
The Human Paraoxonase 2: An Optimized Procedure for Refolding and Stabilization Facilitates Enzyme Analyses and a Proteomics Approach


 ,
,
Abstract
:1. Introduction
2. Results
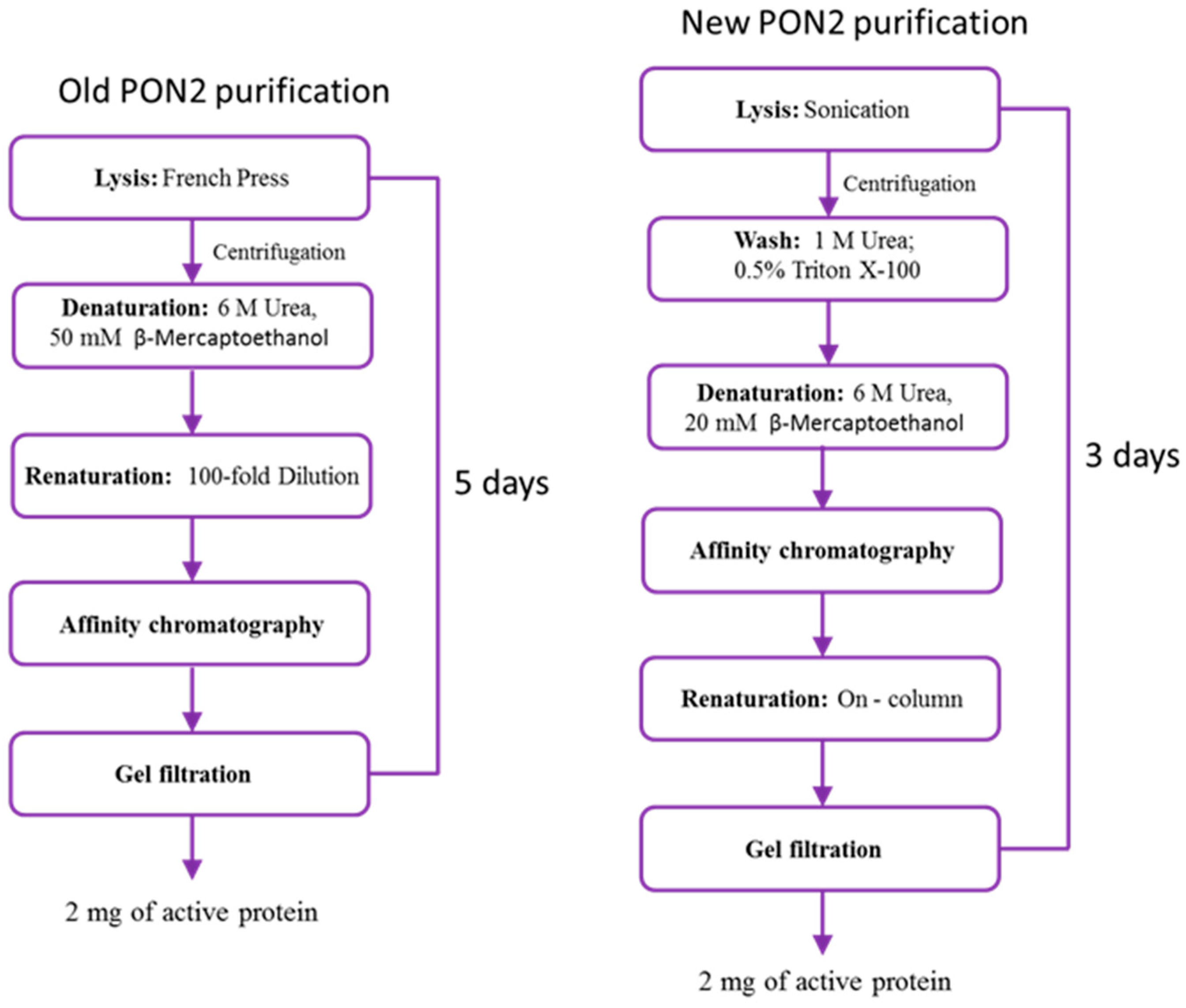
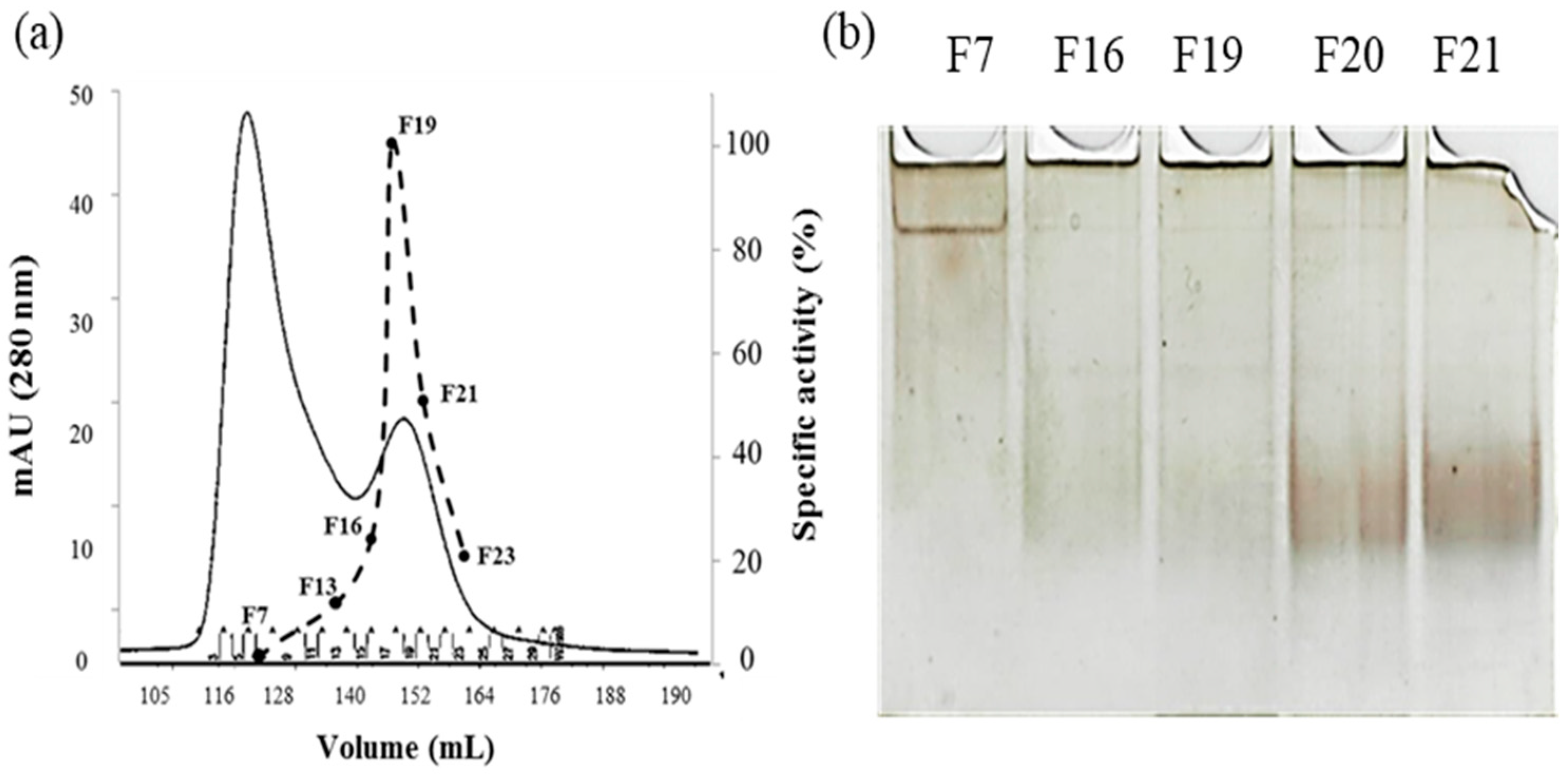
2.1. rPON2 Refolding and Purification
- Cells were lysed by low-amplitude sonication to preserve the inclusion bodies, in the substitution of French press cell disruption.
