
BiLSTM- and CNN-Based m6A Modification Prediction Model for circRNAs

 , , and
, , and
Abstract
:
1. Introduction
2. Results
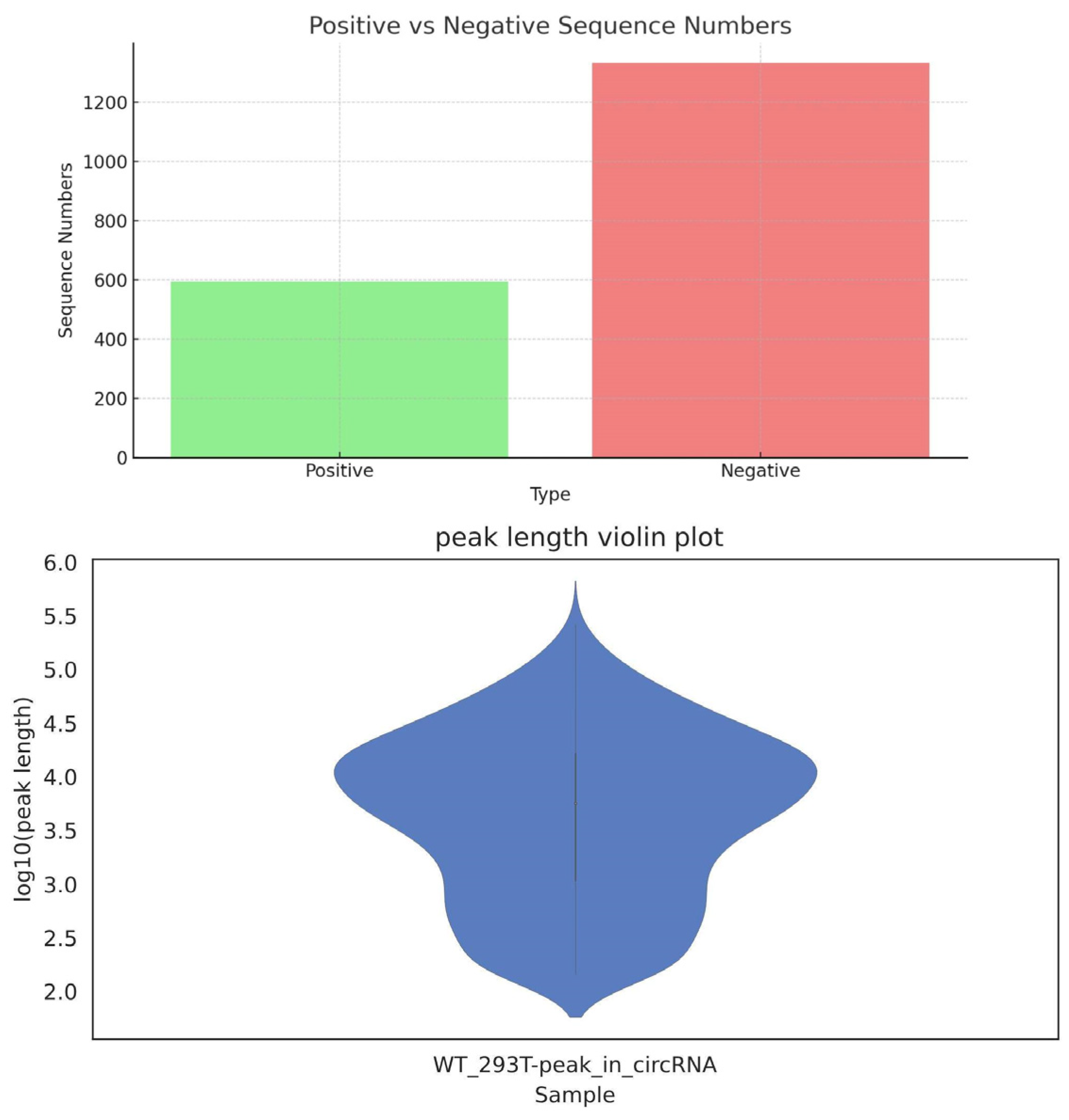
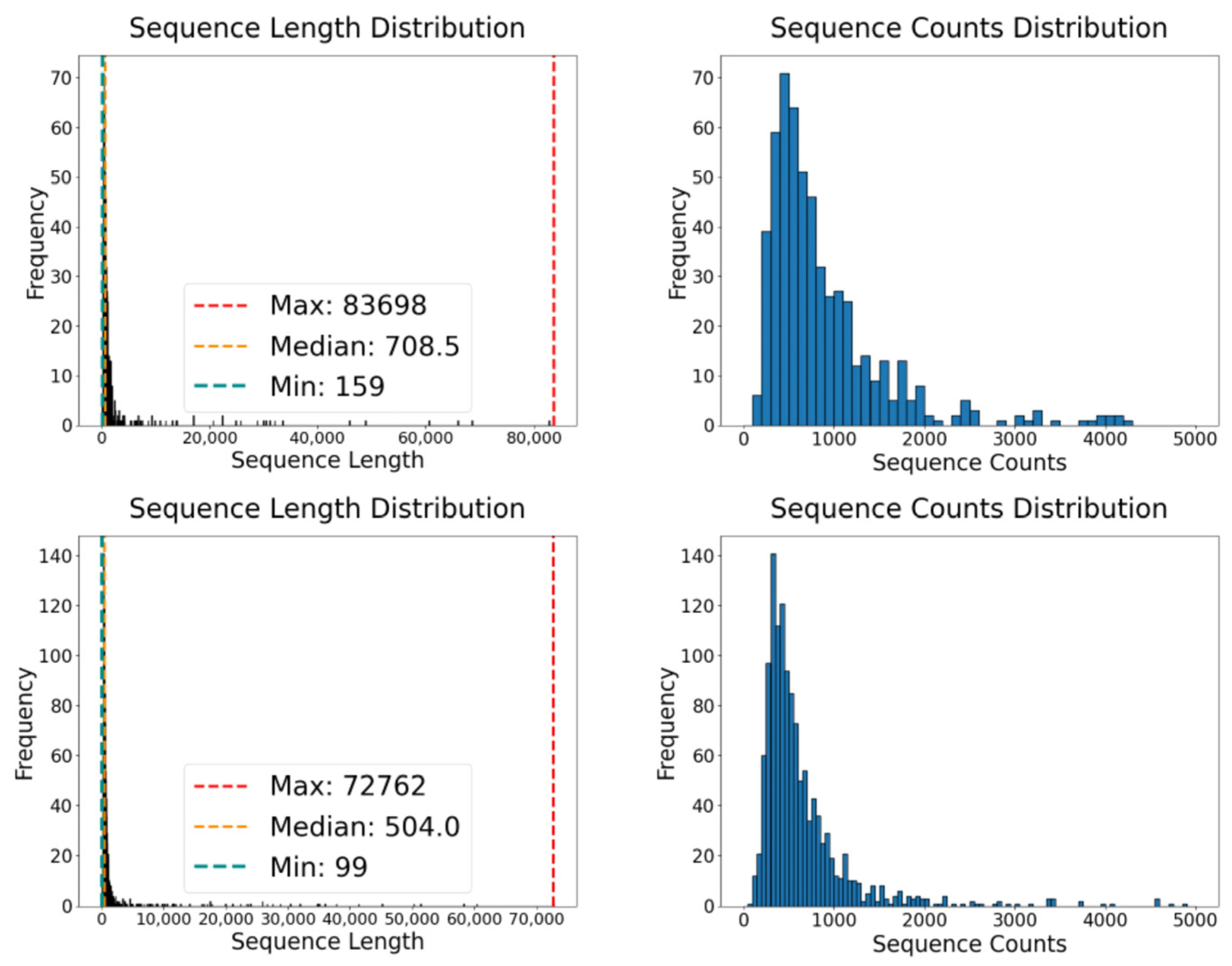
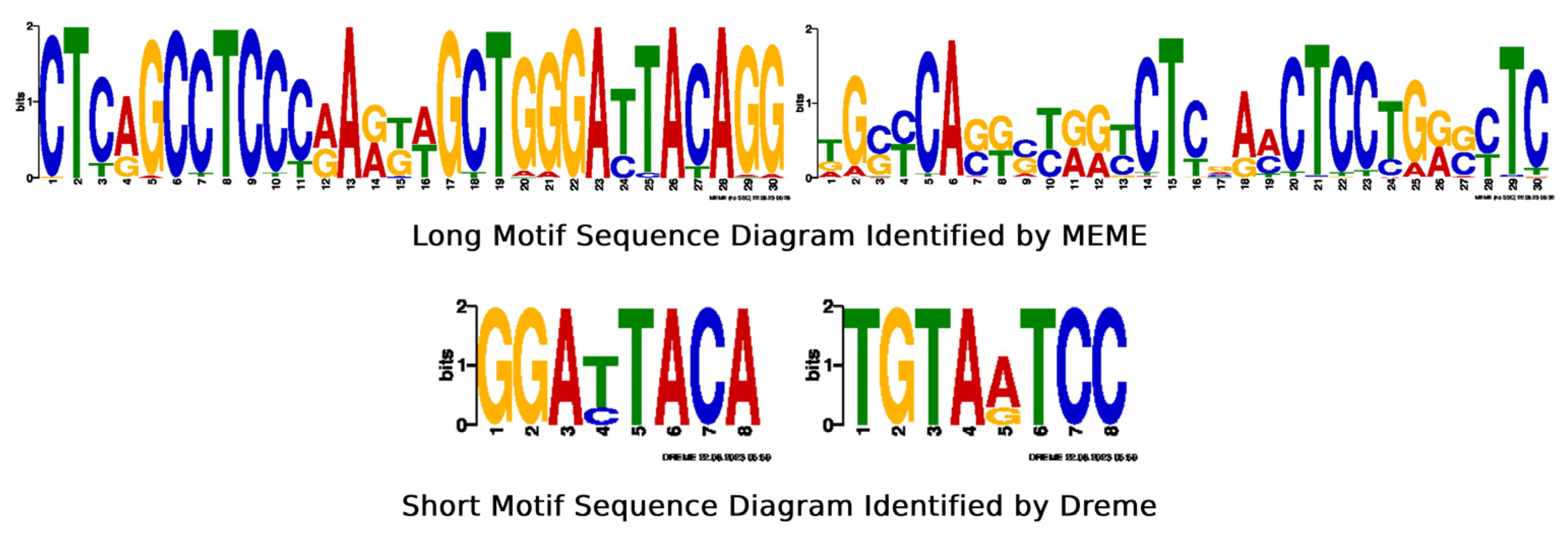
2.1. Predicted Results for Human Genes
2.2. Ten-Fold Cross-Validation of ROC Curves
2.3. Modeling Summary
2.4. Website Construction
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Method
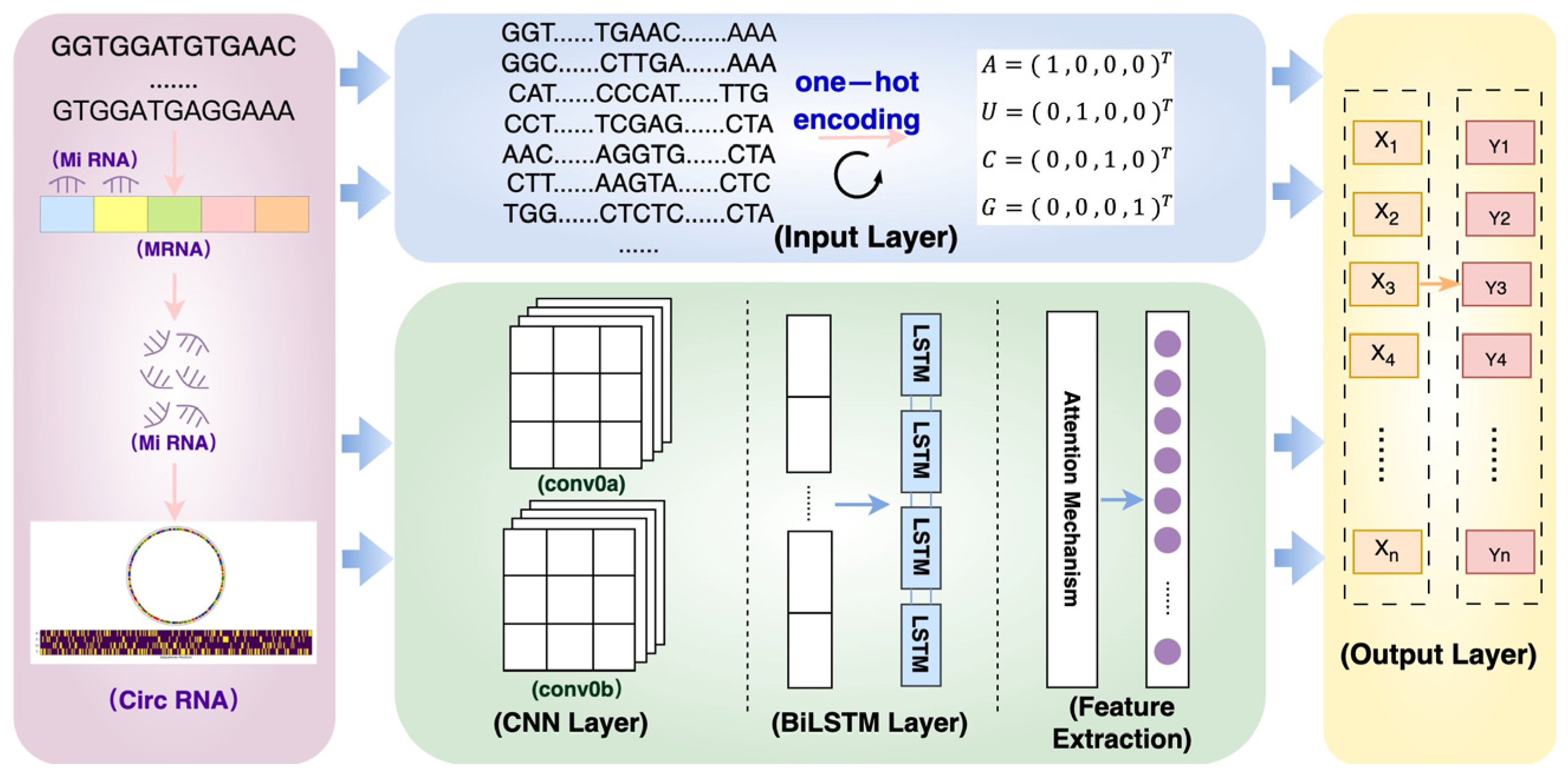
4.2.1. Construction of Predictive Models
4.2.2. Detailed Algorithm Flow
4.2.3. Modeling and Parameterization
4.2.4. Evaluation Indicators
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ChuXiao, L.; LingLing, C. Circular RNAs: Characterization, cellular roles, and applications. Cell 2022, 185, 2016–2034. [Google Scholar]
- Nielsen, A.F.; Bindereif, A.; Bozzoni, I.; Hanan, M.; Hansen, T.B.; Irimia, M.; Kadener, S.; Kristensen, L.S.; Legnini, I.; Morlando, M.; et al. Best practice standards for circular RNA research. Nat. Methods 2022, 19, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Ling-Ling, C. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nature reviews. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Dunin-Horkawicz, S.; Czerwoniec, A.; Gajda, M.J.; Feder, M.; Grosjean, H.; Bujnicki, J.M. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2006, 34, D145–D149. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Jakobsen, T.; Hager, H.; Kjems, J. The emerging roles of circRNAs in cancer and oncology. Nat. Rev. Clin. Oncol. 2021, 19, 188–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Chen, R.; Ahmad, S.; Verma, R.; Kasturi, S.P.; Amaya, L.; Broughton, J.P.; Kim, J.; Cadena, C.; Pulendran, B.; et al. N 6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 2019, 76, 96–109.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.D.; Xie, Y.Y.; Chen, H.X.; Lan, Y.L.; Liu, X.H.; Ji, J.Y.; Wu, F.; Jin, L.; Chen, J.; Mak, D.W.; et al. Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing. Nat. Commun. 2023, 14, 1906. [Google Scholar] [CrossRef] [PubMed]
- Weisheng, C.; Fang, L.; Zhijun, R.; Chen, W.; Chen, Y.; Liu, T.; Ma, Y.; Cao, N.; Wang, J. Parallel functional assessment of m6A sites in human endodermal differentiation with base editor screens. Nat. Commun. 2022, 13, 478. [Google Scholar]
- Zhang, Q.; Zhang, Y.; Chen, H.; Sun, L.N.; Zhang, B.; Yue, D.S.; Wang, C.L.; Zhang, Z.F. METTL3-induced DLGAP1-AS2 promotes non-small cell lung cancer tumorigenesis through m6A/c-Myc-dependent aerobic glycolysis. Cell Cycle 2022, 21, 2602–2614. [Google Scholar] [CrossRef]
- Dao, F.Y.; Lv, H.; Yang, Y.H.; Zulfiqar, H.; Gao, H.; Lin, H. Computational identification of N6-Methyladenosine sites in multiple tissues of mammals. Comput. Struct. Biotechnol. J. 2020, 18, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xie, J.; Xu, S. M6A-BiNP: Predicting N6-methyladenosine sites based on bidirectional position-specific propensities of polynucleotides and pointwise joint mutual information. RNA Biol. 2021, 18, 2498–2512. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cao, L.; Du, P.; Chen, W. im6A-TS-CNN: Identifying the N6-methyladenine site in multiple tissues by using the convolutional neural network. Mol. Ther. Nucleic Acids 2020, 21, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A cross-species method for identifying RNA N6-methyladenosine sites based on deep neural network with multi-information fusion. Genes 2021, 12, 354. [Google Scholar] [CrossRef]
- Zhang, K.; Pan, X.; Yang, Y.; Shen, H.B. CRIP: Predicting circRNAs-RBP-binding sites using a codon-based encoding and hybrid deep neural networks. RNA 2019, 25, 1604–1615. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deng, L.; Lin, W.; Wang, J.; Zhang, J. DeepciRGO: Functional prediction of circular RNAs through hierarchical deep neural networks using heterogeneous network features. BMC Bioinform. 2020, 21, 519. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Wang, J.; Xiao, Y. 3dRNA: 3D Structure Prediction from Linear to Circular RNAs. J. Mol. Biol. 2022, 434, 167452. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, W.; Li, B.; Xie, L.; Tong, Y.; Xu, X. cRNAsp12 Web Server for the Prediction of Circular RNA Secondary Structures and Stabilities. Int. J. Mol. Sci. 2023, 24, 3822. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, G.; Deng, Y.; Liu, Q.; Ye, B.; Dai, Z.; Chen, Y.; Dai, X. Identifying Circular RNA and Predicting Its Regulatory Interactions by Machine Learning. Front. Genet. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Belter, A.; Popenda, M.; Sajek, M.; Woźniak, T.; Naskręt-Barciszewska, M.Z.; Szachniuk, M.; Jurga, S.; Barciszewski, J. A new molecular mechanism of RNA circularization and the microRNA sponge formation. J. Biomol. Struct. Dyn. 2022, 40, 3038–3045. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Jaffrey, S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Krill, P. Vue.js 3.0 Brings More Speed, More TypeScript. InfoWorld.Com 2020. Available online: https://vue3js.cn/ (accessed on 12 May 2024).
