
Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids—An Overview

Abstract
1. Introduction
1.1. The Enigma of Flavonoids’ Antioxidant Activity
- The myriad of reactive species used in the assays varies from ‘simple’ highly reactive oxygen radicals to complex relatively stable synthetic radicals.
- The degradation of the unstable compounds during the experiments.
- Compartmentation, e.g., of lipophilic compounds, in cell culture experiments.
1.2. The Application of Computational Chemistry in Exploring the Antioxidant Activity of Flavonoids
2. Preliminary Statistics on Research Status
3. Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids
3.1. Electronic Structure Analysis
3.1.1. Frontier Molecular Orbitals (FMOs)
3.1.2. Molecular Electrostatic Potential (MEP/ESP)
3.1.3. Global Chemical Reactivity Descriptors (GCRDs)
3.1.4. Natural Bond Orbitals (NBOs)
3.1.5. Natural Transition Orbitals (NTOs)
3.1.6. Spin Density Distribution (SDD)
3.1.7. Polarity and Dipole Moment
3.1.8. Fukui Function (FF)
3.1.9. Atom Charge
3.1.10. Redox Potential
3.1.11. Density-of-States (DOS)
3.1.12. Donator–Acceptor Map (DAM)
3.1.13. Electron Localization Function (ELF) and Localization Orbital Locator (LOL)
3.2. Thermodynamic Analysis
3.2.1. Related Enthalpies of Different Mechanisms for Direct Scavenging of Free Radicals
3.2.2. The Reorganization Enthalpy (RE) and Hydrogen Abstraction Energy (HAE)
3.3. Kinetic Studies
3.4. Interaction Analysis