- A series of washes with low concentrations of Triton X-100 and urea were added to remove contaminants bound to inclusion bodies through non-specific interactions [27].
- The protein–resin binding step was performed with the protein in its unfolded state, in which the his-tag is exposed, to decrease the time of the binding compared to previous conditions.
- Column renaturation was chosen for protein refolding in the substitution of the 100-fold dilution method used before, to overcome the problem of handling huge volumes of sample.
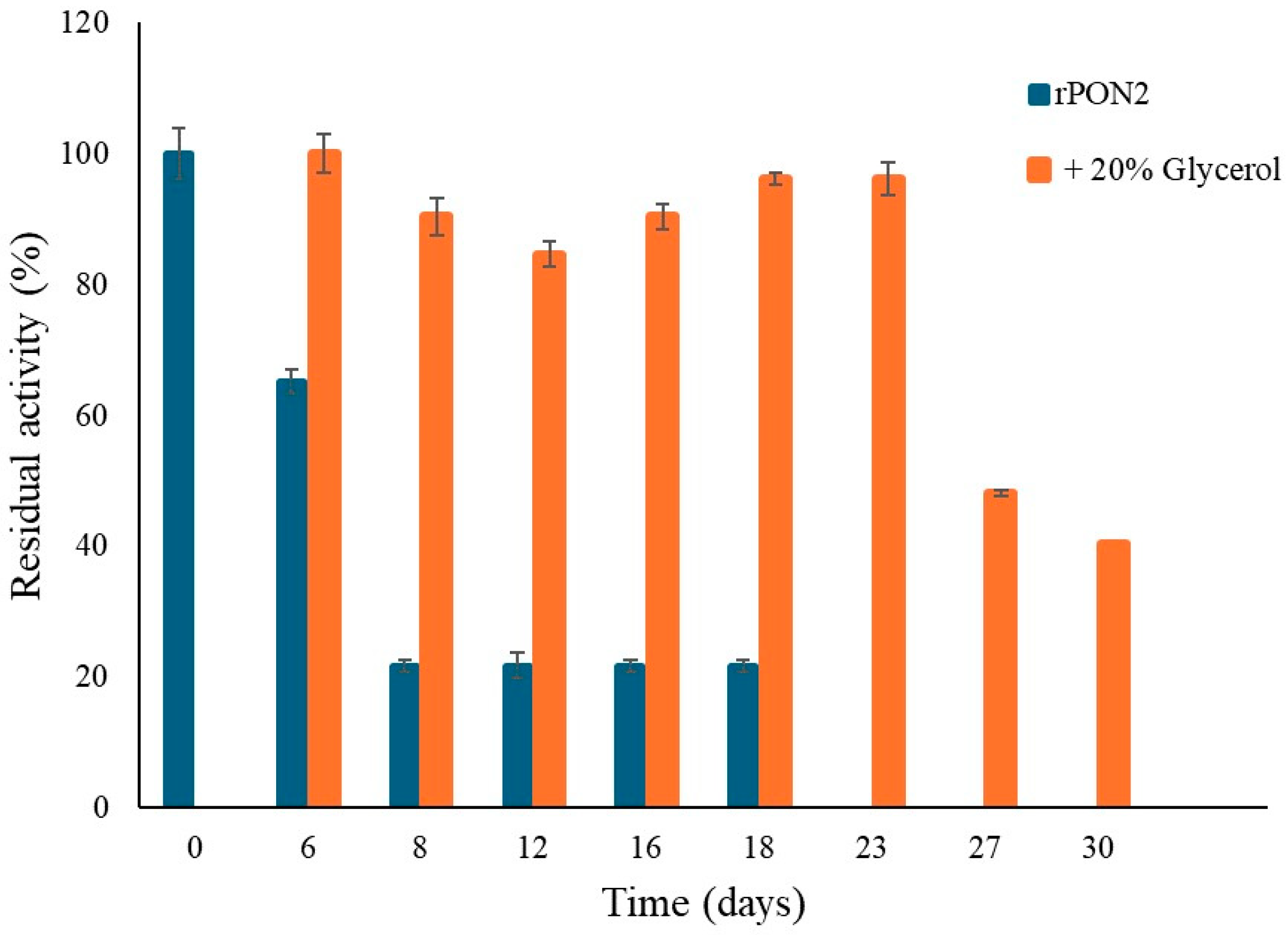
2.2. Protein Stabilization
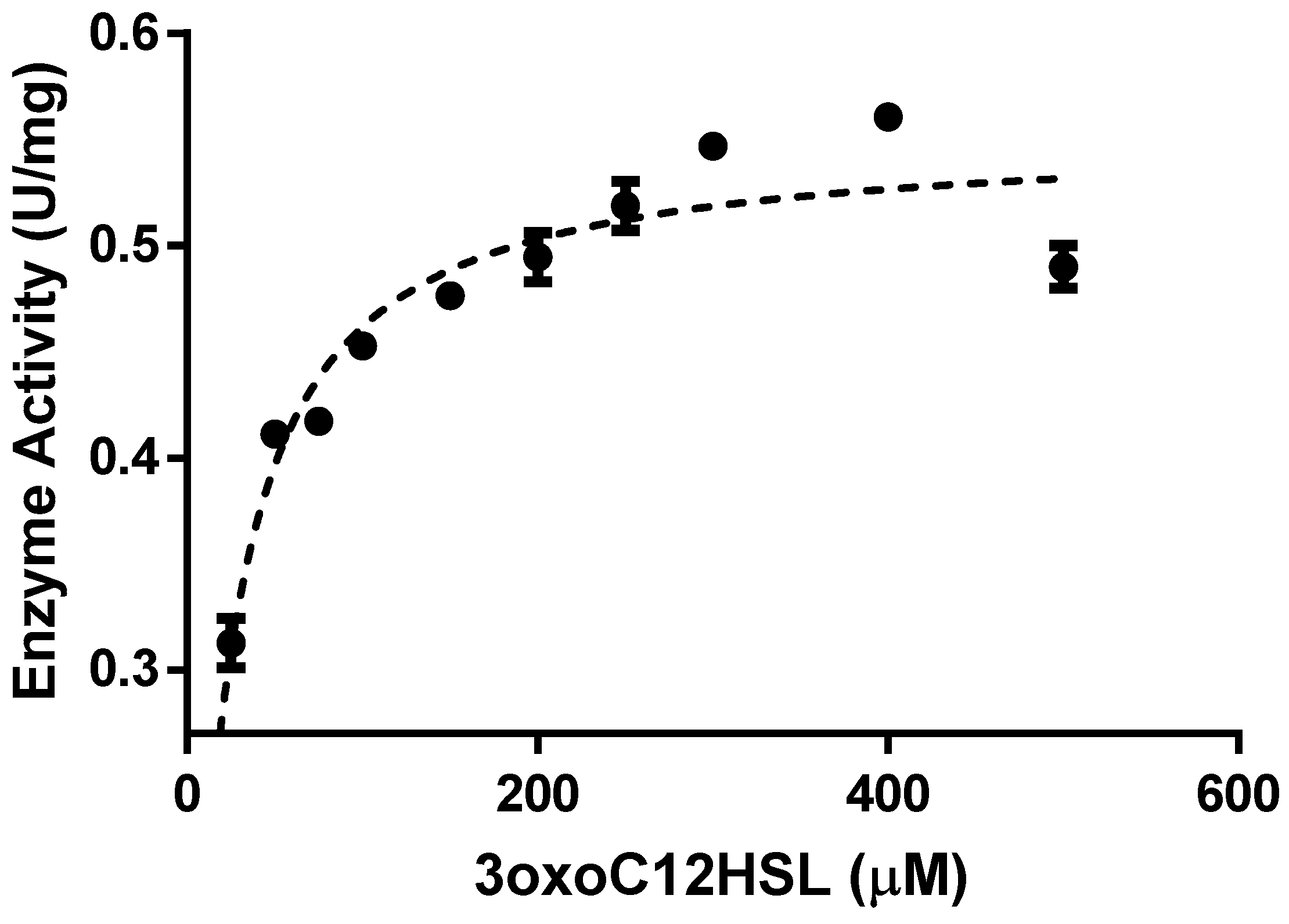
2.3. Enzyme Kinetics
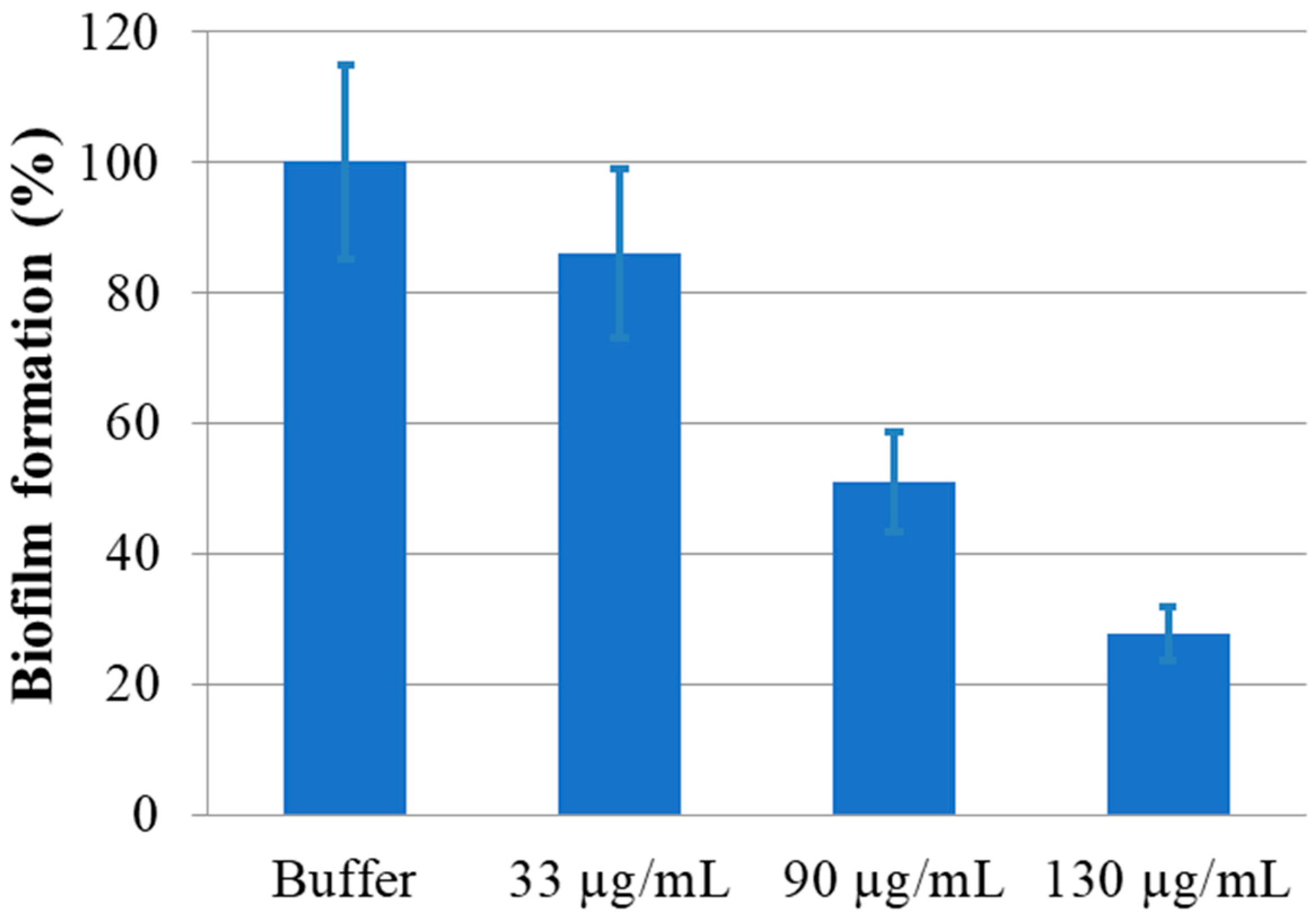
2.4. Anti-Biofilm Assay
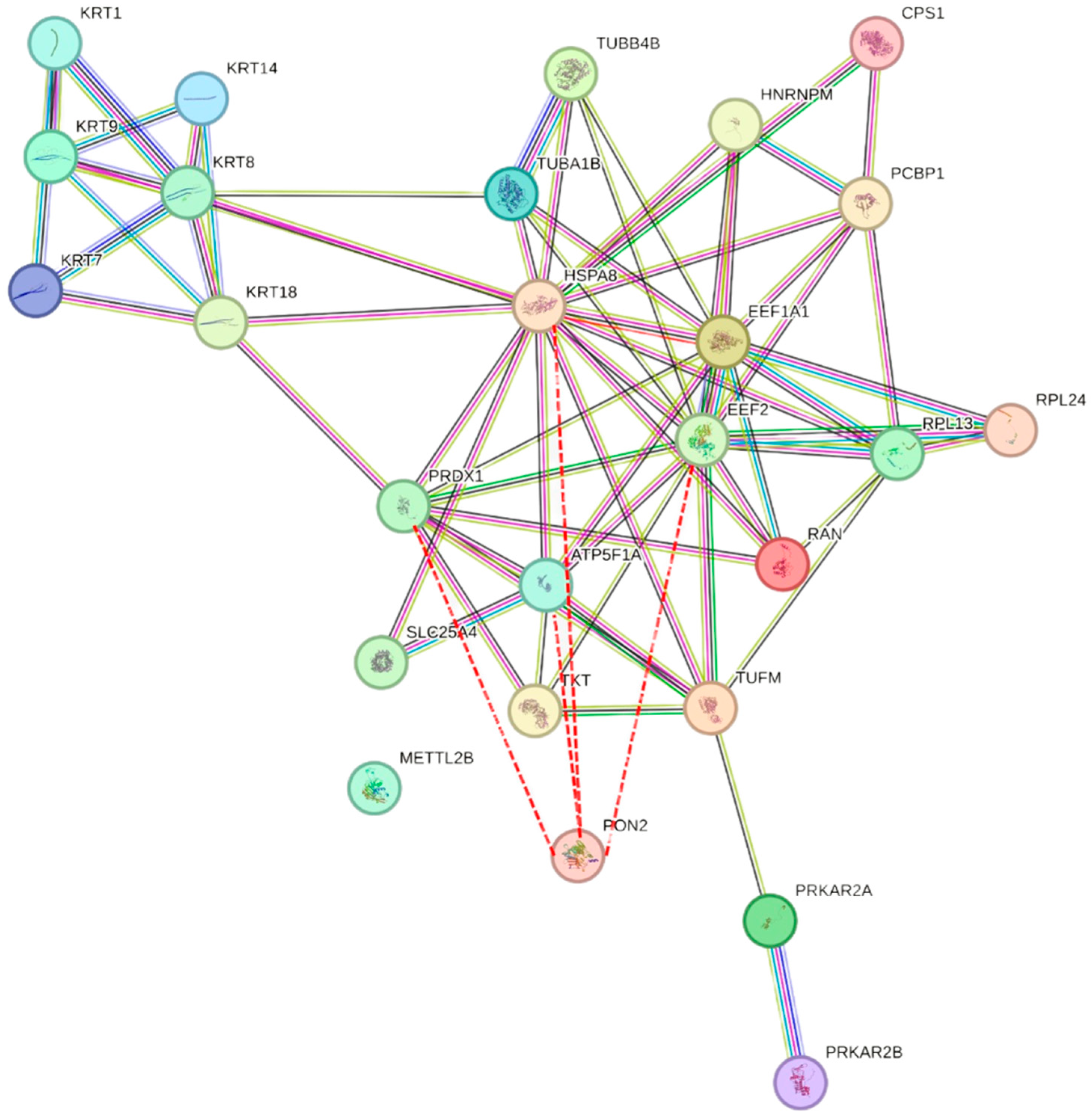
2.5. Proteomic Approach
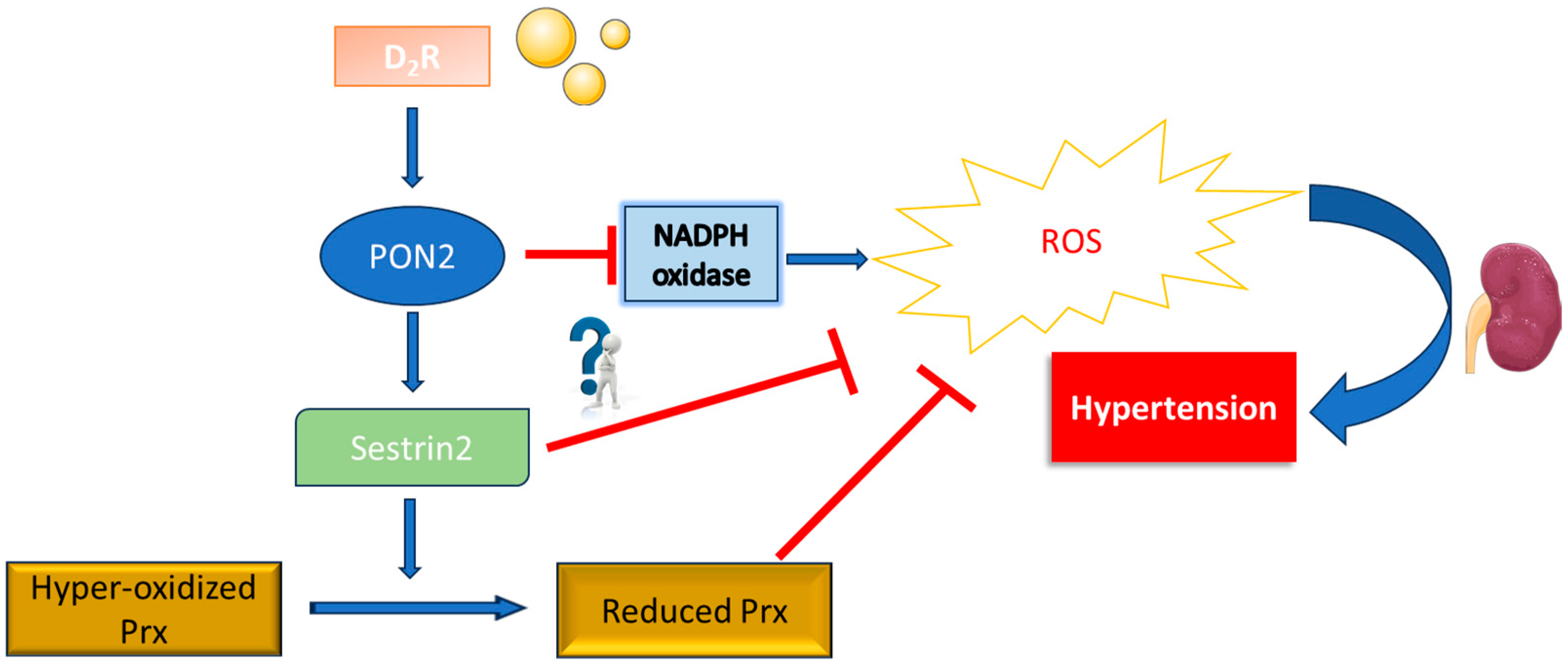
3. Discussion
4. Materials and Methods
4.1. Protein Expression, In Vitro Refolding, and Purification
4.2. Electrophoretic Analysis
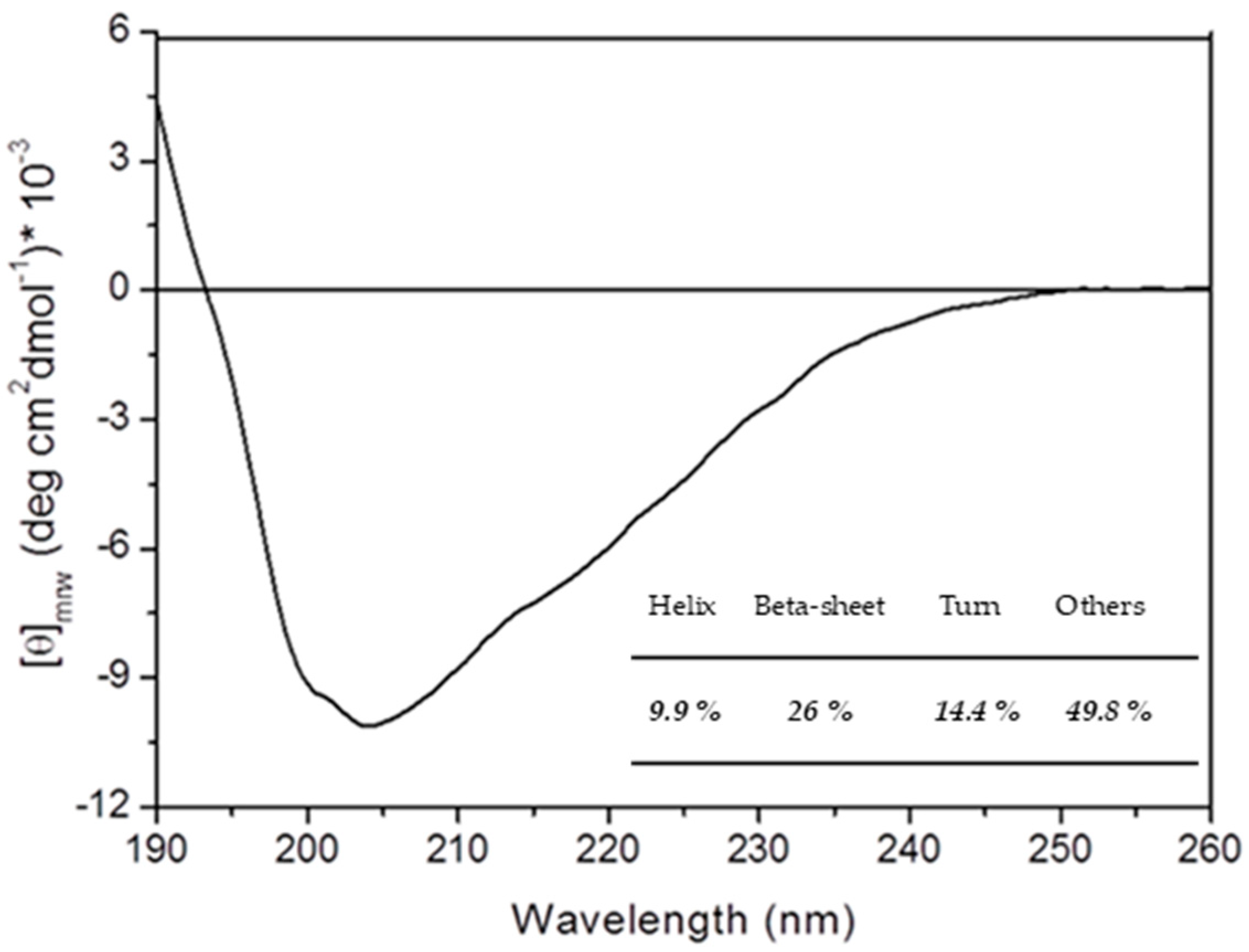
4.3. Circular Dichroism
4.4. Enzymatic Activity Assays
4.5. Inhibition of Biofilm Formation of Pseudomonas aeruginosa by rPON2
4.6. Measurement of Enzyme Stability
4.7. HeLa Cell Extract
4.8. Direct Molecular Fishing
4.9. Sample Preparation and Mass Spectrometry Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Primo-Parmo, S.L.; Sorenson, R.C.; Teiber, J.; Du, B.N.L. The Human Serum Paraoxonase/Arylesterase Gene (PON1) Is One Member of a Multigene Family. Genomics 1996, 33, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Teiber, J.F.; Xiao, J.; Kramer, G.L.; Ogawa, S.; Ebner, C.; Wolleb, H.; Carreira, E.M.; Shih, D.M.; Haley, R.W. Identification of Biologically Active δ-Lactone Eicosanoids as Paraoxonase Substrates. Biochem. Biophys. Res. Commun. 2018, 505, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.I.; La Du, B.N. Pharmacogenetics of Paraoxonases: A Brief Review. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 369, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.J.; Wadleigh, D.J.; Gangopadhyay, A.; Hama, S.; Grijalva, V.R.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Paraoxonase-2 Is a Ubiquitously Expressed Protein with Antioxidant Properties and Is Capable of Preventing Cell-Mediated Oxidative Modification of Low Density Lipoprotein. J. Biol. Chem. 2001, 276, 44444–44449. [Google Scholar] [CrossRef]
- Kulka, M. A Review of Paraoxonase 1 Properties and Diagnostic Applications. Pol. J. Vet. Sci. 2016, 19, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, H.; Kuczkowski, A.; Ruehl, M.; Lamkemeyer, T.; Brodesser, S.; Horke, S.; Dryer, S.; Schermer, B.; Benzing, T.; Brinkkoetter, P.T. Breaking the Chain at the Membrane: Paraoxonase 2 Counteracts Lipid Peroxidation at the Plasma Membrane. FASEB J. 2014, 28, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Horke, S.; Witte, I.; Wilgenbus, P.; Krüger, M.; Strand, D.; Förstermann, U. Paraoxonase-2 Reduces Oxidative Stress in Vascular Cells and Decreases Endoplasmic Reticulum Stress–Induced Caspase Activation. Circulation 2007, 115, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.I.; Teiber, J.F.; Speelman, A.; Osawa, Y.; Sunahara, R.; La Du, B.N. Human Paraoxonases (PON1, PON2, and PON3) Are Lactonases with Overlapping and Distinct Substrate Specificities. J. Lipid Res. 2005, 46, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Witte, I.; Teiber, J.F.; Wilgenbus, P.; Pautz, A.; Li, H.; Daiber, A.; Witan, H.; Clement, A.M.; Förstermann, U.; et al. One Enzyme, Two Functions. J. Biol. Chem. 2010, 285, 24398–24403. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Scherer, S.W.; Xi, T.; Nickle, D.C.; Majer, M.; Huizenga, J.J.; Tsui, L.-C.; Prochazka, M. Human PON2 Gene at 7q21.3: Cloning, Multiple mRNA Forms, and Missense Polymorphisms in the Coding Sequence. Gene 1998, 213, 149–157. [Google Scholar] [CrossRef]