- Lutz, M. Learning Python: Powerful Object-Oriented Programming; O’Reilly Media, Inc.: Newton, MA, USA, 2013. [Google Scholar]
- Uchenna, U.I.; Gregory, U.S.; Virginus, U.N.; Angela, O.A.; Enyioma, C.K.; Ezeora, N.J.; Nneoma, O.C.; Kenechukwu, A.; Michael, I.U. Exploring a Secured Socket Python Flask Framework in Real Time Communication System. Asian, J. Res. Comput. Sci. 2021, 8, 77–87. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. RAMPred: Identifying the N1-methyladenosine sites in eukaryotic transcriptomes. Sci. Rep. 2016, 6, 31080. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-3typeA: Identifying three types of modification at RNA’s adenosine sites. Mol. Ther. Nucleic Acids 2018, 11, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, P.; Li, Y.H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. Rna 2019, 25, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Whistle: A high-accuracy map of the human n 6-methyladenosine (m6a) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019, 47, e41. [Google Scholar]
- Chen, W.; Feng, P.; Ding, H.; Lin, H. Identifying N 6-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol. Genet. Genom. 2016, 291, 2225–2229. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, F.; Zhu, M.; Ahmed, K.; Chen, G.; Yao, X. GPS: A comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005, 33 (Suppl. S2), W184–W187. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Y.; Zhai, Y.F.; Song, J.; Zhang, Z. ZincExplorer: An accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Mol. BioSystems 2013, 9, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Chen, L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural Machine Translation by Jointly Learning to Align and Translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Duchi, C.J.; Hazan, E.; Singer, Y. Adaptive Subgradient Methods for Online Learning and Stochastic Optimization. J. Mach. Learn. Res. 2011, 12, 2121–2159. [Google Scholar]
- Tieleman, T. Lecture 6.5-rmsprop: Divide the gradient by a running average of its recent magnitude. COURSERA Neural Netw. Mach. Learn. 2012, 4, 26. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Altman, D.G.; Bland, J.M. Diagnostic tests. 1: Sensitivity and specificity. BMJ 1994, 308, 1552. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davis, J.; Goadrich, M. The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 233–240. [Google Scholar]
- Saito, T.; Rehmsmeier, M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS ONE 2015, 10, e0118432. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Acc | SN | SP | F1 Score | AUC | MCC |
|---|---|---|---|---|---|---|
| Independent dataset | 78.18% | 81.81% | 74.55% | 0.79 | 79.34% | 0.57 |
| TransCirc dataset | 77.03% | 66.67% | 85.37% | 0.72 | 82.19% | 0.53 |
| Term | All | Positive | Negative | ≥500 nt | ≥100 nt | N50 | Max_ Length | Min_ Length | Average_ Length |
|---|---|---|---|---|---|---|---|---|---|
| circRNAs | 1927 | 594 | 1333 | 1093 | 426 | 16,318 | 83,693 | 99 | 1837.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Tang, X.; Li, H.; Lang, X.; Song, Y.; Yang, Y.; Zhou, Z. BiLSTM- and CNN-Based m6A Modification Prediction Model for circRNAs. Molecules 2024, 29, 2429. https://doi.org/10.3390/molecules29112429
Yuan Y, Tang X, Li H, Lang X, Song Y, Yang Y, Zhou Z. BiLSTM- and CNN-Based m6A Modification Prediction Model for circRNAs. Molecules. 2024; 29(11):2429. https://doi.org/10.3390/molecules29112429
Chicago/Turabian StyleYuan, Yuqian, Xiaozhu Tang, Hongyan Li, Xufeng Lang, Yihua Song, Ye Yang, and Zuojian Zhou. 2024. "BiLSTM- and CNN-Based m6A Modification Prediction Model for circRNAs" Molecules 29, no. 11: 2429. https://doi.org/10.3390/molecules29112429
APA StyleYuan, Y., Tang, X., Li, H., Lang, X., Song, Y., Yang, Y., & Zhou, Z. (2024). BiLSTM- and CNN-Based m6A Modification Prediction Model for circRNAs. Molecules, 29(11), 2429. https://doi.org/10.3390/molecules29112429