3.4.1. Quantum Theory of Atoms in Molecules (QTAIM)
3.4.2. Reduced Density Gradient (RDG) and Noncovalent Interactions (NCI) Analysis
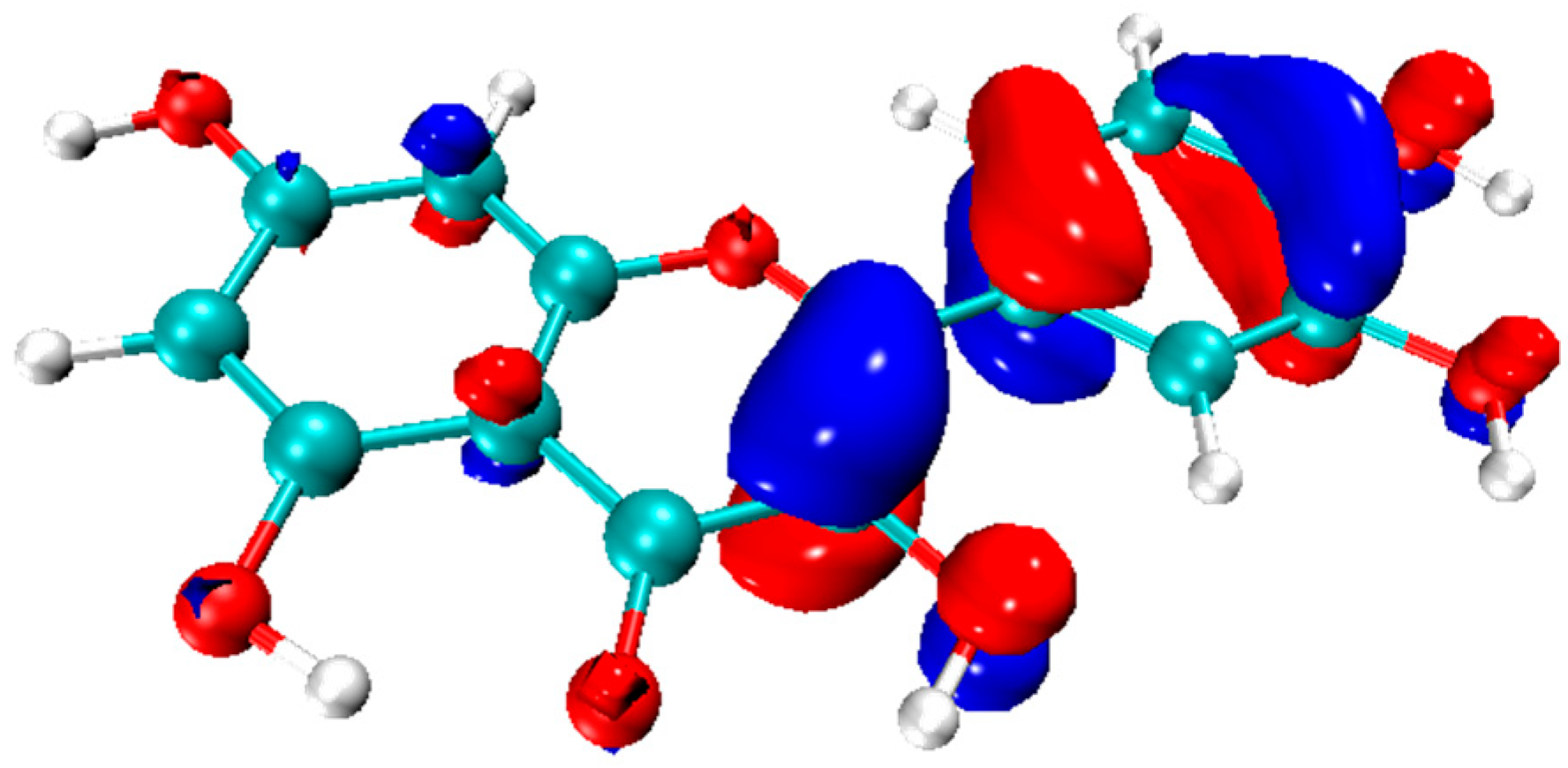
- RDG is a dimensionless parameter utilized to characterize electronic nonuniformity in density functional theory. In 2010, Professor Yang introduced the noncovalent interaction (NCI) analysis method based on RDG, to study the interactions between atoms or molecules [190]. While the electron localization function and QTAIM theory are suitable for analyzing chemical bonds, they possess certain limitations when examining non-covalent interactions like hydrogen bonds, steric hindrance, and π–π stacking. NCI analysis is a potent tool to reveal these non-covalent interactions.
- NCI analysis can intuitively and comprehensively depict the distribution of weak interactions of flavonoid molecules [27,58]. In NCI analysis, the interaction types are visualized using a color gradient, varying from blue to green indicating a gradual weakening of weak interactions, while red denotes increased resistance. An example is given in Figure 7.
3.4.3. Molecular Docking (MD) and Molecular Dynamics Simulations (MDS)
- Rigid docking: both the conformation of ligands and the receptors are fixed.
- Semiflexible docking: only the conformation of the ligands can change freely.
- Flexible docking: the conformation of both the ligands and the receptors can change freely.