- Carusone, T.M.; Cardiero, G.; Cerreta, M.; Mandrich, L.; Moran, O.; Porzio, E.; Catara, G.; Lacerra, G.; Manco, G. WTAP and BIRC3 Are Involved in the Posttranscriptional Mechanisms That Impact on the Expression and Activity of the Human Lactonase PON2. Cell Death Dis. 2020, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Suzuki, Y.; Nishikawa, T.; Otsuki, T.; Sugiyama, T.; Irie, R.; Wakamatsu, A.; Hayashi, K.; Sato, H.; Nagai, K.; et al. Complete Sequencing and Characterization of 21,243 Full-Length Human cDNAs. Nat. Genet. 2004, 36, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fan, Z.; Huang, J.; Su, S.; Yu, Q.; Zhao, J.; Hui, R.; Yao, Z.; Shen, Y.; Qiang, B.; et al. Extensive Association Analysis Between Polymorphisms of PON Gene Cluster with Coronary Heart Disease in Chinese Han Population. ATVB 2003, 23, 328–334. [Google Scholar] [CrossRef]
- Martinelli, N.; Girelli, D.; Olivieri, O.; Stranieri, C.; Trabetti, E.; Pizzolo, F.; Friso, S.; Tenuti, I.; Cheng, S.; Grow, M.A.; et al. Interaction between Smoking and PON2 Ser311Cys Polymorphism as a Determinant of the Risk of Myocardial Infarction. Eur. J. Clin. Investig. 2004, 34, 14–20. [Google Scholar] [CrossRef]
- Sanghera, D.K.; Aston, C.E.; Saha, N.; Kamboh, M.I. DNA Polymorphisms in Two Paraoxonase Genes (PON1 and PON2) Are Associated with the Risk of Coronary Heart Disease. Am. J. Hum. Genet. 1998, 62, 36–44. [Google Scholar] [CrossRef]
- Janka, Z.; Juhász, A.; Rimanóczy, Á.; Boda, K.; Márki-Zay, J.; Kálmán, J. Codon 311 (Cys → Ser) Polymorphism of Paraoxonase-2 Gene Is Associated with Apolipoprotein E4 Allele in Both Alzheimer’s and Vascular Dementias. Mol. Psychiatry 2002, 7, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, S.; Tang, M.; Liu, X.; Li, T.; Han, H.; Wang, Y.; Guo, Y.; Zhao, J.; Li, H.; et al. Possible Association between Cys311Ser Polymorphism of Paraoxonase 2 Gene and Late-Onset Alzheimer’s Disease in Chinese. Mol. Brain Res. 2004, 120, 201–204. [Google Scholar] [CrossRef]
