Vivo. J. Med. Chem. 2015, 58, 8683–8693. [Google Scholar] [CrossRef]
- Lai, Y.-T. Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition. Viruses 2021, 13, 843. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef]
- Wang, T.; Ueda, Y.; Zhang, Z.; Yin, Z.; Matiskella, J.; Pearce, B.C.; Yang, Z.; Zheng, M.; Parker, D.D.; Yamanaka, G.A.; et al. Discovery of the Human Immunodeficiency Virus Type 1 (HIV-1) Attachment Inhibitor Temsavir and Its Phosphonooxymethyl Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 6308–6327. [Google Scholar] [CrossRef]
- Soundararajan, N.; Qiu, Y.; Hu, W.; Kronenthal, D.R.; Sirard, P.; Lajeunesse, J.; Droghini, R.; Chidambaram, R.; Qian, X.; Natalie, K.J.; et al. Process for Preparing Triazole Substituted Azaindoleoxoacetic Piperazine Derivatives and Novel Salt Forms Produced Therein. U.S. Patent US2006293304A1, 28 December 2006. [Google Scholar]
- Saha, D.; Ryan, K.R.; Lakkaniga, N.R.; Acharya, B.; Garcia, N.G.; Smith, E.L.; Frett, B. Targeting Rearranged during Transfection in Cancer: A Perspective on Small-Molecule Inhibitors and Their Clinical Development. J. Med. Chem. 2021, 64, 11747–11773. [Google Scholar] [CrossRef]
- Subbiah, V.; Shen, T.; Terzyan, S.S.; Liu, X.; Hu, X.; Patel, K.P.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol. 2021, 32, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.W.; Aronow, S.; Blake, J.F.; Brandhuber, B.J.; Cook, A.; Haas, J.; Jiang, Y.; Kolakowski, G.R.; Mcfaddin, E.A.; Mckenney, M.L.; et al. Substituted Pyrazolo[1,5-a]pyridine Compounds as RET Kinase Inhibitors. Patent WO2018071447A1, 19 April 2018. [Google Scholar]
- Eary, C.T.; Spencer, S.; Crane, Z.; Chando, K.; Asselin, S.; Liu, W.; Welch, M.; Cook, A.; Kolakowski, G.R.; Metcalf, A.T.; et al. Process for the Preparation of 6-(2-Hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile. Patent. US2019106438A1, 11 April 2019. [Google Scholar]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed]
- Campagne, S.; Boigner, S.; Rüdisser, S.; Moursy, A.; Gillioz, L.; Knörlein, A.; Hall, J.; Ratni, H.; Cléry, A.; Allain, F.H.T. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat. Chem. Biol. 2019, 15, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.-M.; Fantasia, S.M.; Fishlock, D.V.; Hoffmann-Emery, F.; Moine, G.; Pfleger, C.; Moessner, C. Process for the Prepration of 7-(4,7-Diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one Derivatives. Patent WO2019057740A1, 28 March 2019. [Google Scholar]