3.5. Bioavailability Analysis
3.5.1. Dissociation Constant (pKa)
3.5.2. Lipid Water Partition Coefficient (LogP and LogD)
3.5.3. Polar Surface Area (PSA)
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Munir, S.; Badshah, S.L.; Khan, N.; Ghani, L.; Poulson, B.G.; Emwas, A.-H.; Jaremko, M. Important Flavonoids and Their Role as a Therapeutic Agent. Molecules 2020, 25, 5243. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Fraser, T.R. V.—On the connection between chemical constitution and physiological action. part. i.—On the physiological action of the salts of the ammonium bases, derived from Strychnia, Brucia, Thebaia, Codeia, Morphia, and Nicotia. Earth Environ. Sci. Trans. R. Soc. Edinb. 1868, 25, 151–203. [Google Scholar] [CrossRef]
- Li, Z.; Moalin, M.; Zhang, M.; Vervoort, L.; Hursel, E.; Mommers, A.; Haenen, G.R.M.M. The Flow of the Redox Energy in Q during Its Antioxidant Activity in Water. Int. J. Mol. Sci. 2020, 21, 6015. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, M.; Haenen, G.R.M.M.; Vervoort, L.; Moalin, M. Flavonoids Seen through the Energy Perspective. Int. J. Mol. Sci. 2022, 23, 187. [Google Scholar] [CrossRef] [PubMed]
- Gorbachev, M.; Gorinchoy, N.; Balan, I.; Arsene, I. Electronic Structure-Antioxidant Action Relationships for Chemical Compounds: A Quantum Chemical Study. In Fundamental and Biomedical Aspects of Redox Processes; IGI Global: Hershey, PA, USA, 2023; pp. 143–158. [Google Scholar]
- Spiegel, M. Current Trends in Computational Quantum Chemistry Studies on Antioxidant Radical Scavenging Activity. J. Chem. Inf. Model. 2022, 62, 2639–2658. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Irfan, A.; Khalid, M.; Khalid, N.; Uddin, J.; Hussain, R.; Ali, B.; Hussien, M.; Assiri, M.A.; Al-Sehemi, A.G. In-Vitro and in-Silico Antioxidant, α-Glucosidase Inhibitory Potentials of Abutilins C and D, New Flavonoide Glycosides from Abutilon Pakistanicum. Arab. J. Chem. 2021, 14, 103021. [Google Scholar] [CrossRef]
- Roquete Amparo, T.; Cherem Peixoto da Silva, A.; Brandão Seibert, J.; dos Santos da Silva, D.; Martins Rebello dos Santos, V.; Melo de Abreu Vieira, P.; Célio Brandão, G.; Henrique Bianco de Souza, G.; Aloise Maneira Corrêa Santos, B. In Vitro and in Silico Investigation of the Photoprotective and Antioxidant Potential of Protium Spruceanum Leaves and Its Main Flavonoids. J. Photochem. Photobiol. Chem. 2022, 431, 114037. [Google Scholar] [CrossRef]
- Anbazhakan, K.; Praveena, R.; Sadasivam, K.; Salgado, G.; Cardona, W.; Gerli, L.; Alvarado-Soto, L.; Ramirez-Tagle, R. Theoretical Insight on Structural Activities and Targets of Kaempferol Glycosides. Afinidad J. Chem. Eng. Theor. Appl. Chem. 2021, 78, 236–239. [Google Scholar]
- Ninh The, S.; Do Minh, T.; Nguyen Van, T. Isoflavones and Isoflavone Glycosides: Structural-Electronic Properties and Antioxidant Relations—A Case of DFT Study. J. Chem. 2019, 2019, 4360175. [Google Scholar] [CrossRef]
- Yang, X.; Wang, T.; Guo, J.; Sun, M.; Wong, M.W.; Huang, D. Dietary Flavonoids Scavenge Hypochlorous Acid via Chlorination on A- and C-Rings as Primary Reaction Sites: Structure and Reactivity Relationship. J. Agric. Food Chem. 2019, 67, 4346–4354. [Google Scholar] [CrossRef] [PubMed]