- Manco, G.; Porzio, E.; Carusone, T.M. Human Paraoxonase-2 (PON2): Protein Functions and Modulation. Antioxidants 2021, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, J.; Feng, B.; Lu, W.; Zhao, C.; Li, L. Mendelian Randomization Analysis Identified Genes Pleiotropically Associated with the Risk and Prognosis of COVID-19. J. Infect. 2021, 82, 126–132. [Google Scholar] [CrossRef]
- Mandrich, L.; Cerreta, M.; Manco, G. An Engineered Version of Human PON2 Opens the Way to Understand the Role of Its Post-Translational Modifications in Modulating Catalytic Activity. PLoS ONE 2015, 10, e0144579. [Google Scholar] [CrossRef]
- Pearson, J.P.; Gray, K.M.; Passador, L.; Tucker, K.D.; Eberhard, A.; Iglewski, B.H.; Greenberg, E.P. Structure of the Autoinducer Required for Expression of Pseudomonas aeruginosa Virulence Genes. Proc. Natl. Acad. Sci. USA 1994, 91, 197–201. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Hodgkinson, J.T.; Bowden, S.D.; Welch, M.; Spring, D.R. Quorum Sensing in Gram-Negative Bacteria: Small-Molecule Modulation of AHL and AI-2 Quorum Sensing Pathways. Chem. Rev. 2011, 111, 28–67. [Google Scholar] [CrossRef]
- Singh, S.; Singh, S.K.; Chowdhury, I.; Singh, R. Understanding the Mechanism of Bacterial Biofilms Resistance to Antimicrobial Agents. Open Microbiol. J. 2017, 11, 53–62. [Google Scholar] [CrossRef]
- Porzio, E.; Andrenacci, D.; Manco, G. Thermostable Lactonases Inhibit Pseudomonas aeruginosa Biofilm: Effect In Vitro and in Drosophila Melanogaster Model of Chronic Infection. Int. J. Mol. Sci. 2023, 24, 17028. [Google Scholar] [CrossRef]
- Rather, M.A.; Saha, D.; Bhuyan, S.; Jha, A.N.; Mandal, M. Quorum Quenching: A Drug Discovery Approach Against Pseudomonas aeruginosa. Microbiol. Res. 2022, 264, 127173. [Google Scholar] [CrossRef]