- Adam, J.-M.; Pfleger, C.; Wuitschik, G. Novel Process. Patent WO2022194909A2, 22 September 2022. [Google Scholar]
- Crump, B.R.; Goss, C.; Lovelace, T.; Lewis, R.; Peterson, J. Influence of Reaction Parameters on the First Principles Reaction Rate Modeling of a Platinum and Vanadium Catalyzed Nitro Reduction. Org. Process Res. Dev. 2013, 17, 1277–1286. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, Y.; Yi, F.; Zhao, X.; Lu, K. Recent advances in the development of inhibitors targeting KRAS-G12C and its related pathways. Eur. J. Med. Chem. 2023, 259, 115698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Griffin, D.J.; Beaver, M.G.; Blue, L.E.; Borths, C.J.; Brown, D.B.; Caille, S.; Chen, Y.; Cherney, A.H.; Cochran, B.M.; et al. Development of a Commercial Manufacturing Process for Sotorasib, a First-in-Class KRASG12C Inhibitor. Org. Process Res. Dev. 2022, 26, 3115–3125. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef]
- Parsons, A.T.; Caille, S.; Caporini, M.A.; Griffin, D.J.; Lovette, M.A.; Powazinik, W.I.V.; St-Pierre, G. Axial Chirality in the Sotorasib Drug Substance, Part 1: Development of a Classical Resolution to Prepare an Atropisomerically Pure Sotorasib Intermediate. Org. Process Res. Dev. 2022, 26, 2629–2635. [Google Scholar] [CrossRef]
- Blake, J.F.; Burgess, L.E.; Chicarelli, M.J.; Christensen, J.G.; Cook, A.; Fell, J.B.; Fischer, J.P.; Marx, M.A.; Mejia, M.J.; Savechenkov, P.; et al. KRAS G12C Inhibitors. U.S. Patent US2019144444A1, 16 May 2019. [Google Scholar]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef]
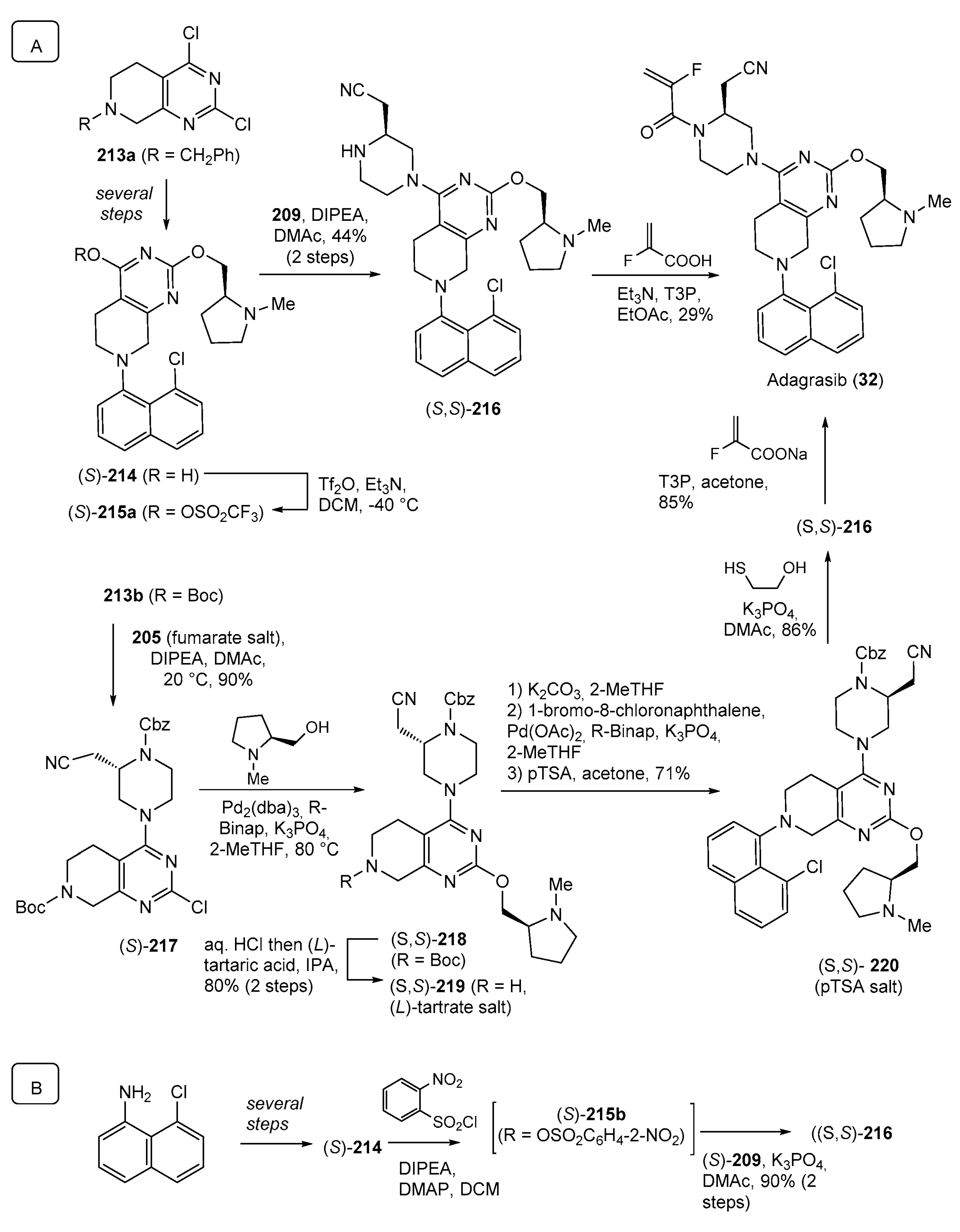
- Snead, D.R.; Gan, Y.; Scattolin, T.; Paymode, D.J.; Achmatowicz, M.; Rudisill, D.E.; Vidal, E.S.; Gharbaoui, T.; Roberts, P.; Yang, J.; et al. Development of Adagrasib’s Commercial Manufacturing Route. Org. Process Res. Dev. 2023, 27, 530–538. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lu, Z.; Scattolin, T.; Chen, C.; Gan, Y.; Mclaughlin, M. Synthesis of Adagrasib (MRTX849), a Covalent KRASG12C Inhibitor Drug for the Treatment of Cancer. Org. Lett. 2023, 25, 944–949. [Google Scholar] [CrossRef] [PubMed]