- de Souza Farias, S.A.; da Costa, K.S.; Martins, J.B.L. Analysis of Conformational, Structural, Magnetic, and Electronic Properties Related to Antioxidant Activity: Revisiting Flavan, Anthocyanidin, Flavanone, Flavonol, Isoflavone, Flavone, and Flavan-3-Ol. ACS Omega 2021, 6, 8908–8918. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Ahuja, D. QSAR Studies of Flavonoids Derivatives for Antioxidant and Antimicrobial Activity. J. Drug Deliv. Ther. 2019, 9, 765–773. [Google Scholar]
- Bitew, M.; Desalegn, T.; Demissie, T.B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R. Pharmacokinetics and Drug-Likeness of Antidiabetic Flavonoids: Molecular Docking and DFT Study. PLoS ONE 2021, 16, e0260853. [Google Scholar] [CrossRef] [PubMed]
- Messaadia, L.; Bekkar, Y.; Benamira, M.; Lahmar, H. Predicting the Antioxidant Activity of Some Flavonoids of Arbutus Plant: A Theoretical Approach. Chem. Phys. Impact 2020, 1, 100007. [Google Scholar] [CrossRef]
- Huy, N.T.; Trang, N.V.; Xuyen, N.T.; Hong, C.T.; Anh, V.T.K.; Thai, V.Q.; Cuong, B.V.; Van, T.T.T.; Trang, N.V.; Lam, T.D.; et al. Studies on the Antioxidant Activity of Apigenin, Luteolin and Nevadensin Using DFT. Vietnam. J. Sci. Technol. 2021, 59, 19–29. [Google Scholar] [CrossRef]
- Irfan, A.; Imran, M.; Khalid, M.; Sami Ullah, M.; Khalid, N.; Assiri, M.A.; Thomas, R.; Muthu, S.; Raza Basra, M.A.; Hussein, M.; et al. Phenolic and Flavonoid Contents in Malva Sylvestris and Exploration of Active Drugs as Antioxidant and Anti-COVID19 by Quantum Chemical and Molecular Docking Studies. J. Saudi Chem. Soc. 2021, 25, 101277. [Google Scholar] [CrossRef]
- Elsharkawy, E.R.; Almalki, F.; Ben Hadda, T.; Rastija, V.; Lafridi, H.; Zgou, H. DFT Calculations and POM Analyses of Cytotoxicity of Some Flavonoids from Aerial Parts of Cupressus Sempervirens: Docking and Identification of Pharmacophore Sites. Bioorg. Chem. 2020, 100, 103850. [Google Scholar] [CrossRef]
- Meshram, R.J.; Bagul, K.T.; Pawnikar, S.P.; Barage, S.H.; Kolte, B.S.; Gacche, R.N. Known Compounds and New Lessons: Structural and Electronic Basis of Flavonoid-Based Bioactivities. J. Biomol. Struct. Dyn. 2020, 38, 1168–1184. [Google Scholar] [CrossRef]
- Boulebd, H. The Role of Benzylic-Allylic Hydrogen Atoms on the Antiradical Activity of Prenylated Natural Chalcones: A Thermodynamic and Kinetic Study. J. Biomol. Struct. Dyn. 2021, 39, 1955–1964. [Google Scholar] [CrossRef]
- Bartella, L.; Mazzotti, F.; Talarico, I.R.; De Luca, G.; Santoro, I.; Prejanò, M.; Riccioni, C.; Marino, T.; Di Donna, L. Structural Characterization of Peripolin and Study of Antioxidant Activity of HMG Flavonoids from Bergamot Fruit. Antioxidants 2022, 11, 1847. [Google Scholar] [CrossRef]
- Mustafa, S.R.; Al-Ani, H.N. Calculation of Vibrational Frequencies, Energetic and Some Other Quantum Chemical Parameters for Some Flavonoids. J. Phys. Conf. Ser. 2021, 1999, 012018. [Google Scholar] [CrossRef]