- Horke, S.; Witte, I.; Altenhöfer, S.; Wilgenbus, P.; Goldeck, M.; Förstermann, U.; Xiao, J.; Kramer, G.L.; Haines, D.C.; Chowdhary, P.K.; et al. Paraoxonase 2 Is Down-Regulated by the Pseudomonas aeruginosa Quorumsensing Signal N-(3-Oxododecanoyl)-L-Homoserine Lactone and Attenuates Oxidative Stress Induced by Pyocyanin. Biochem. J. 2010, 426, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Jürgen, B.; Breitenstein, A.; Urlacher, V.; Büttner, K.; Lin, H.; Hecker, M.; Schweder, T.; Neubauer, P. Quality Control of Inclusion Bodies in Escherichia coli. Microb. Cell Fact. 2010, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Moussong, É.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.-H.; Molnár, T.; Réfrégiers, M.; Goto, Y.; et al. BeStSel: Webserver for Secondary Structure and Fold Prediction for Protein CD Spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef] [PubMed]
- Alibolandi, M.; Mirzahoseini, H. Chemical Assistance in Refolding of Bacterial Inclusion Bodies. Biochem. Res. Int. 2011, 2011, 631607. [Google Scholar] [CrossRef]
- Jain, N.K.; Roy, I. Effect of Trehalose on Protein Structure. Protein Sci. 2009, 18, 24–26. [Google Scholar] [CrossRef]
- Mari, E.; Ricci, C.; Pieraccini, S.; Spinozzi, F.; Mariani, P.; Ortore, M.G. Trehalose Effect on The Aggregation of Model Proteins into Amyloid Fibrils. Life 2020, 10, 60. [Google Scholar] [CrossRef]
- Dominy, B.N.; Perl, D.; Schmid, F.X.; Brooks, C.L. The Effects of Ionic Strength on Protein Stability: The Cold Shock Protein Family. J. Mol. Biol. 2002, 319, 541–554. [Google Scholar] [CrossRef]
- Lindman, S.; Xue, W.-F.; Szczepankiewicz, O.; Bauer, M.C.; Nilsson, H.; Linse, S. Salting the Charged Surface: pH and Salt Dependence of Protein G B1 Stability. Biophys. J. 2006, 90, 2911–2921. [Google Scholar] [CrossRef]
- Mao, Y.-J.; Sheng, X.-R.; Pan, X.-M. The Effects of NaCl Concentration and pH on the Stability of Hyperthermophilic Protein Ssh10b. BMC Biochem. 2007, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Maurer, R.W.; Sandler, S.I.; Lenhoff, A.M. Salting-in Characteristics of Globular Proteins. Biophys. Chem. 2011, 156, 72–78. [Google Scholar] [CrossRef]