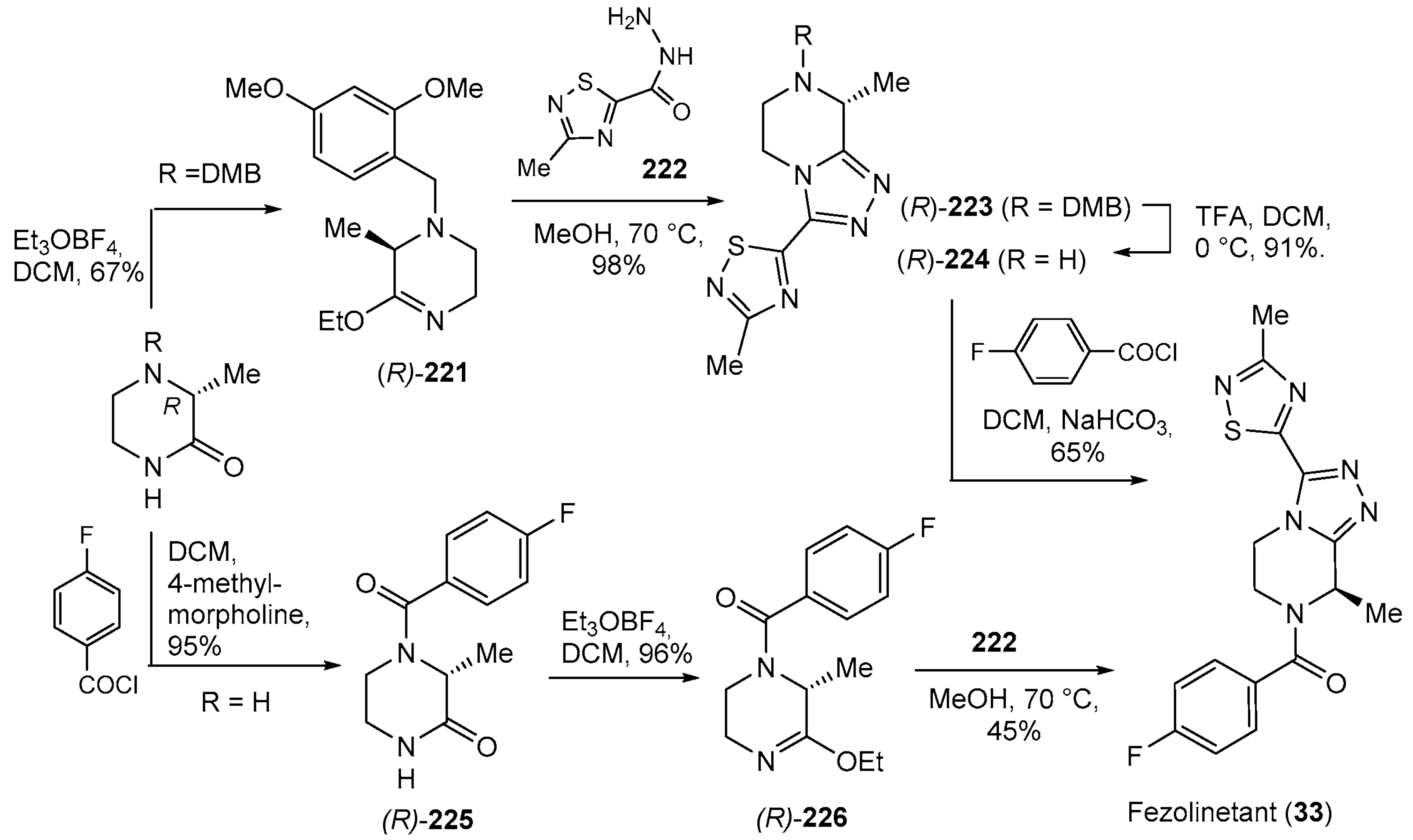
- Lee, A. Fezolinetant: First Approval. Drugs 2023, 83, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Hoveyda, H.R.; Fraser, G.L.; Roy, M.-O.; Dutheuil, G.; Batt, F.; El Bousmaqui, M.; Korac, J.; Lenoir, F.; Lapin, A.; Noël, S.; et al. Discovery and Optimization of Novel Antagonists to the Human Neurokinin-3 Receptor for the Treatment of Sex-Hormone Disorders (Part I). J. Med. Chem. 2015, 58, 3060–3082. [Google Scholar] [CrossRef] [PubMed]
- Hoveyda, H.R.; Fraser, G.L.; Dutheuil, G.; El Bousmaqui, M.; Korac, J.; Lenoir, F.; Lapin, A.; Noël, S. Optimization of Novel Antagonists to the Neurokinin-3 Receptor for the Treatment of Sex-Hormone Disorders (Part II). ACS Med. Chem. Lett. 2015, 6, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Hoveyda, H.R.; Dutheuil, G. Novel Chiral Synthesis of N-Acyl-(3-substituted)-(8-substituted)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazines. Patent WO2016046398A1, 31 March 2016. [Google Scholar]
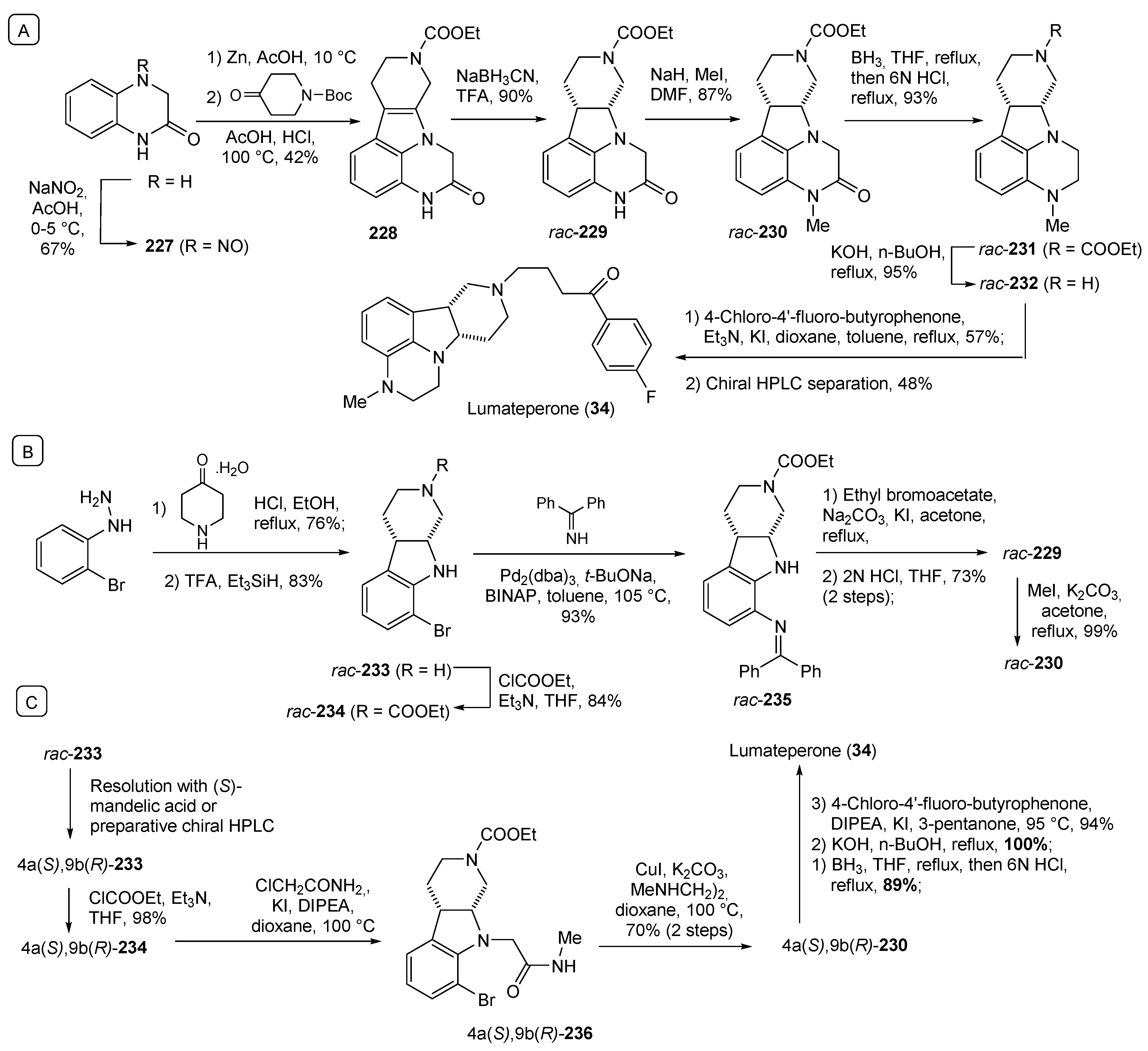
- Blair, H.A. Lumateperone: First Approval. Drugs 2020, 80, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, Q.; Robichaud, A.J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J.D.; Snyder, G.L.; Zhu, H.; Peng, Y.; et al. Discovery of a Tetracyclic Quinoxaline Derivative as a Potent and Orally Active Multifunctional Drug Candidate for the Treatment of Neuropsychiatric and Neurological Disorders. J. Med. Chem. 2014, 57, 2670–2682. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, Q. Substituted Heterocycle Fused Gamma-Carbolines Synthesis. Patent WO2019241278A1, 19 December 2019. [Google Scholar]
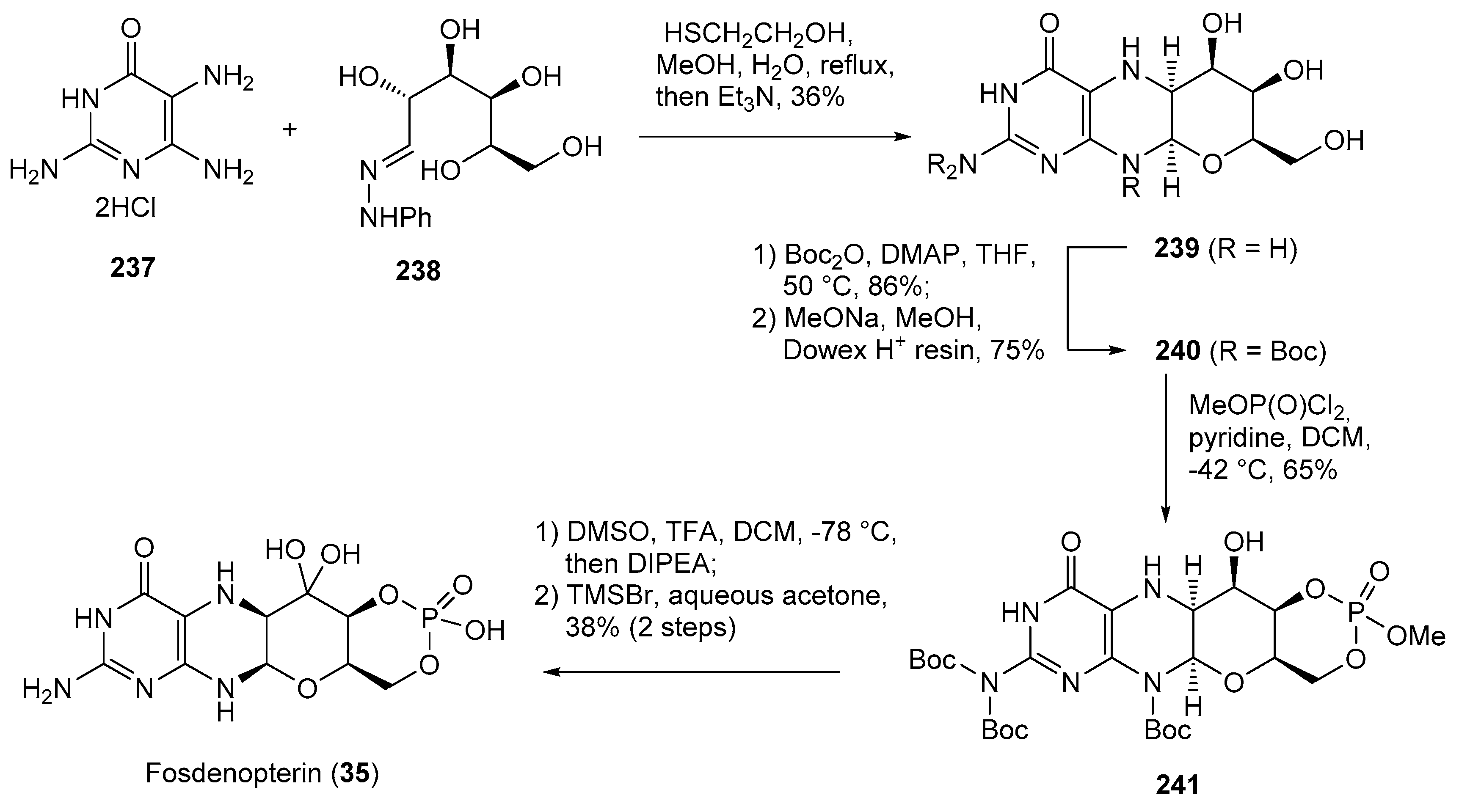
- Kang, C. Fosdenopterin: First Approval. Drugs 2021, 81, 953–956. [Google Scholar] [CrossRef]
- Clinch, K.; Watt, D.K.; Dixon, R.A.; Baars, S.M.; Gainsford, G.J.; Tiwari, A.; Schwarz, G.; Saotome, Y.; Storek, M.; Belaidi, A.A.; et al. Synthesis of Cyclic Pyranopterin Monophosphate, a Biosynthetic Intermediate in the Molybdenum Cofactor Pathway. J. Med. Chem. 2013, 56, 1730–1738. [Google Scholar] [CrossRef]