- Jalezadeh, A.; Mirjafary, Z.; Rouhani, M.; Saeidian, H. Investigation of Structural, Electronic, and Antioxidant Properties of Calycopetrin and Xanthomicrol as Two Polymethoxylated Flavones Using DFT Calculations. Struct. Chem. 2022, 33, 1241–1250. [Google Scholar] [CrossRef]
- Veiko, A.G.; Lapshina, E.A.; Zavodnik, I.B. Comparative Analysis of Molecular Properties and Reactions with Oxidants for Q, Catechin, and Naringenin. Mol. Cell. Biochem. 2021, 476, 4287–4299. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Q.; Li, Y.; Wang, S.; Yang, F.; Zhao, X. How the Functional Group Substitution and Solvent Effects Affect the Antioxidant Activity of (+)-Catechin? J. Mol. Liq. 2021, 327, 114818. [Google Scholar] [CrossRef]
- Deviani, V.; Hardianto, A.; Farabi, K.; Herlina, T. Flavanones from Erythrina Crista-Galli Twigs and Their Antioxidant Properties Determined through In Silico and In Vitro Studies. Molecules 2022, 27, 6018. [Google Scholar] [CrossRef]
- Santos, K.L.B.; Queiroz, A.N.; Lobato, C.C.; Vale, J.K.L.; Santos, C.B.R.; Borges, R.S. A Comparative Theoretical Mechanism on Simplified Flavonoid Derivatives and Isoxazolone Analogous as Michael System Inhibitor. J. Mol. Model. 2021, 27, 26. [Google Scholar] [CrossRef]
- Faria, E.C.M.; Duarte, V.S.; Oliveira, A.M. A trimethoxy-chalcone applied as antioxidant and antibacterial additive for diesel and biodiesel blend. In Proceedings of the XXIX International Symposium on Automotive Engineering—SIMEA 2022, São Paulo, Brazil, 17–18 August 2022. [Google Scholar]
- Moreira, C.A.; Faria, E.C.; Queiroz, J.E.; Duarte, V.S.; Gomes, M.D.N.; da Silva, A.M.; de Paula, R.L.G.; Franco, C.H.; Cavalcanti, E.H.D.S.; de Aquino, G.L.; et al. Structural Insights and Antioxidant Analysis of a Tri-Methoxy Chalcone with Potential as a Diesel-Biodiesel Blend Additive. Fuel Process. Technol. 2022, 227, 107122. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, Y.; Zhang, L. Antioxidant and spectral properties of chalcones and analogous aurones: Theoretical insights. Int. J. Quantum Chem. 2019, 119, e25808. [Google Scholar] [CrossRef]
- Shang, C.; Zhang, Y.; Sun, C.; Wang, L. Tactfully Improve the Antioxidant Activity of 2′-Hydroxychalcone with the Strategy of Substituent, Solvent and Intramolecular Hydrogen Bond Effects. J. Mol. Liq. 2022, 362, 119748. [Google Scholar] [CrossRef]
- Siddiqa, A.; Tajammal, A.; Irfan, A.; Azam, M.; Munawar, M.A.; Hardy, R.S.; Basra, M.A.R. Synthesis, Molecular Docking, Bio-Evaluation and Quantitative Structure Activity Relationship of New Chalcone Derivatives as Antioxidants. J. Mol. Struct. 2023, 1277, 134814. [Google Scholar] [CrossRef]
- Khalili, A.; Baei, M.T.; Ghaboos, S.H.H. Chrysin Flavonoid Adsorbed on B12N12 Nanocage—A Novel Antioxidant Nanomaterial. Vietnam J. Chem. 2021, 59, 211–220. [Google Scholar] [CrossRef]
