Antiproliferative and Pro-Apoptotic Activity and Tubulin Dynamics Modulation of 1H-Benzimidazol-2-yl Hydrazones in Human Breast Cancer Cell Line MDA-MB-231

 ,
,  , , and
, , and 
Abstract
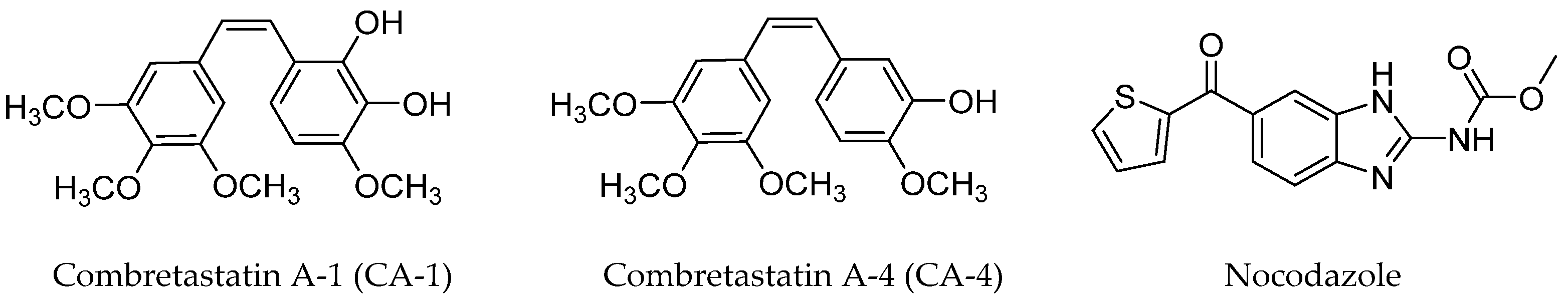
1. Introduction
2. Results and Discussion
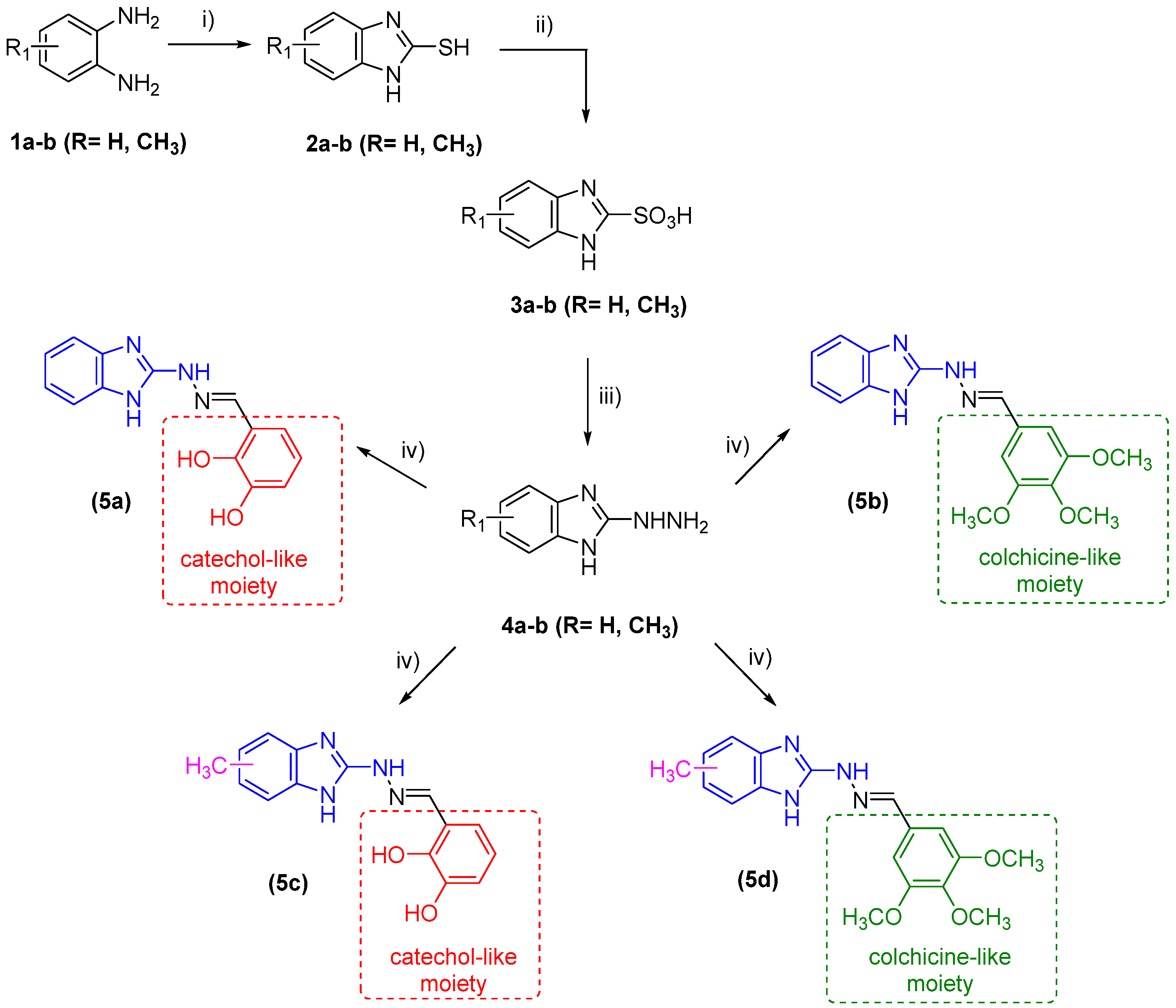
2.1. Synthesis of Target Compounds
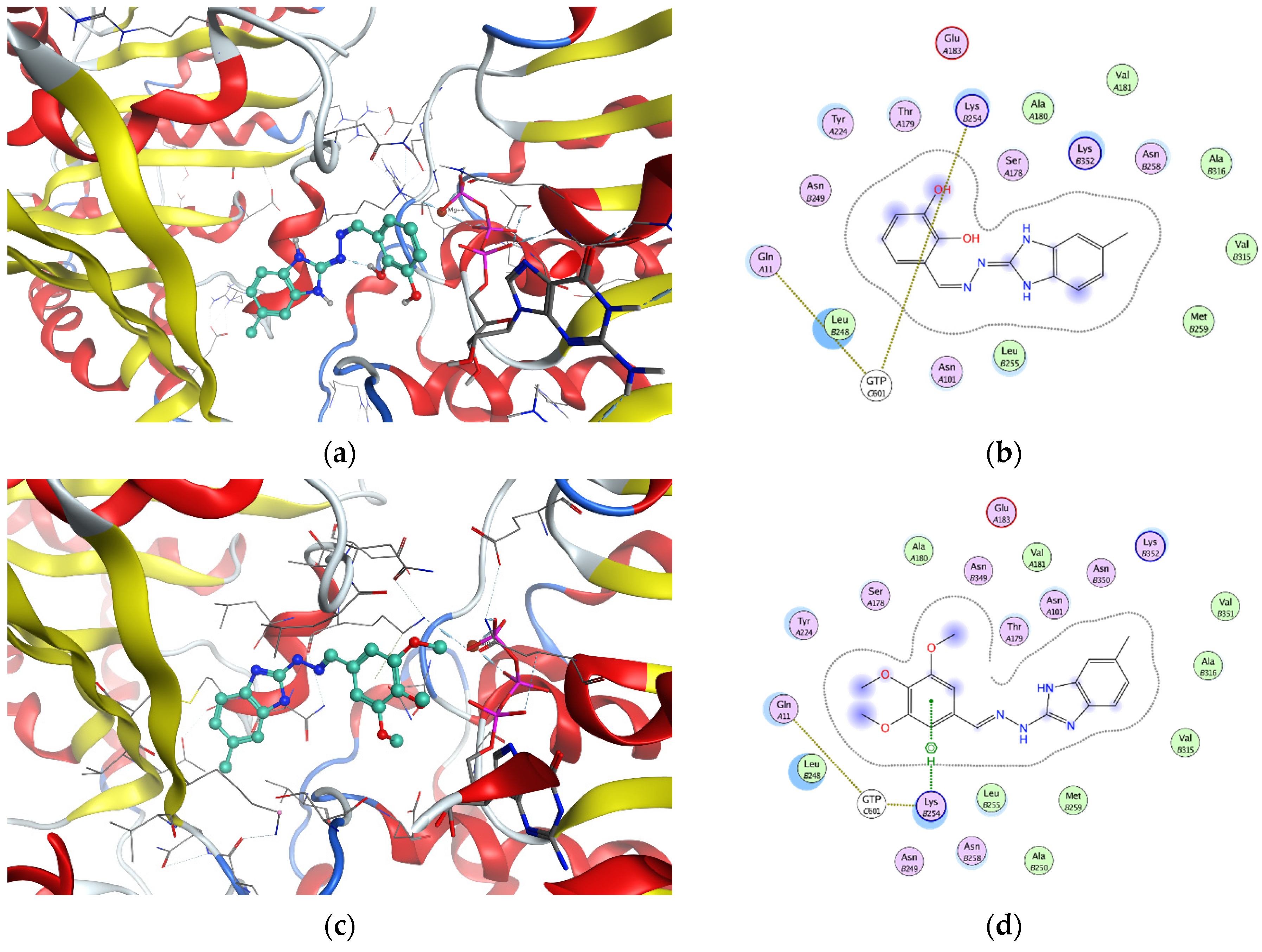
2.2. In Vitro Effect on Tubulin Polymerization and Docking Study of Tubulin–Ligand Interactions
2.3. Determining Cell Viability by MTT Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | |
|---|---|---|
| 48 h | 72 h | |
| Unsubstituted Benzimidazole Ring | ||
| 5a | 43.93 ± 9.3 | 16.04 ± 2.2 |
| 5b | 60.08 ± 15.3 | 20.30 ± 4.3 1 |
| 5(6)-Methyl Benzimidazole Ring | ||
| 5c | 47.28 ± 8.0 | 14.83 ± 3.8 |
| 5d | 40.3 ± 4.1 | 12.65 ± 2.5 |
| Reference Compound | ||
| Nocodazole | 0.38 2 | |
2.4. Investigation of Microtubule Organization and Nuclear Morphology
3. Materials and Methods
3.1. Reagents and Materials
3.2. Synthesis
3.3. In Vitro Tubulin Polymerization Assay
3.4. Cell Lines
3.5. Cell Viability Assay
3.6. Immunofluorescence
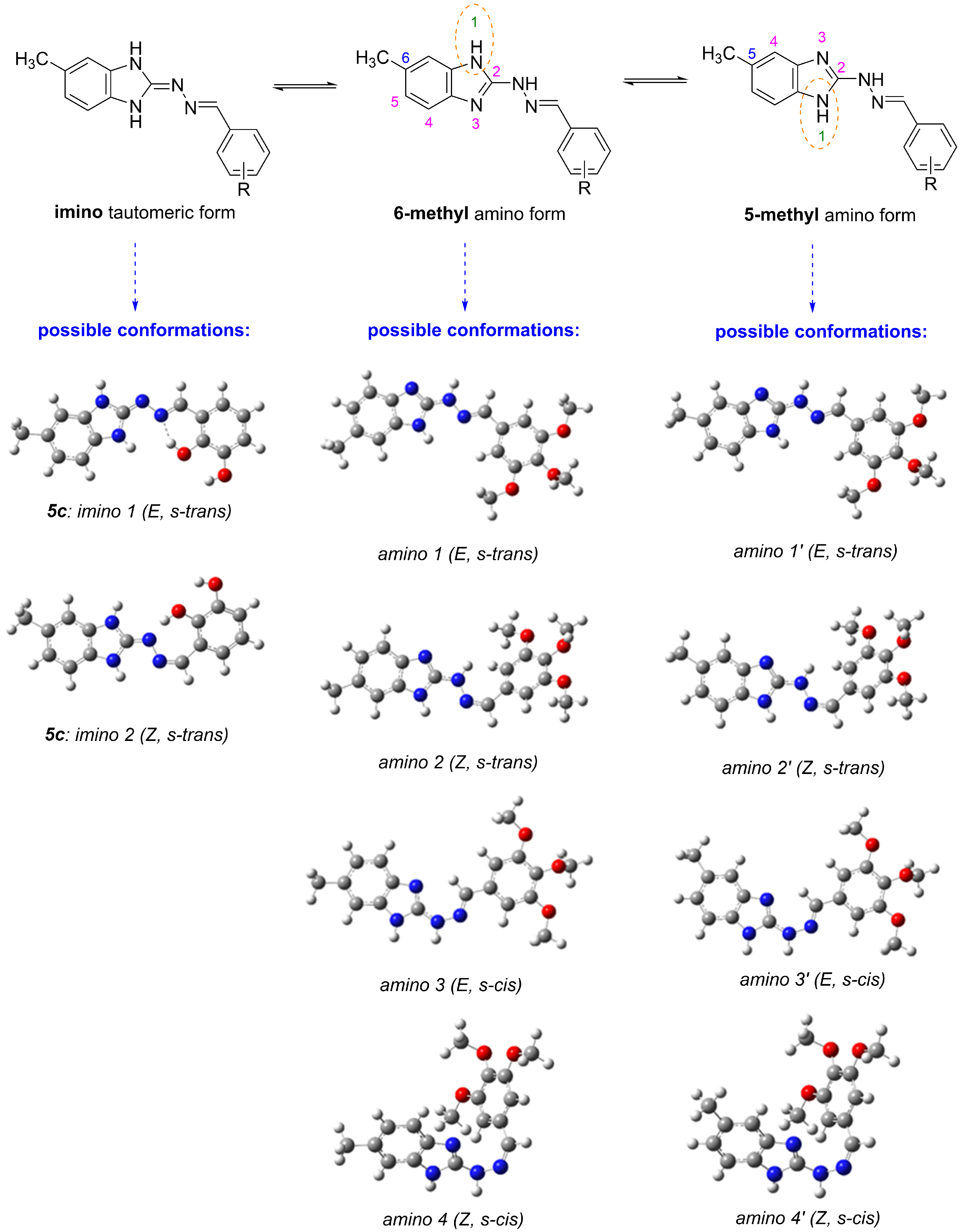
3.7. Molecular Docking and DFT Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKean, P.G.; Vaughan, S.; Gull, K. The extended tubulin superfamily. J. Cell Sci. 2001, 114, 2723–2733. [Google Scholar] [CrossRef]