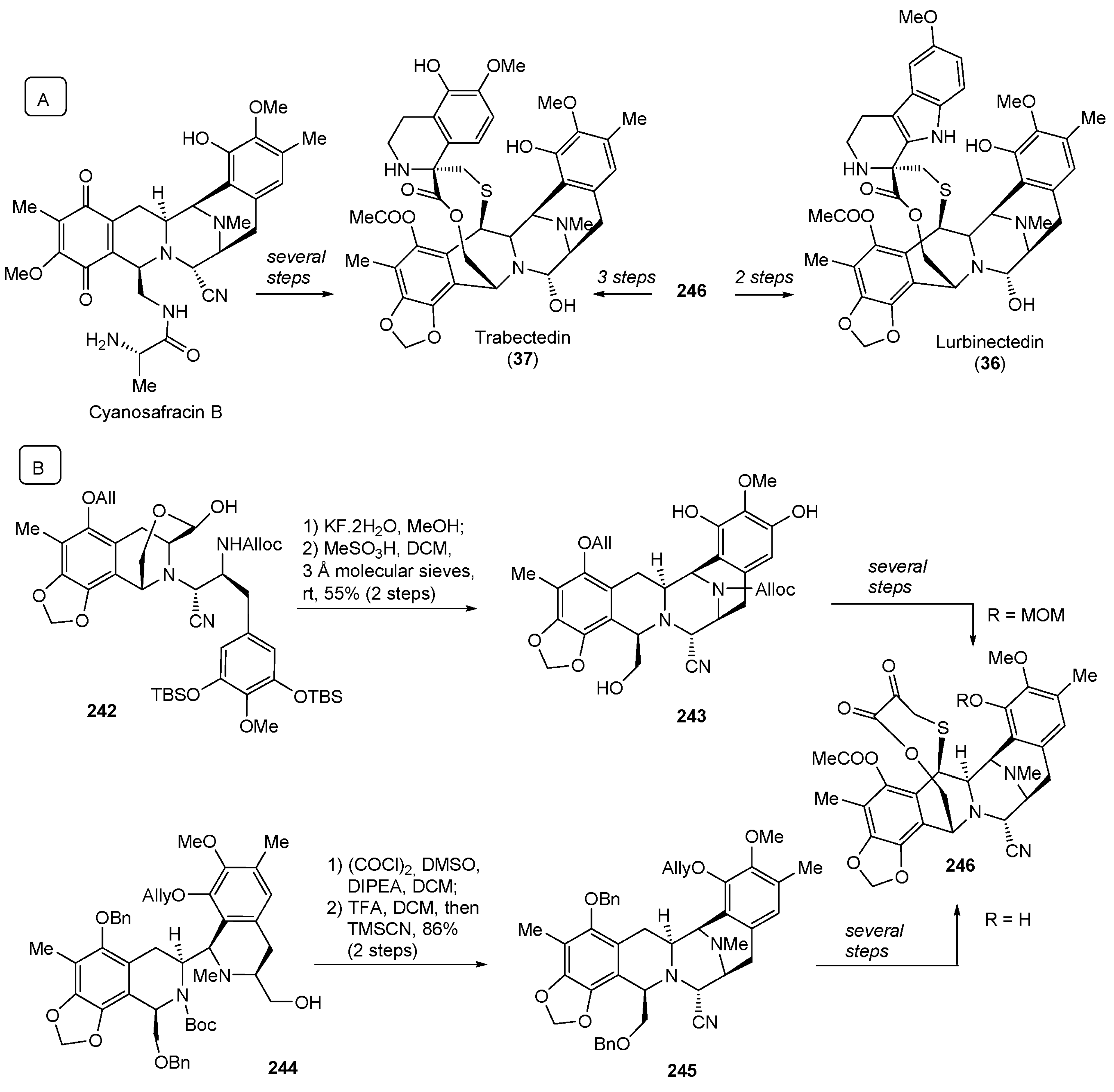
- De Sanctis, R.; Jacobs, F.; Benvenuti, C.; Gaudio, M.; Franceschini, R.; Tancredi, R.; Pedrazzoli, P.; Santoro, A.; Zambelli, A. From seaside to bedside: Current evidence and future perspectives in the treatment of breast cancer using marine compounds. Front. Pharmacol. 2022, 13, 909566. [Google Scholar] [CrossRef]
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA Sequence- and Structure-Selective Alkylation of Guanine N2 in the DNA Minor Groove by Ecteinascidin 743, a Potent Antitumor Compound from the Caribbean Tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef]
- He, W.; Zhang, Z.; Ma, D. A Scalable Total Synthesis of the Antitumor Agents Et-743 and Lurbinectedin. Angew. Chem. Int. Ed. 2019, 58, 3972–3975. [Google Scholar] [CrossRef] [PubMed]
- Martin Lopez, M.J.; Francesch Solloso, A.; Cuevas Marchante, M.D.C. Synthetic Process for the Manufacture of Ecteinascidin Compounds. Patent WO2011147828A1, 1 December 2011. [Google Scholar]
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective Total Synthesis of Ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors 3. A Diastereomeric Approach to Chiral Nonracemic Tricyclic Ring Systems and the Discovery of Dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.V.; Crutchley, R.D.; Guduru, R.C.; Ton, K.; Lam, T.; Min, A.C. A clinical review of HIV integrase strand transfer inhibitors (INSTIs) for the prevention and treatment of HIV-1 infection. Retrovirology 2022, 19, 22. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Taishi, T.; Taoda, Y. Polycyclic Carbamoylpyridone Derivative Having Hiv Integrase Inhibitory Activity. Patent WO2006116764A1, 2 November 2006. [Google Scholar]
- Jin, H.; Lazerwith, S.E.; Martin, T.A.T.; Bacon, E.M.; Cottell, J.J.; Cai, Z.R.; Pyun, H.-J.; Morganelli, P.A.; Ji, M.; Taylor, J.G.; et al. Polycyclic-Carbamoylpyridone Compounds and Their Pharmaceutical Use. Patent WO2014100323A1, 26 June 2014. [Google Scholar]
- Huang, H.-C.; Tremont, S.J.; Lee, L.F.; Keller, B.T.; Carpenter, A.J.; Wang, C.-C.; Banerjee, S.C.; Both, S.R.; Fletcher, T.; Garland, D.J.; et al. Discovery of Potent, Nonsystemic Apical Sodium-Codependent Bile Acid Transporter Inhibitors (Part 2). J. Med. Chem. 2005, 48, 5853–5868. [Google Scholar] [CrossRef]