- Szewczuk, N.A.; Duchowicz, P.R.; Pomilio, A.B.; Lobayan, R.M. Resonance Structure Contributions, Flexibility, and Frontier Molecular Orbitals (HOMO–LUMO) of Pelargonidin, Cyanidin, and Delphinidin throughout the Conformational Space: Application to Antioxidant and Antimutagenic Activities. J. Mol. Model. 2022, 29, 2. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, F.; Cao, Y.; Cai, G.; Wei, Q.; Shi, S.; Guo, Y. Screening of Potent α-Glucosidase Inhibitory and Antioxidant Polyphenols in Prunella vulgaris L. by Bioreaction–HPLC–Quadrupole-Time-of-Flight-MS/MS and in Silico Analysis. J. Sep. Sci. 2022, 45, 3393–3403. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ma, Y.; Li, H.; Chen, X.; Zhang, C.; Wang, H.; Deng, Z. The Antioxidant Activity and Active Sites of Delphinidin and Petunidin Measured by DFT, in Vitro Chemical-Based and Cell-Based Assays. J. Food Biochem. 2019, 43, e12968. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Fu, F.; Wei, Y.; Shi, S.; Shan, Y. Online Extraction–DPPH–HPLC–DAD–QTOF-MS System for Efficient Screening and Identification of Antioxidants from Citrus aurantium L. Var. Amara (Rutaceae): Integrating Sample Preparation and Antioxidants Profiling. Antioxidants 2022, 11, 1014. [Google Scholar] [CrossRef] [PubMed]
- Boulmokh, Y.; Belguidoum, K.; Meddour, F.; Amira-Guebailia, H. Investigation of Antioxidant Activity of Epigallocatechin Gallate and Epicatechin as Compared to Resveratrol and Ascorbic Acid: Experimental and Theoretical Insights. Struct. Chem. 2021, 32, 1907–1923. [Google Scholar] [CrossRef]
- Boulebd, H. Structure-Activity Relationship of Antioxidant Prenylated (Iso)Flavonoid-Type Compounds: Quantum Chemistry and Molecular Docking Studies. J. Biomol. Struct. Dyn. 2022, 40, 10373–10382. [Google Scholar] [CrossRef]
- Menacer, R.; Rekkab, S.; Kabouche, Z. Fisetin and Robinetin Antiradical Activity under Solvent Effect: Density Functional Theory Study. J. Mol. Model. 2022, 28, 240. [Google Scholar] [CrossRef]
- Pop, R. Evaluation of the Radical-Scavenging Properties of Various Flavonols in Ethanol Environment: An Ab Initio Study. Croat. Chem. Acta 2019, 92, 337–346. [Google Scholar] [CrossRef]
- Ugodi, G.W.; Asogwa, F.C.; Paul, E.; Asogwa, C.J. Reactivity Indexes of Antioxidant Molecules from Psorospermum Febrifugum. J. Pharmacogn. Phytochem. 2023, 12, 14–18. [Google Scholar] [CrossRef]
- Ji, W.; Li, Z.; Gu, L.; Zou, X.; Wu, J.; Zhang, S.; Deng, H. Differences in the Composition of Archidendron Clypearia at Different Harvest Periods and Spectrum–Effect Relationship and Structure–Activity Analysis of Antioxidant Components. SSRN Electron. J. 2022, preprint. [Google Scholar] [CrossRef]
- Razooqi, M.S.; Al-Ani, H.N. Quantum Mechanical Calculations and Electrochemical Study of Vibrational Frequencies, Energies in Some Flavonoids Molecules. Iraqi J. Sci. 2022, 63, 2331–2344. [Google Scholar] [CrossRef]
- Lewandowski, W.; Lewandowska, H.; Golonko, A.; Świderski, G.; Świsłocka, R.; Kalinowska, M. Correlations between Molecular Structure and Biological Activity in “Logical Series” of Dietary Chromone Derivatives. PLoS ONE 2020, 15, e0229477. [Google Scholar] [CrossRef]
- Rammohan, A.; Bhaskar, B.V.; Camilo, A.; Gunasekar, D.; Gu, W.; Zyryanov, G.V. In Silico, in Vitro Antioxidant and Density Functional Theory Based Structure Activity Relationship Studies of Plant Polyphenolics as Prominent Natural Antioxidants. Arab. J. Chem. 2020, 13, 3690–3701. [Google Scholar] [CrossRef]