- Meiring, J.C.M.; Shneyer, B.I.; Akhmanova, A. Generation and regulation of microtubule network asymmetry to drive cell polarity. Curr. Opin. Cell Biol. 2020, 62, 86–95. [Google Scholar] [CrossRef]
- Pellegrini, F.; Budman, D.R. Review: Tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest. 2005, 23, 264–2735. [Google Scholar] [CrossRef]
- Barlan, K.; Gelfand, V.I. Microtubule-Based Transport and the Distribution, Tethering, and Organization of Organelles. Cold Spring Harb. Perspect. Biol. 2017, 9, 025817. [Google Scholar] [CrossRef]
- Kline-Smith, S.L.; Walczak, C.E. Mitotic Spindle Assembly and Chromosome Segregation: Refocusing on Microtubule Dynamics. Mol. Cell 2004, 15, 317–327. [Google Scholar] [CrossRef]
- Schaefer, K.L. PPARγ Inhibitors as Novel Tubulin-Targeting Agents. PPAR Res. 2008, 2008, 785405. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calò, V.; Castiglia, M.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef]
- Avendaño, C.; Carlos Menéndez, J. Anticancer Drugs Targeting Tubulin and Microtubules. In Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Avendaño, C., Carlos Menéndez, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 359–390. [Google Scholar]
- Rowinsky, E.; Donehower, R. Antimicrotubule agents. In DeVita VT, 5th ed.; Hellmann, S., Rosenberg, S.A., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1997. [Google Scholar]