- Wendt, M.D.; Shen, W.; Kunzer, A.; Mcclellan, W.J.; Bruncko, M.; Oost, T.K.; Ding, H.; Joseph, M.K.; Zhang, H.; Nimmer, P.M.; et al. Discovery and Structure−Activity Relationship of Antagonists of B-Cell Lymphoma 2 Family Proteins with Chemopotentiation Activity in Vitro and in Vivo. J. Med. Chem. 2006, 49, 1165–1181. [Google Scholar] [CrossRef]
- Soskic, V.; Sukalovic, V.; Kostic-Rajacic, S. Exploration of N-arylpiperazine Binding Sites of D2 Dopaminergic Receptor. Mini-Rev. Med. Chem. 2015, 15, 988–1001. [Google Scholar] [CrossRef]
- Möller, D.; Salama, I.; Kling, R.C.; Hübner, H.; Gmeiner, P. 1,4-Disubstituted aromatic piperazines with high 5-HT2A/D2 selectivity: Quantitative structure-selectivity investigations, docking, synthesis and biological evaluation. Bioorg. Med. Chem. 2015, 23, 6195–6209. [Google Scholar] [CrossRef]
- Partyka, A.; Kurczab, R.; Canale, V.; Satała, G.; Marciniec, K.; Pasierb, A.; Jastrzębska-Więsek, M.; Pawłowski, M.; Wesołowska, A.; Bojarski, A.J.; et al. The impact of the halogen bonding on D2 and 5-HT1A/5-HT7 receptor activity of azinesulfonamides of 4-[(2-ethyl)piperidinyl-1-yl]phenylpiperazines with antipsychotic and antidepressant properties. Bioorg. Med. Chem. 2017, 25, 3638–3648. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | Name, Year of Approval, Mechanism of Action and Therapeutic Indication | Structure |
|---|---|---|
| 1 | Palbociclib (2015): Cyclin-Dependent Kinase 4/6 inhibitor (treatment of metastatic breast cancer) | 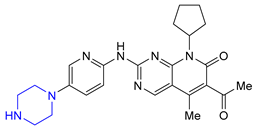 |
| 2 | Ribociclib (2017): Cyclin-Dependent Kinase 4/6 inhibitor (treatment of metastatic breast cancer) | 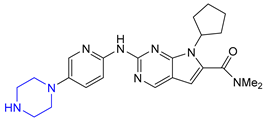 |
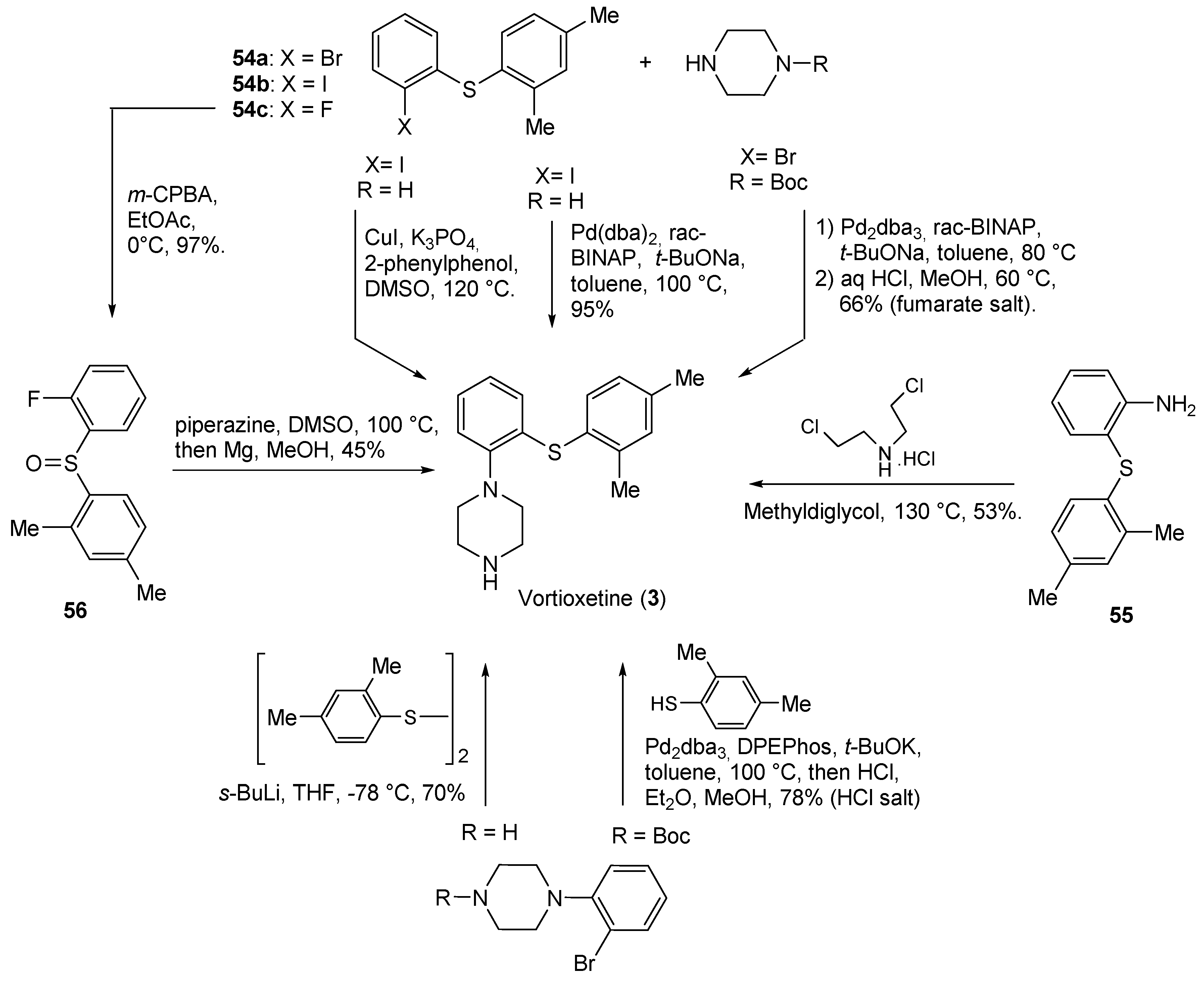
| 3 | Vortioxetine (2013): serotoninergic modulator (treatment of major depressive disorder) | 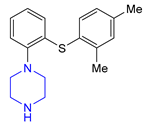 |
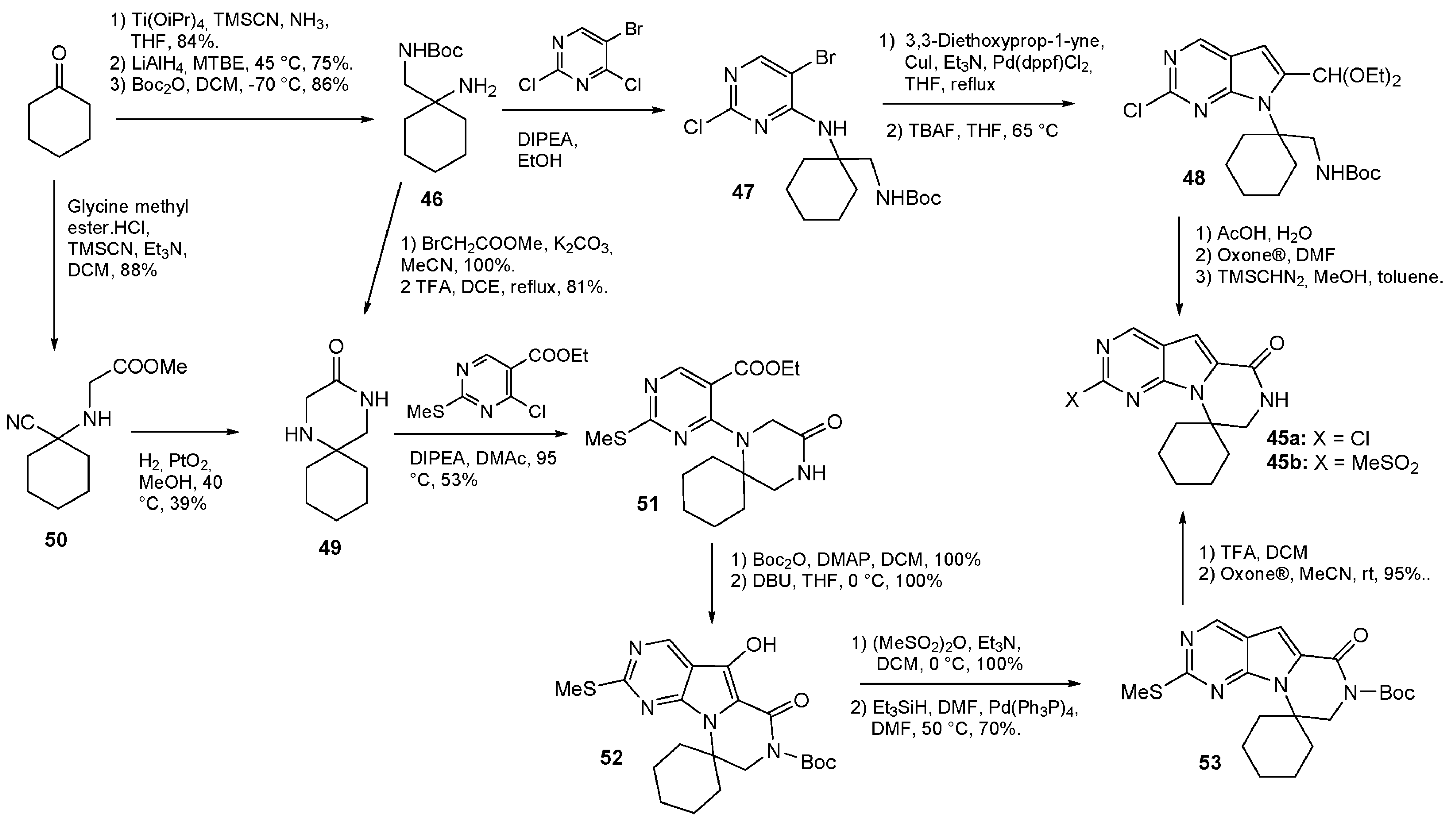
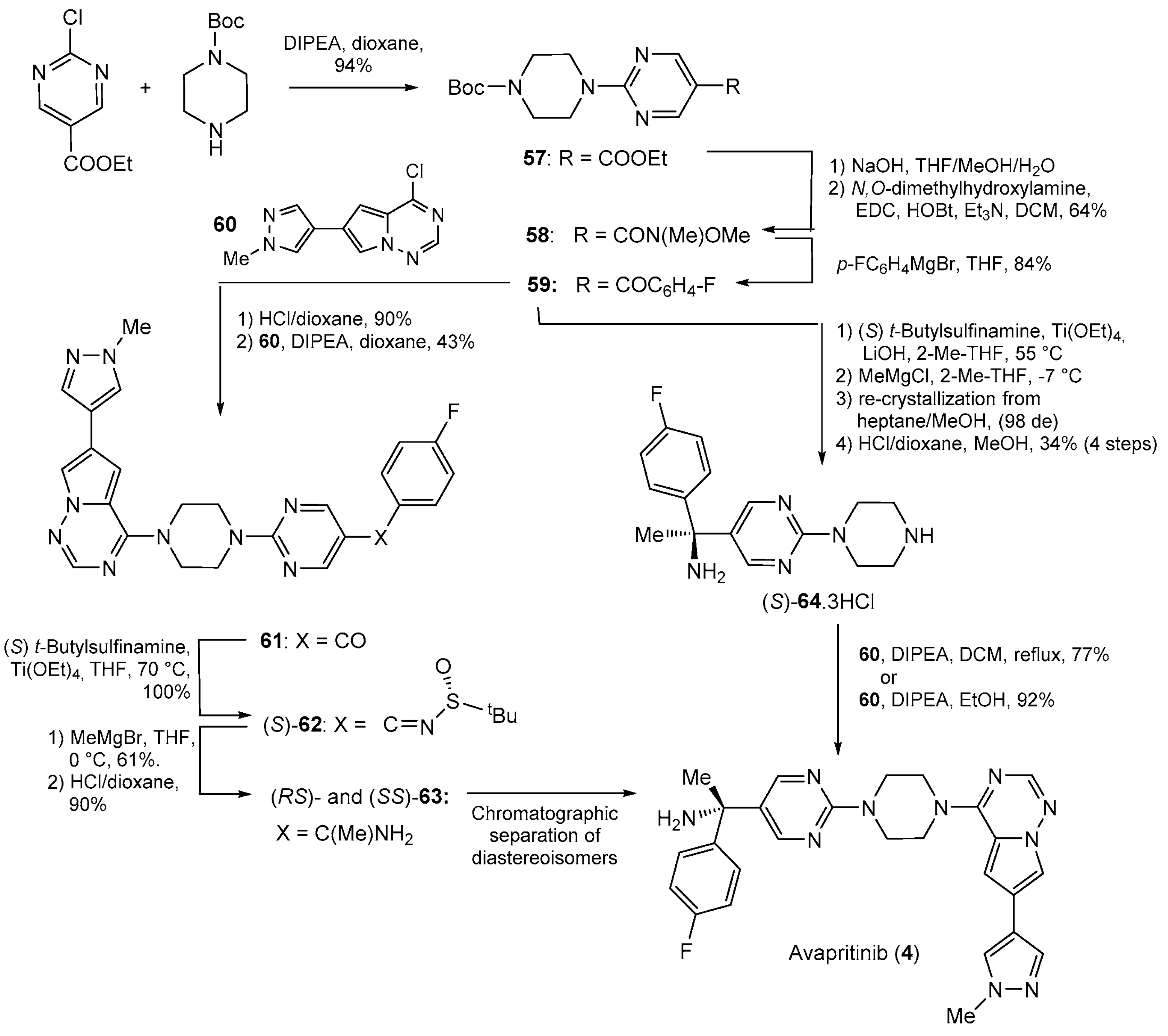
| 4 | Avapritinib (2020): platelet-derived growth factor receptor alpha inhibitor (treatment of Gastrointestinal Stromal Tumor) | 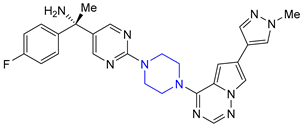 |
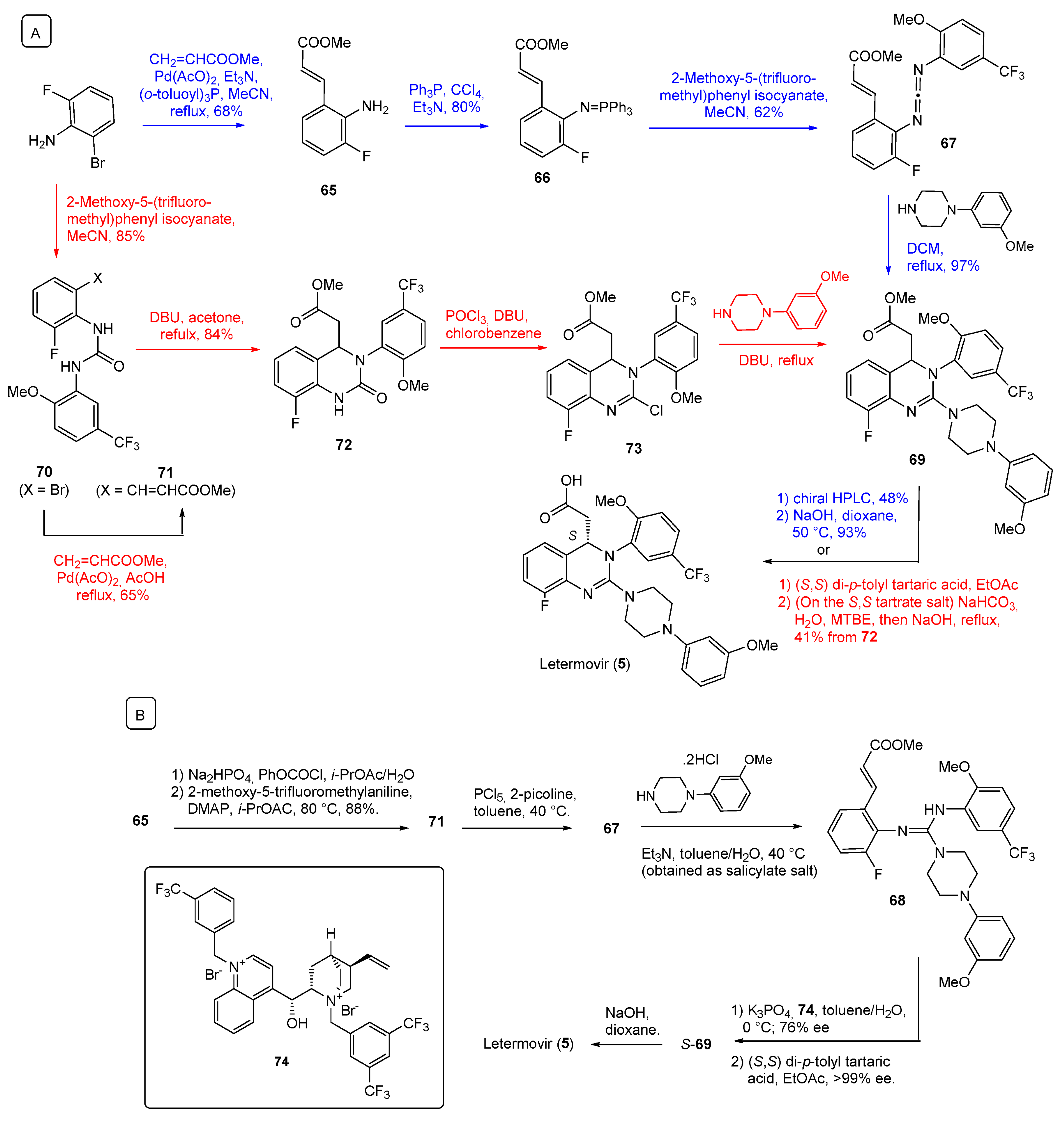