- Bağlan, M.; Yildiko, Ü.; Gören, K. Computational Investigation of 5.5’’,7’’-Trihydroxy-3,7-Dimethoxy-4’-4’’-O-Biflavone from Flavonoids Using DFT Calculations and Molecular Docking. Adıyaman Univ. J. Sci. 2022, 12, 283–298. [Google Scholar] [CrossRef]
- Anbazhakan, K.; Praveena, R.; Sadasivam, K. Theoretical Insight on Antioxidant Potency of Kanzakiflavone-2 and Its Derivatives. Struct. Chem. 2021, 32, 1451–1458. [Google Scholar] [CrossRef]
- Praveena, A.; Prabu, S.; Madi, F.; Rajamohan, R. Theoretical Investigation of Inclusion Complexes of 3-Hydroxyflavone and Q as Guests with Native and Modified β-Cyclodextrins as Hosts. Polycycl. Aromat. Compd. 2023, 43, 141–153. [Google Scholar] [CrossRef]
- El-Hadidy, E.; Ali, M. Theoretical Study, Antioxidant Activity and Anti-Cancer Studies of Galangal (Alpinia Galangal). Int. J. Curr. Res. 2020, 7, 101–145. [Google Scholar]
- Balanescu, F.; Busuioc, A.C.; Botezatu, A.V.D.; Gosav, S.; Avramescu, S.M.; Furdui, B.; Dinica, R.M. Comparative Study of Natural Antioxidants from Glycine Max, Anethum Graveolensand Pimpinella Anisum Seed and Sprout Extracts Obtained by Ultrasound-Assisted Extraction. Separations 2022, 9, 152. [Google Scholar] [CrossRef]
- Son, N.T.; Mai Thanh, D.T.; Van Trang, N. Flavone Norartocarpetin and Isoflavone 2′-Hydroxygenistein: A Spectroscopic Study for Structure, Electronic Property and Antioxidant Potential Using DFT (Density Functional Theory). J. Mol. Struct. 2019, 1193, 76–88. [Google Scholar] [CrossRef]
- Wang, R.; Li, W.; Fang, C.; Zheng, X.; Liu, C.; Huang, Q. Identification of New Flavonoid Compounds in Dandelion Taraxacum Mongolicum Hand.-Mazz. and Evaluation of Their Antioxidant Activities. Res. Sq. 2022. submitted. [Google Scholar]
- Wang, R.; Li, W.; Fang, C.; Zheng, X.; Liu, C.; Huang, Q. Extraction and Identification of New Flavonoid Compounds in Dandelion Taraxacum Mongolicum Hand.-Mazz. with Evaluation of Antioxidant Activities. Sci. Rep. 2023, 13, 2166. [Google Scholar] [CrossRef]
- Sulaiman, G.M.; Waheeba, H.M.; AL-Shmgani, H.; Eassa, H.A.; Al-Amiery, A.A.; Jabir, M.S.; Dewir, Y.H.; Alwahibi, M.S.; Soliman, D.A. Synthesis, Molecular Modeling, DNA Damage Interaction, and Antioxidant Potential of Hesperidin Loaded on Gold Nanoparticles. J. Biomim. Biomater. Biomed. Eng. 2022, 54, 17–29. [Google Scholar] [CrossRef]
- Ragi, C.; Muraleedharan, K. Antioxidant Activity of Hibiscetin and Hibiscitrin: Insight from DFT, NCI, and QTAIM. Theor. Chem. Acc. 2023, 142, 30. [Google Scholar] [CrossRef]
- Hu, Y.; Liang, P.; Wang, Z.; Jiang, C.; Zeng, Q.; Shen, C.; Wu, Y.; Liu, L.; Yi, Y.; Zhu, H.; et al. Explore the Effect of the Structure-Activity Relationship and Dose-Effect Relationship on the Antioxidant Activity of Licorice Flavonoids. J. Mol. Struct. 2023, 1292, 136101. [Google Scholar] [CrossRef]
- Enisoğlu Atalay, V.; Ölüç, İ.B. Antioxidant Activity of the Hazelnut Plant Determination by Computational Chemistry Methods. Main Group Chem. 2021, 19, 273–282. [Google Scholar] [CrossRef]