- Martin, R.J.; Robertson, A.P.; Bjorn, H. Target sites of anthelmintics. Parasitology 1997, 114, 111–124. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Anticancer Agents from Natural Products, 2nd ed.; Cragg, G.M., Kingston, D.G.I., Newman, D.J., Eds.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Van Den Bossche, H.; Rochette, F.; Hörig, C. Advances in Pharmacology and Chemotherapy; Academic Press: New York, NY, USA, 1982; Volume 19, pp. 67–127. [Google Scholar]
- Bukhari, S.N.A.; Kumar, G.B.; Revankar, H.M.; Qin, H.-L. Development of combretastatins as potent tubulin polymerization inhibitors. Bioorg. Chem. 2017, 72, 130–147. [Google Scholar] [CrossRef]
- Uckun, F.M.; Cogle, C.R.; Lin, T.L.; Qazi, S.; Trieu, V.N.; Schiller, G.; Watts, J.M. A Phase 1B Clinical Study of Combretastatin A1 Diphosphate (OXi4503) and Cytarabine (ARA-C) in Combination (OXA) for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Cancers 2020, 12, 74. [Google Scholar] [CrossRef]
- Grisham, R.; Ky, B.; Tewari, K.S.; Chaplin, D.J.; Walker, J. Clinical trial experience with CA4P anticancer therapy: Focus on efficacy, cardiovascular adverse events, and hypertension management. Gynecol. Oncol. Res. Pract. 2018, 5, 1. [Google Scholar] [CrossRef]
- Blay, J.Y.; Pápai, Z.; Tolcher, A.W.; Italiano, A.; Cupissol, D.; López-Pousa, A.; Chawla, S.P.; Bompas, E.; Babovic, N.; Penel, N.; et al. Ombrabulin plus cisplatin versus placebo plus cisplatin in patients with advanced soft-tissue sarcomas after failure of anthracycline and ifosfamide chemotherapy: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 531–540. [Google Scholar] [CrossRef]