| 5 | Letermovir (2017): cytomegalovirus DNA terminase inhibitor (to prevent infection in bone marrow transplant) | 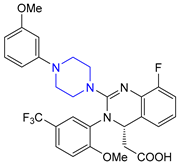 |
| 6 | Trilaciclib (2021): Cyclin-Dependent Kinase 4/6 inhibitor (mitigation of chemotherapy-induced myelosuppression in small cell lung cancer) | 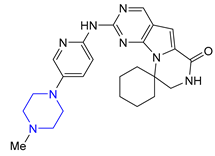 |
| 7 | Infigratinib (2021): fibroblast growth factor receptor inhibitor (treatment of cholangiocarcinoma) | 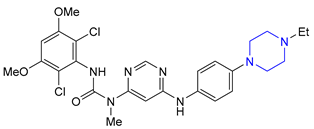 |
| 8 | Entrectinib (2019): ALK, ROS1 and Trk kinase inhibitor (treatment of metastatic non-small cell lung cancer) | 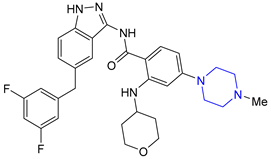 |
| 9 | Avatrombopag (2018): thrombopoietin receptor agonist (treatment of thrombocytopenia) | 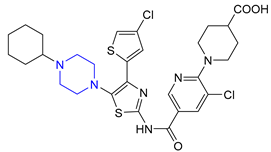 |
| 10 11 | Netupitant (2014) and Fosnetupitant (2018): NK1 receptor antagonists (treatment of nausea and vomiting in patients undergoing cancer chemotherapy, in combination with palosetron) | 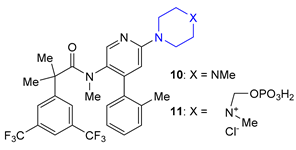 |
| 12 | Venetoclax (2016): Bcl-2 blocker (treatment of chronic lymphocytic leukemia in patients with a specific chromosomal abnormality) |  |
| 13 | Brexpiprazole (2015): (treatment of schizophrenia and major depressive disorder) | 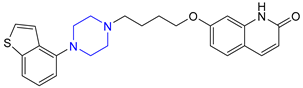 |
| 14 | Vilazodone (2011): serotoninergic modulator (treatment of major depressive disorder) | 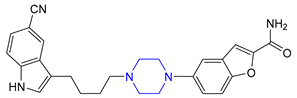 |
| 15 | Flibanserin (2015): 5-HT1A agonist (treatment of acquired, generalized hypoactive sexual desire disorder in premenopausal women) | 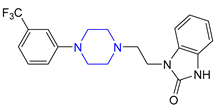 |
| 16 | Aripiprazole Lauroxil (2015): long-acting antipsychotic (treatment of schizophrenia) | 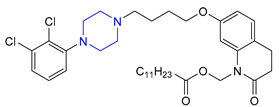 |
| 17 | Cariprazine (2015): D2/D3 receptors partial agonist (treatment of schizophrenia and bipolar disorder in adults) | 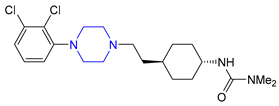 |
| 18 | Bosutinib (2012): Bcr-Abl tyrosine-kinase inhibitor (treatment of chronic myelogenous leukemia) | 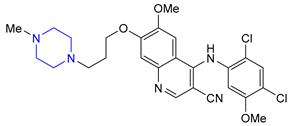 |
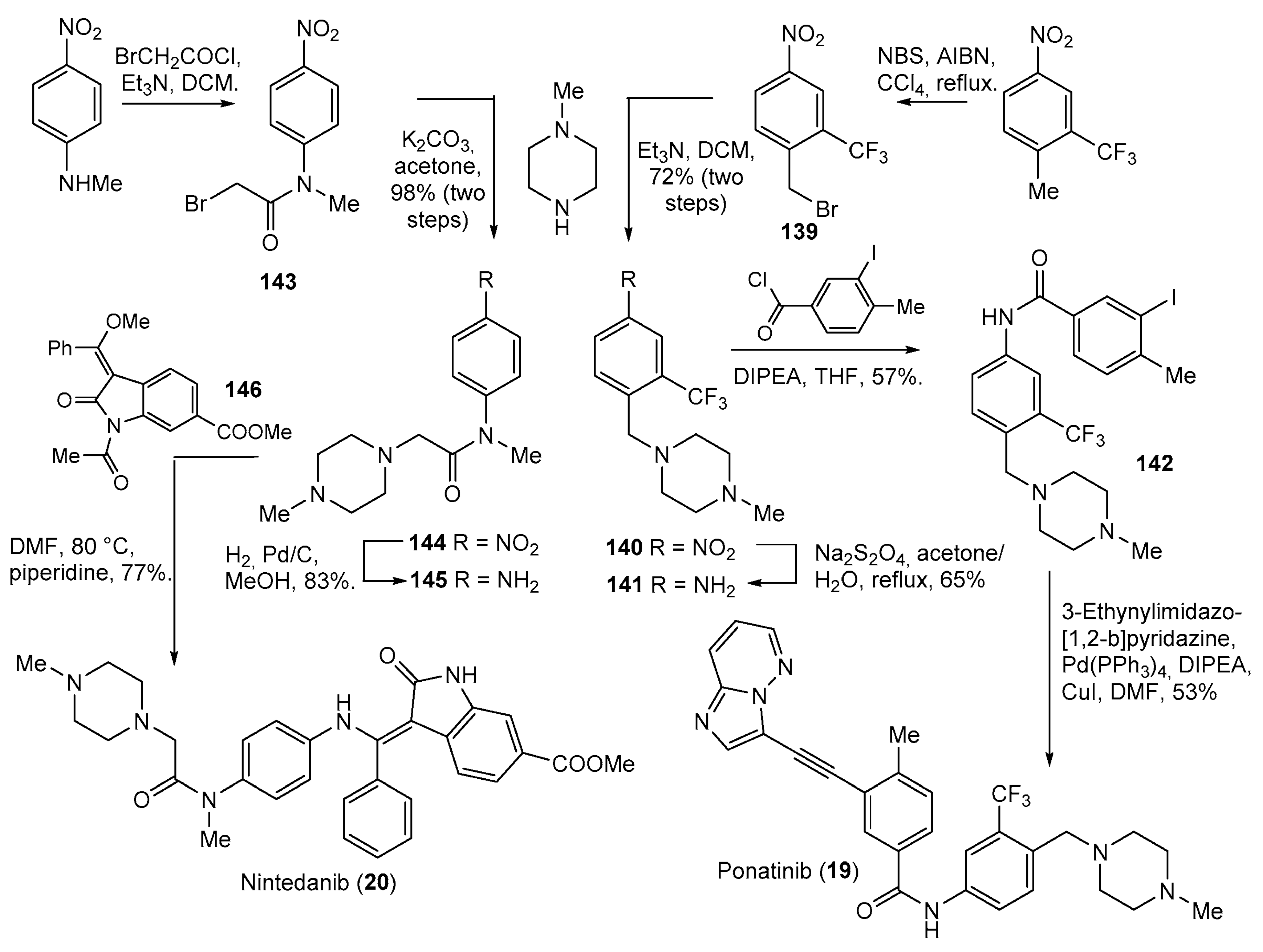
| 19 | Ponatinib (2012): Bcr-Abl tyrosine-kinase inhibitor (treatment of chronic myeloid leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia) | 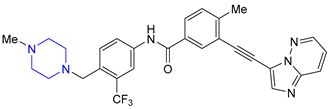 |