- Vagenende, V.; Yap, M.G.S.; Trout, B.L. Mechanisms of Protein Stabilization and Prevention of Protein Aggregation by Glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Tyagi, R. Investigations of Mixed Surfactant Systems of Lauryl Alcohol-Based Bissulfosuccinate Anionic Gemini Surfactants with Conventional Surfactants: A Fluorometric Study. J. Taibah Univ. Sci. 2015, 9, 477–489. [Google Scholar] [CrossRef]
- The Protein Protocols Handbook, 3rd ed.; Walker, J.M. (Ed.) Humana Press: New York, NY, USA, 2009; ISBN 978-1-60327-474-6. [Google Scholar]
- Naidu, K.T.; Rao, D.K.; Prabhu, N.P. Cryo vs. Thermo: Duality of Ethylene Glycol on the Stability of Proteins. J. Phys. Chem. B 2020, 124, 10077–10088. [Google Scholar] [CrossRef]
- Damjanovich, S.; Bot, J.; Somogyi, B.; Sümegi, J. Effect of Glycerol on Some Kinetic Parameters of Phosphorylase b. Biochim. Biophys. Acta (BBA)—Enzymol. 1972, 284, 345–348. [Google Scholar] [CrossRef]
- Sampedro, J.G.; Rivera-Moran, M.A.; Uribe-Carvajal, S. Kramers’ Theory and the Dependence of Enzyme Dynamics on Trehalose-Mediated Viscosity. Catalysts 2020, 10, 659. [Google Scholar] [CrossRef]
- Paz-Alfaro, K.J.; Ruiz-Granados, Y.G.; Uribe-Carvajal, S.; Sampedro, J.G. Trehalose-Mediated Thermal Stabilization of Glucose Oxidase from Aspergillus Niger. J. Biotechnol. 2009, 141, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meng, Z.; Shi, R.; Zhan, L.; Hu, W.; Xiang, H.; Xie, Q. Effects of Temperature and Additives on the Thermal Stability of Glucoamylase from Aspergillus Niger. J. Microbiol. Biotechnol. 2015, 25, 33–43. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, G.A. Microtiter Dish Biofilm Formation Assay. JoVE 2011, 30, 2437. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BIOGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Teiber, J. Defense Technical Information Center. Available online: https://apps.dtic.mil/sti/tr/pdf/AD1003001.pdf (accessed on 16 May 2024).