- Rashid, M.; Husain, A.; Siddiqui, A.A.; Mishra, R. Benzimidazole clubbed with triazolo-thiadiazoles and triazolo-thiadiazines: New anticancer agents. Eur. J. Med. Chem. 2013, 62, 785–798. [Google Scholar]
- Tahlan, S.; Kumar, S.; Kakkar, S.; Narasimhan, B. Benzimidazole scaffolds as promising antiproliferative agents: A review. BMC Chem. 2019, 15, 66. [Google Scholar] [CrossRef]
- Kamal, A.; Reddy, T.S.; Vishnuvardhan, M.V.P.S.; Nimbarte, V.D.; Subba Rao, A.V.; Srinivasulu, V.; Shankaraiah, N. Synthesis of 2-aryl-1, 2, 4-oxadiazolo-benzimidazoles: Tubulin polymerization inhibitors and apoptosis inducing agents. Bioorg. Med. Chem. 2015, 23, 4608–4623. [Google Scholar] [CrossRef]
- Miao, T.T.; Tao, X.B.; Li, D.D.; Chen, H.; Jin, X.Y.; Geng, Y.; Wang, S.F.; Gu, W. Synthesis and biological evaluation of 2-aryl-benzimidazole derivatives of dehydroabietic acid as novel tubulin polymerization inhibitors. RSC Adv. 2018, 8, 17511–17526. [Google Scholar] [CrossRef]
- Ricart, A.D.; Ashton, E.A.; Cooney, M.M. A phase I study of MN-029 (denibulin), a novel vascular-disrupting agent, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2011, 68, 959–970. [Google Scholar] [CrossRef]
- de la Roche, N.M.; Mühlethaler, T.; Di Martino, R.M.C.; Ortega, J.A.; Gioia, D.; Royd, B.; Prota, A.E.; Steinmetz, M.O.; Cavalli, A. Novel fragment-derived colchicine-site binders as microtubule-destabilizing agents. Eur. J. Med. Chem. 2022, 241, 114614. [Google Scholar] [CrossRef]