| 20 | Nintedanib (2014): receptor and non-receptor tyrosine kinase inhibitor (treatment of idiopathic pulmonary fibrosis) |  |
| 21 | Abemaciclib (2017): Cyclin-Dependent Kinase 4/6 inhibitor (treatment of metastatic breast cancer) | 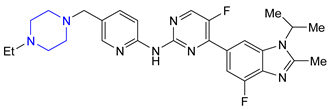 |
| 22 | Gilteritinib (2018): FMS-like tyrosine kinase 3 inhibitor (treatment of Acute Myeloid Leukemia) |  |
| 23 | Brigatinib (2017): anaplastic lymphoma kinase/epidermal growth factor receptor inhibitor (treatment of non-small cell lung cancer) | 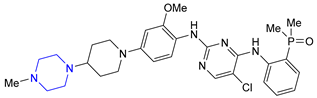 |
| 24 | Maralixibat (2021): ileal bile acid transporter inhibitor (treatment of cholestatic pruritus associated with Alagille syndrome) | 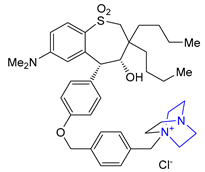 |
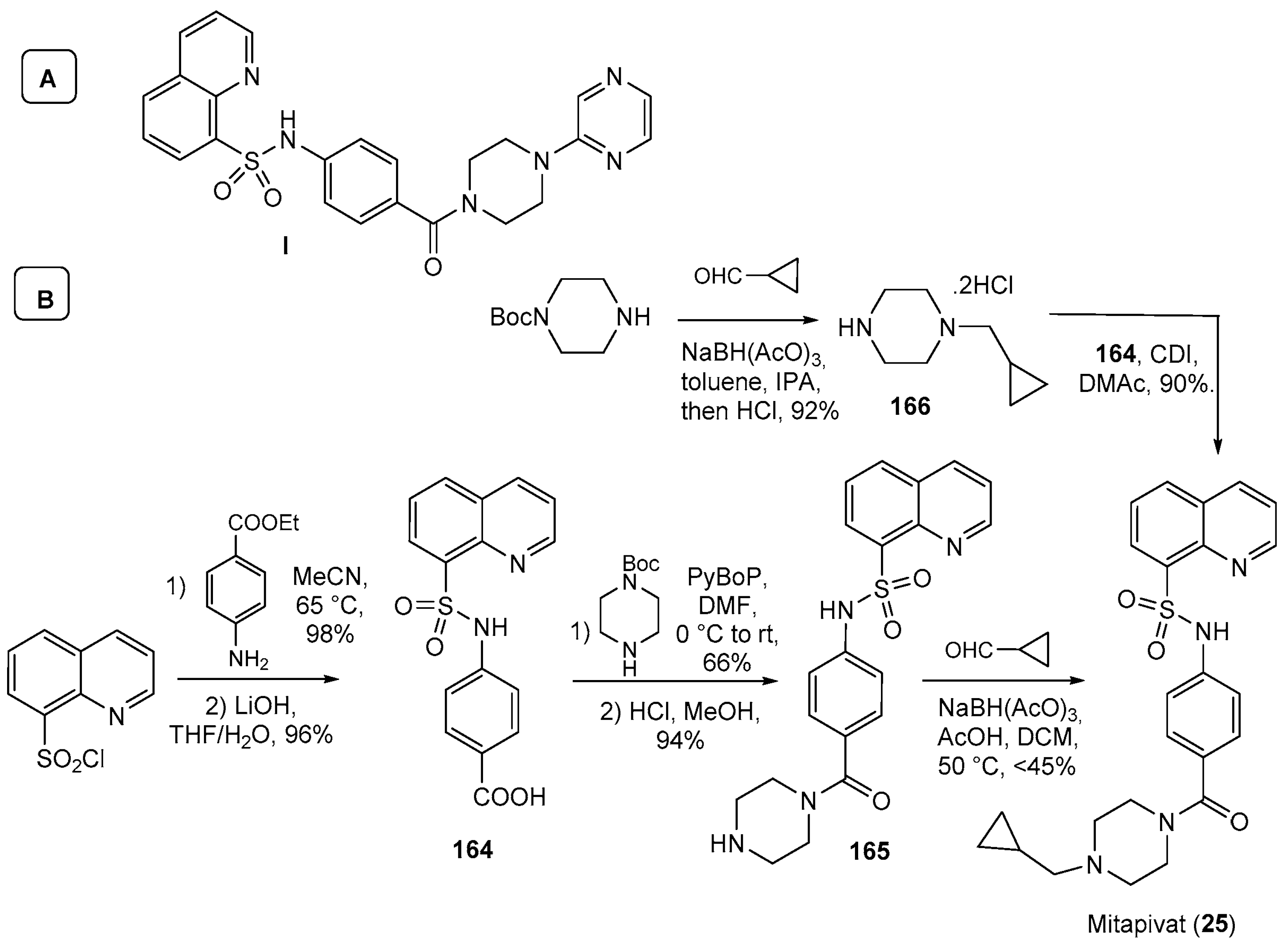
| 25 | Mitapivat (2022): pyruvate kinase activator (treatment of hemolytic anemia in pyruvate kinase deficiency) | 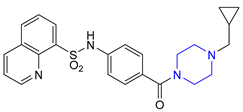 |
| 26 | Zavegepant (2023): calcitonin gene-related peptide receptor antagonist (treatment of migraines) |  |
| 27 | Olaparib (2014): poly ADP ribose polymerase inhibitor (treatment of advanced ovarian cancer) | 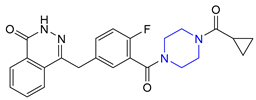 |
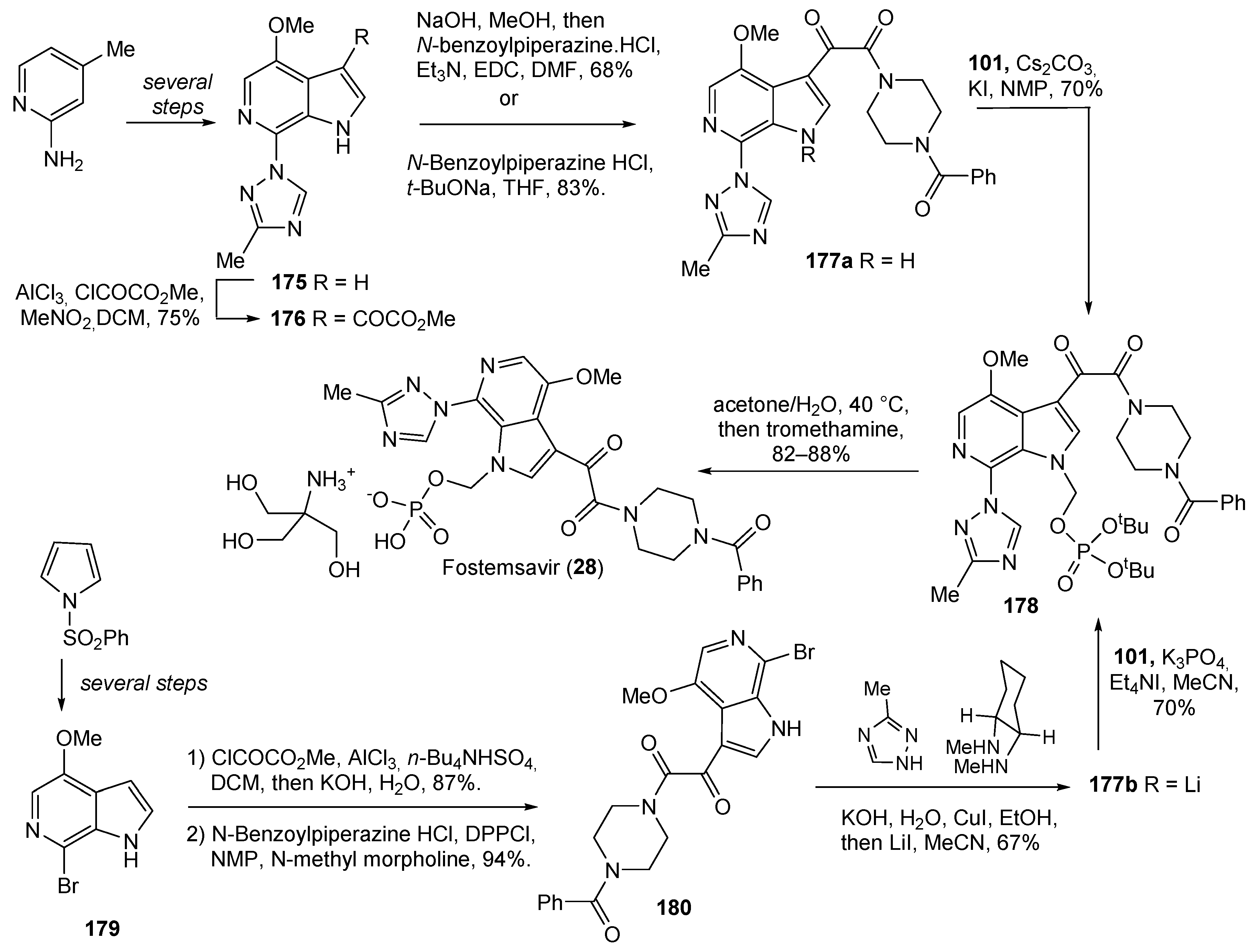
| 28 | Fostemsavir (2020): HIV attachment inhibitor (treatment of HIV infection) | 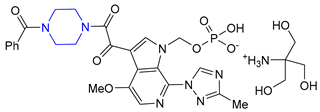 |
| Compound Number | Name, Year of Approval, Mechanism of Action and Therapeutic Indication | Structure |
|---|---|---|
| 29 | Selpercatinib (2020): Rearranged during Transfection (RET) inhibitor (treatment of lung and thyroid cancers) | 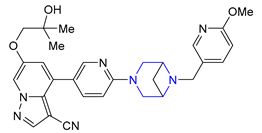 |
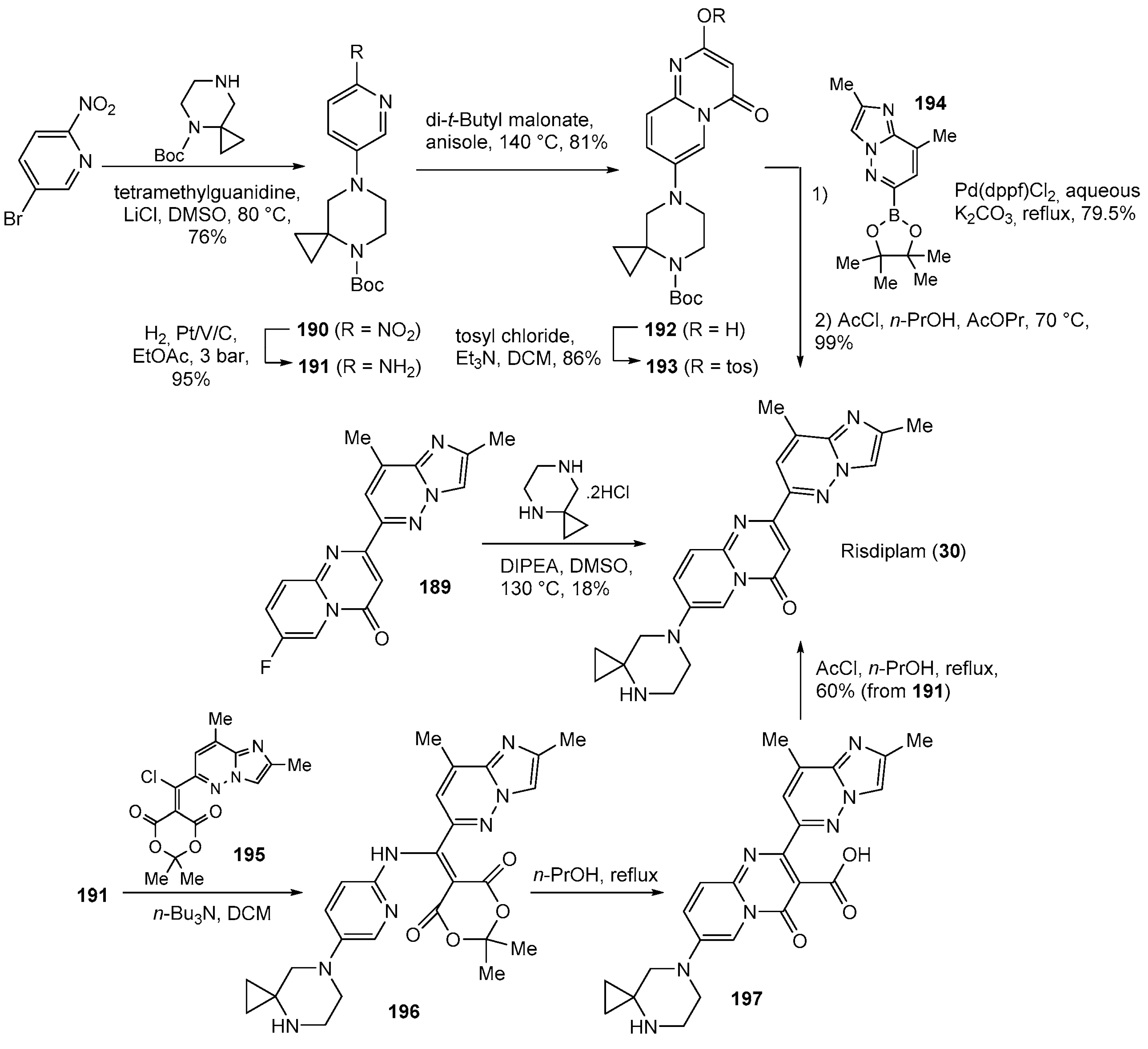
| 30 | Risdiplam (2020): Survival Motor Neuron-2 RNA splicing modifier (treatment of spinal muscular atrophy) | 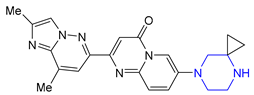 |
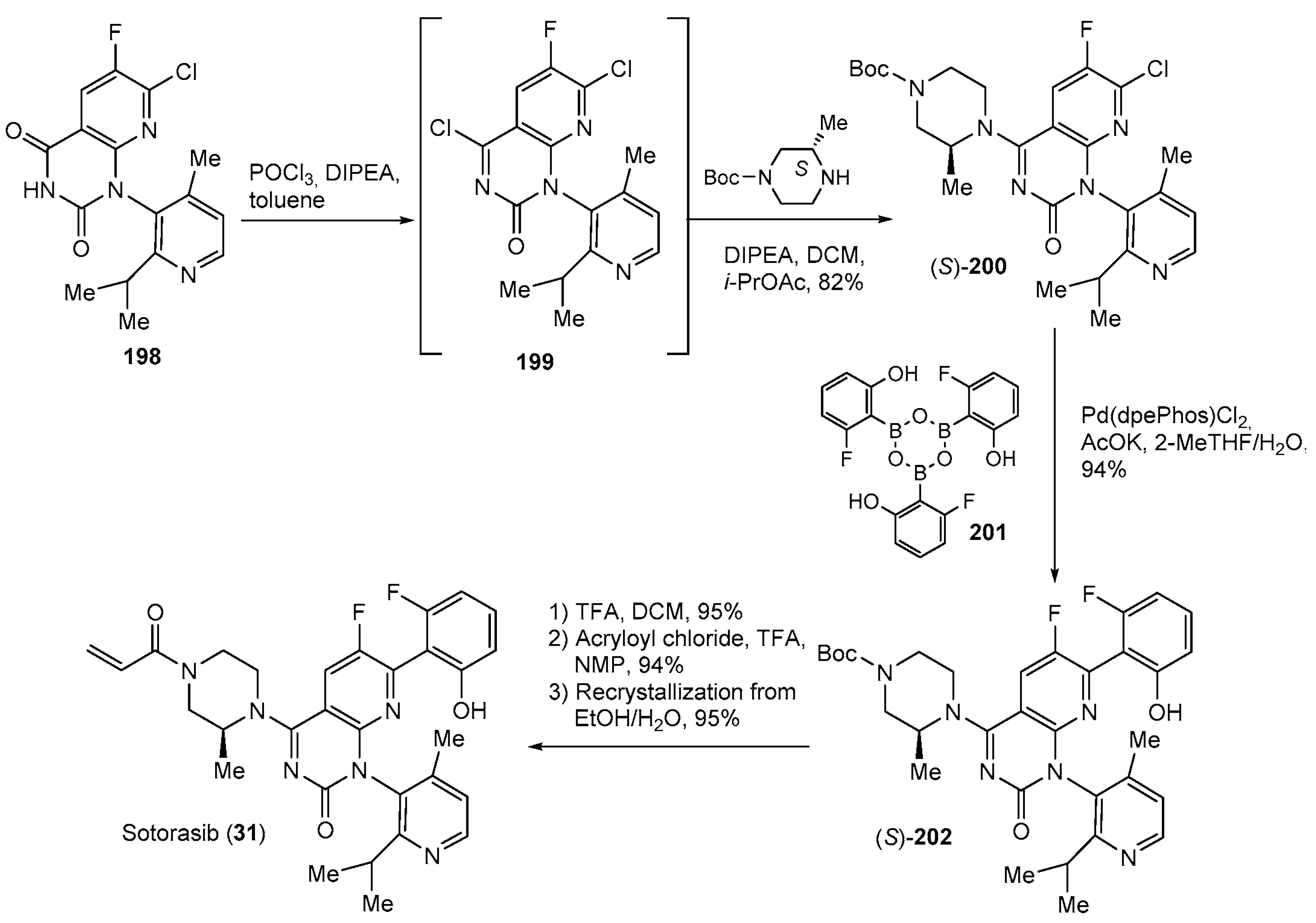
| 31 | Sotorasib (2021): KRASG12C inhibitor (treatment of non-small cell lung cancer) |  |
| 32 | Adagrasib (2022): KRASG12C inhibitor (treatment of locally advanced or metastatic non-small cell lung cancer) | 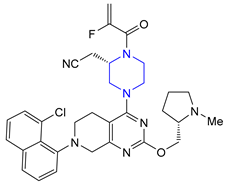 |