- Yang, Y.; Cuevas, S.; Yang, S.; Villar, V.A.; Escano, C.; Asico, L.; Yu, P.; Jiang, X.; Weinman, E.J.; Armando, I.; et al. Sestrin2 Decreases Renal Oxidative Stress, Lowers Blood Pressure, and Mediates Dopamine D 2 Receptor–Induced Inhibition of Reactive Oxygen Species Production. Hypertension 2014, 64, 825–832. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Meilin, E.; Aviram, M.; Hayek, T. Paraoxonase 2 (PON2) Decreases High Glucose-Induced Macrophage Triglycerides (TG) Accumulation, via Inhibition of NADPH-Oxidase and DGAT1 Activity: Studies in PON2-Deficient Mice. Atherosclerosis 2010, 208, 390–395. [Google Scholar] [CrossRef]
- Rosenblat, M.; Coleman, R.; Reddy, S.T.; Aviram, M. Paraoxonase 2 Attenuates Macrophage Triglyceride Accumulation via Inhibition of Diacylglycerol Acyltransferase 1. J. Lipid Res. 2009, 50, 870–879. [Google Scholar] [CrossRef]
- Qujeq, D.; Mahrooz, A.; Alizadeh, A.; Boorank, R. Paraoxonase-2 Variants Potentially Influence Insulin Resistance, Beta-Cell Function, and Their Interrelationships with Alanine Aminotransferase in Type 2 Diabetes. J. Res. Med. Sci. 2018, 23, 107. [Google Scholar] [CrossRef]
- Kielkopf, C.L.; Bauer, W.; Urbatsch, I.L. Bradford Assay for Determining Protein Concentration. Cold Spring Harb. Protoc. 2020, 2020, 102269. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J. Disc Electrophoresis—II Method and Application to Human Serum Proteins. Ann. N. Y. Acad. Sci. 2006, 121, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to Study Proteins by Circular Dichroism. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Moussong, É.; Murvai, N.; Tantos, Á.; Tőke, O.; Réfrégiers, M.; Wien, F.; Kardos, J. Disordered–Ordered Protein Binary Classification by Circular Dichroism Spectroscopy. Front. Mol. Biosci. 2022, 9, 863141. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.; Wong, C.-H. A pH Sensitive Colorometric Assay for the High-Throughput Screening of Enzyme Inhibitors and Substrates. Bioorg. Med. Chem. 2002, 10, 551–555. [Google Scholar] [CrossRef]
- Marone, M.; Porzio, E.; Lampitella, E.A.; Manco, G. A Mesophilic Phosphotriesterase-like Lactonase Shows High Stability and Proficiency as Quorum Quenching Enzyme. Chem.-Biol. Interact. 2023, 383, 110657. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Gene ID | Protein Name |
|---|---|---|
| 1 | PON2 | Paraoxonase2 |
| 2 | CPS1 | Carbamoyl-phosphate synthase [ammonia], mitochondrial |
| 3 | ATP5A1 | ATP synthase F1 subunit alpha |
| 4 | HNRNPM | Heterogeneous nuclear ribonucleoprotein M |
| 5 | PRDX1 | Peroxiredoxin1 |
| 6 | KRT1 | Keratin type II cytoskeletal 1 |
| 7 | KRT7 | Keratin type II cytoskeletal 7 |
| 8 | KRT8 | Keratin type II cytoskeletal 8 |
| 9 | KRT9 | Keratin type II cytoskeletal 9 |
| 10 | KRT14 | Keratin type II cytoskeletal 14 |
| 11 | KRT18 | Keratin type II cytoskeletal 18 |
| 12 | RPL13 | 60S ribosomal protein L13 |
| 13 | RPL24 | 60S ribosomal protein L24 |
| 14 | EEF1A1 | Elongation factor 1°1 |
| 15 | EEF2 | Elongation factor 2 |
| 16 | EFTU | Tu translation elongation factor, mitochondrial |
| 17 | HSPA8 | Hsp70-binding protein 1 |
| 18 | RAN | GTP-binding nuclear protein Ran |
| 19 | TKT | Transketolase |
| 20 | PCBP1 | Poly(rC)-binding protein 1 |
| 21 | TUBA1B | Tubulin A1B |
| 22 | TUBB4B | Tubulin 4B |
| 23 | PRKAR2A | cAMP-dependent protein kinase type II alpha regulatory subunit |
| 24 | PRKAR2B | cAMP-dependent protein kinase type II beta regulatory subunit |
| 25 | METTL2B | tRNA N(3)-methylcytidine methyltransferase |
| 26 | SLC25A4 | ADP/ATP translocase 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lampitella, E.A.; Marone, M.; Achanta, N.S.K.; Porzio, E.; Trepiccione, F.; Manco, G. The Human Paraoxonase 2: An Optimized Procedure for Refolding and Stabilization Facilitates Enzyme Analyses and a Proteomics Approach. Molecules 2024, 29, 2434. https://doi.org/10.3390/molecules29112434
Lampitella EA, Marone M, Achanta NSK, Porzio E, Trepiccione F, Manco G. The Human Paraoxonase 2: An Optimized Procedure for Refolding and Stabilization Facilitates Enzyme Analyses and a Proteomics Approach. Molecules. 2024; 29(11):2434. https://doi.org/10.3390/molecules29112434
Chicago/Turabian StyleLampitella, Eros A., Maria Marone, Nagendra S. K. Achanta, Elena Porzio, Francesco Trepiccione, and Giuseppe Manco. 2024. "The Human Paraoxonase 2: An Optimized Procedure for Refolding and Stabilization Facilitates Enzyme Analyses and a Proteomics Approach" Molecules 29, no. 11: 2434. https://doi.org/10.3390/molecules29112434