- González, L.C.; Espinosa-Mendoza, J.D.; Matadamas-Martínez, F.; Romero-Velásquez, A.; Flores-Ramos, M.; Colorado-Pablo, L.F.; Cerbón-Cervantes, M.A.; Castillo, R.; González-Sánchez, I.; Yépez-Mulia, L.; et al. Structure-Based Optimization of Carbendazim-Derived Tubulin Polymerization Inhibitors through Alchemical Free Energy Calculations. J. Chem. Inf. Model. 2023, 63, 7228–7238. [Google Scholar] [CrossRef]
- Anichina, K.; Argirova, M.; Tzoneva, R.; Uzunova, V.; Mavrova, A.; Vuchev, D.; Popova-Daskalova, G.; Fratev, F.; Guncheva, M.; Yancheva, D. 1H-benzimidazole-2-yl hydrazones as tubulin-targeting agents: Synthesis, structural characterization, an-thelmintic activity and antiproliferative activity against MCF-7 breast carcinoma cells and molecular docking studies. Chem. Biol. Interact. 2021, 345, 109540. [Google Scholar] [CrossRef]
- Argirova, M.A.; Georgieva, M.K.; Hristova-Avakumova, N.G.; Vuchev, D.I.; Popova-Daskalova, G.V.; Anichina, K.K.; Yancheva, D.Y. New 1H-benzimidazole-2-yl hydrazones with combined antiparasitic and antioxidant activity. RSC Adv. 2021, 11, 39848–39868. [Google Scholar] [CrossRef]
- Argirova, M.; Guncheva, M.; Momekov, G.; Cherneva, E.; Mihaylova, R.; Rangelov, M.; Todorova, N.; Denev, P.; Anichina, K.; Mavrova, A.; et al. Modulation Effect on Tubulin Polymerization, Cytotoxicity and Antioxidant Activity of 1H-Benzimidazole-2-Yl Hydrazones. Molecules 2023, 28, 291. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2022.