| 33 | Fezolinetant (2023): NK3 receptor antagonist (treatment of moderate to severe hot flashes caused by menopause) | 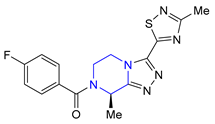 |
| 34 | Lumateperone (2019): 5-HT2A/D2 antagonist (schizophrenia) | 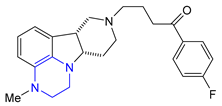 |
| 35 | Fosdenopterin (2021): molybdenum cofactor precursor (to reduce the risk of mortality in patients with molybdenum cofactor deficiency Type A) | 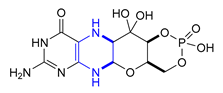 |
| 36 | Lurbinectedin (2020): DNA minor groove binder (treatment of metastatic small cell lung cancer) | 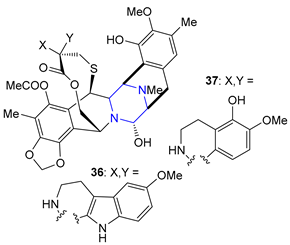 |
| 37 | Trabectedin (2015): DNA minor groove binder (treatment of specific soft tissue sarcomas: liposarcoma and leiomyosarcoma) | |
| 38 | Dolutegravir (2013): Integrase inhibitor (treatment of HIV infection) | 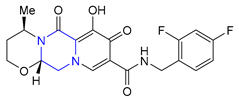 |
| 39 | Bictegravir (2018): Integrase inhibitor, approved in combination with emtricitabine and tenofovir alafenamide (treatment of HIV infection) | 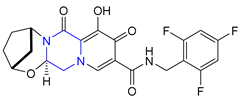 |
| 40 | Cabotegravir (2021): Integrase inhibitor, approved in combination with rilpivirine (treatment of HIV infection) | 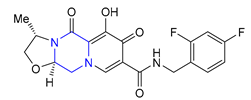 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanelli, M.N.; Braconi, L.; Gabellini, A.; Manetti, D.; Marotta, G.; Teodori, E. Synthetic Approaches to Piperazine-Containing Drugs Approved by FDA in the Period of 2011–2023. Molecules 2024, 29, 68. https://doi.org/10.3390/molecules29010068
Romanelli MN, Braconi L, Gabellini A, Manetti D, Marotta G, Teodori E. Synthetic Approaches to Piperazine-Containing Drugs Approved by FDA in the Period of 2011–2023. Molecules. 2024; 29(1):68. https://doi.org/10.3390/molecules29010068
Chicago/Turabian StyleRomanelli, Maria Novella, Laura Braconi, Alessio Gabellini, Dina Manetti, Giambattista Marotta, and Elisabetta Teodori. 2024. "Synthetic Approaches to Piperazine-Containing Drugs Approved by FDA in the Period of 2011–2023" Molecules 29, no. 1: 68. https://doi.org/10.3390/molecules29010068
APA StyleRomanelli, M. N., Braconi, L., Gabellini, A., Manetti, D., Marotta, G., & Teodori, E. (2024). Synthetic Approaches to Piperazine-Containing Drugs Approved by FDA in the Period of 2011–2023. Molecules, 29(1), 68. https://doi.org/10.3390/molecules29010068