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Knossow, M.; Campanacci, V.; Khodja, L.A.; Gigant, B. The Mechanism of Tubulin Assembly into Microtubules: Insights from Structural Studies. iScience 2020, 23, 101511. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef]
- Othman, D.I.A.; Hamdi, A.; Tawfik, S.S.; Elgazar, A.A.; Mostafa, A.S. Identification of new benzimidazole-triazole hybrids as anticancer agents: Multi-target recognition, in vitro and in silico studies. J. Enz. Inhibit. Med. Chem. 2023, 38, 2166037. [Google Scholar] [CrossRef]
- Husain, A.; Bhutani, M.; Parveen, S.; Khan, S.A.; Ahmad, A.; Iqbal, M.A. Synthesis, in vitro cytotoxicity, ADME, and molecular docking studies of benzimidazole-bearing furanone derivatives. J. Chin. Chem. Soc. 2021, 68, 362–373. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, Y.; Deng, S.; Chen, T. Microwave-assisted syntheses of benzimidazole-containing selenadiazole derivatives that induce cell-cycle arrest and apoptosis in human breast cancer cells by activation of the ROS/AKT pathway. Chem. Med. Chem. 2016, 11, 2339–2346. [Google Scholar] [CrossRef]
- Hranjec, M.; Starčević, K.; Pavelić, S.K.; Lučin, P.; Pavelić, K.; Karminski Zamola, G. Synthesis, spectroscopic characterization and antiproliferative evaluation in vitro of novel Schiff bases related to benzimidazoles. Eur. J. Med. Chem. 2011, 46, 2274–2279. [Google Scholar] [CrossRef]
- Kamal, A.; Subba Rao, A.V.S.; Lakshma Nayak, V.L.; Subba Reddy, N.V.S.; Swapna, K.; Ramakrishna, G.; Alvala, M. Synthesis and biological evaluation of imidazo[1,5-a]pyridine-benzimidazole hybrids as inhibitors of both tubulin polymerization and PI3K/Akt pathway. Org. Biom. Chem. 2014, 12, 9864–9880. [Google Scholar] [CrossRef]
- Kumar, A.; Banerjee, S.; Roy, P.; Sondhi, S.M.; Sharma, A. Solvent-free synthesis and anticancer activity evaluation of benzimidazole and perimidine derivatives. Mol. Diver. 2018, 22, 113–127. [Google Scholar] [CrossRef]
- Abbade, Y.; Kisla, M.M.; Hassan, M.A.; Celik, I.; Dogan, T.S.; Mutlu, P.; Ates-Alagoz, Z. Synthesis, Anticancer Activity, and In Silico Modeling of Alkylsulfonyl Benzimidazole Derivatives: Unveiling Potent Bcl-2 Inhibitors for Breast Cancer. ACS Omega 2024, 9, 9547–9563. [Google Scholar] [CrossRef]
- Beč, A.; Cindrić, M.; Persoons, L.; Banjanac, M.; Radovanović, V.; Daelemans, D.; Hranjec, M. Novel Biologically Active N-Substituted Benzimidazole Derived Schiff Bases: Design, Synthesis, and Biological Evaluation. Molecules 2023, 28, 3720. [Google Scholar] [CrossRef]
- Blajeski, A.L.; Phan, V.A.; Kottke, T.J.; Kaufmann, S.H. G(1) and G(2) cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J. Clin. Investig. 2002, 110, 91–99. [Google Scholar] [CrossRef]
- Bryaskova, R.; Georgiev, N.; Philipova, N.; Bakov, V.; Anichina, K.; Argirova, M.; Apostolova, S.; Georgieva, I.; Tzoneva, R. Novel Fluorescent Benzimidazole- Hydrazone-Loaded Micellar Carriers for Controlled Release: Impact on Cell Toxicity, Nuclear and Microtubule Alterations in Breast Cancer Cells. Pharmaceutics 2023, 15, 1753. [Google Scholar] [CrossRef]
- Haar, E.T.; Day, B.W.; Rosenkranz, H.S. Direct tubulin polymerization perturbation contributes significantly to the induction of micronuclei in vivo. Mutat. Res. Mol. Mech. Mutagen. 1996, 350, 331–337. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. I. 2nd row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent Perturbation Theory of Diamagnetism. I. A Gauge-Invariant LCAO (Linear Combination of Atomic Orbitals) Method for NMR Chemical Shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
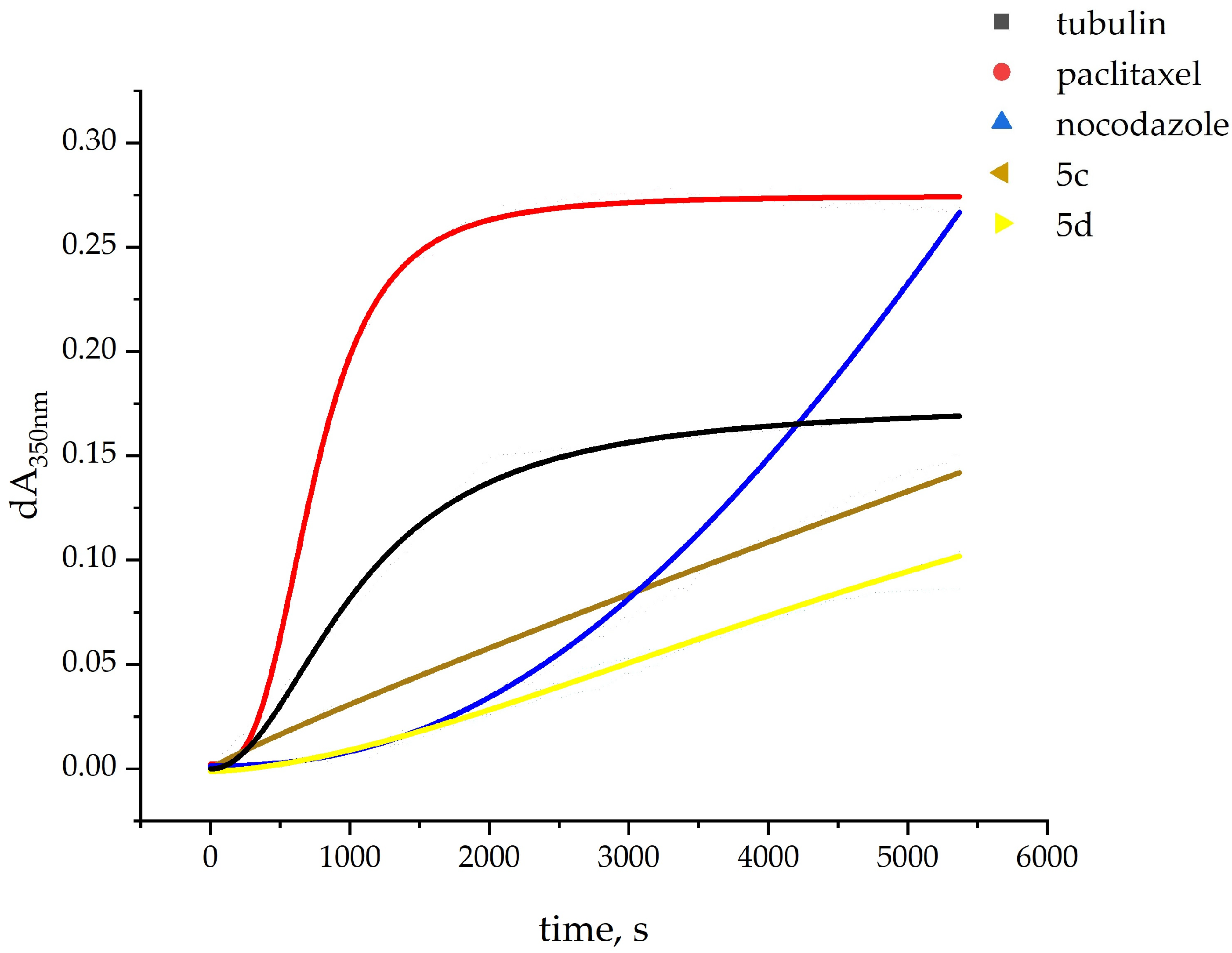

| Compound | Tubulin Polymerization 1 | |
|---|---|---|
| Lag Time, s | Initial Rate 2 | |
| Tubulin (spontaneous polymerization) | no lag phase | 75.3 |
| Unsubstituted benzimidazole ring | ||
| 5a 3 | 1461 | 13.4 |
| 5b 3 | 988 | 20.6 |
| 5(6)-Methyl benzimidazole ring | ||
| 5c | no lag phase | 27.0 |
| 5d | 600 | 12.9 |
| Reference compounds | ||
| Paclitaxel 3 | 151 | 167 |
| Nocodazole 3 | 935 | 52 |
| Compound | Characteristics | |||
|---|---|---|---|---|
| Mitosis | Morphology | Tubulin | ||
| Nucleus | Cell | |||
| Unsubstituted benzimidazole ring | ||||
| 5a | Until 6 h | Abnormal shape | Abnormal shape | Abnormal mitotic spindle |
| 5b | Until 24 h | Micronuclei | Not affected | Not directly observed |
| 5(6)-Methyl benzimidazole ring | ||||
| 5c | Until 12 h | Not affected | Not affected | Elongated polar microtubules, cytoplasmic granules |
| 5d | Arrested in mitosis | Abnormal shape | Abnormal shape | Cytoplasmic granules |
| Reference compound | ||||
| Nocodazole | Arrested in mitosis 1 | Abnormal shape | Abnormal shape | Abnormal mitotic spindle |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yancheva, D.; Argirova, M.; Georgieva, I.; Milanova, V.; Guncheva, M.; Rangelov, M.; Todorova, N.; Tzoneva, R. Antiproliferative and Pro-Apoptotic Activity and Tubulin Dynamics Modulation of 1H-Benzimidazol-2-yl Hydrazones in Human Breast Cancer Cell Line MDA-MB-231. Molecules 2024, 29, 2400. https://doi.org/10.3390/molecules29102400
Yancheva D, Argirova M, Georgieva I, Milanova V, Guncheva M, Rangelov M, Todorova N, Tzoneva R. Antiproliferative and Pro-Apoptotic Activity and Tubulin Dynamics Modulation of 1H-Benzimidazol-2-yl Hydrazones in Human Breast Cancer Cell Line MDA-MB-231. Molecules. 2024; 29(10):2400. https://doi.org/10.3390/molecules29102400
Chicago/Turabian StyleYancheva, Denitsa, Maria Argirova, Irina Georgieva, Vanya Milanova, Maya Guncheva, Miroslav Rangelov, Nadezhda Todorova, and Rumiana Tzoneva. 2024. "Antiproliferative and Pro-Apoptotic Activity and Tubulin Dynamics Modulation of 1H-Benzimidazol-2-yl Hydrazones in Human Breast Cancer Cell Line MDA-MB-231" Molecules 29, no. 10: 2400. https://doi.org/10.3390/molecules29102400
APA StyleYancheva, D., Argirova, M., Georgieva, I., Milanova, V., Guncheva, M., Rangelov, M., Todorova, N., & Tzoneva, R. (2024). Antiproliferative and Pro-Apoptotic Activity and Tubulin Dynamics Modulation of 1H-Benzimidazol-2-yl Hydrazones in Human Breast Cancer Cell Line MDA-MB-231. Molecules, 29(10), 2400. https://doi.org/10.3390/molecules29102400