- Vilas-Boas, I.T.; da Silva, A.C.P.; de AF Accioli, C.; Amorim, J.M.; Leite, P.M.; Faraco, A.A.G.; Santos, B.A.M.C.; Scopel, M.; Castilho, R.O. Optimized Baccharis Dracunculifolia Extract as Photoprotective and Antioxidant: In Vitro and in Silico Assessment. J. Photochem. Photobiol. Chem. 2023, 440, 114654. [Google Scholar] [CrossRef]
- Santos, S.C.; Fortes, G.A.C.; Camargo, L.T.F.M.; Camargo, A.J.; Ferri, P.H. Antioxidant Effects of Polyphenolic Compounds and Structure-Activity Relationship Predicted by Multivariate Regression Tree. LWT 2021, 137, 110366. [Google Scholar] [CrossRef]
- Babiaka, S.B.; Nia, R.; Abuga, K.O.; Mbah, J.A.; Vincent de Paul, N.N.; Paper, D.H.; Ntie-Kang, F. Antioxidant Potential of Flavonoid Glycosides from Manniophyton Fulvum Müll. (Euphorbiaceae): Identification and Molecular Modeling. Sci. Afr. 2020, 8, e00423. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, Z.; Shen, C.; Jiang, C.; Zhu, Z.; Liang, P.; Li, H.; Zeng, Q.; Xue, Y.; Wu, Y.; et al. Influence of the pKa Value on the Antioxidant Activity of Licorice Flavonoids under Solvent-Mediated Effects. Arch. Pharm. 2023, 356, 2200470. [Google Scholar] [CrossRef] [PubMed]
- Kenouche, S.; Sandoval-Yañez, C.; Martínez-Araya, J.I. The Antioxidant Capacity of Myricetin. A Molecular Electrostatic Potential Analysis Based on DFT Calculations. Chem. Phys. Lett. 2022, 801, 139708. [Google Scholar] [CrossRef]
- Rajan, V.K.; Ragi, C.; Muraleedharan, K. A Computational Exploration into the Structure, Antioxidant Capacity, Toxicity and Drug-like Activity of the Anthocyanidin “Petunidin”. Heliyon 2019, 5, e02115. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, M. Evaluation of Structural Properties and Antioxidant Capacity of Proxison: A DFT Investigation. Comput. Theor. Chem. 2021, 1195, 113096. [Google Scholar] [CrossRef]
- Thuy, P.T.; Quan, P.M.; Duc, D.X.; Son, N.T. The Antioxidative Potential of Procyanidin B1: DFT (Density Functional Theory) and Docking Approaches. J. Mol. Model. 2022, 28, 356. [Google Scholar] [CrossRef] [PubMed]
- da Silva Pantoja, L.V.P.; Trindade, S.S.A.; da Silva Carneiro, A.; Silva, J.P.B.; da Paixão, T.P.; Romeiro, C.F.R.; de Moraes, C.S.P.; Pinto, A.C.G.; Raposo, N.R.B.; de Andrade, M.A. Computational Study of the Main Flavonoids from Chrysobalanus icaco L. against NADPH-Oxidase and in Vitro Antioxidant Activity. Res. Soc. Dev. 2022, 11, e5011628542. [Google Scholar] [CrossRef]
- Borges, R.S.; Aguiar, C.P.O.; Oliveira, N.L.L.; Amaral, I.N.A.; Vale, J.K.L.; Chaves Neto, A.M.J.; Queiroz, A.N.; da Silva, A.B.F. Antioxidant Capacity of Simplified Oxygen Heterocycles and Proposed Derivatives by Theoretical Calculations. J. Mol. Model. 2023, 29, 232. [Google Scholar] [CrossRef] [PubMed]
- Kiraz, A.Ö.; Yalçın, F. Structure-Activity and Antioxidant Properties of Q and Its Co2+ Chelate. Int. J. Second. Metab. 2021, 8, 414–424. [Google Scholar] [CrossRef]
- Thao, T.H.D.; Dung, V.T.N.; Dao, D.Q. Antioxidant vs. pro-Oxidant Activities of Q in Aqueous Phase: A Density Functional Theory Study. Vietnam J. Chem. 2019, 57, 696–701. [Google Scholar] [CrossRef]
- Aydın, L.; Özpozan, T. NBO, HOMO–LUMO, Conformational and Vibrational Spectroscopic Study of 2-(3,4-Dihydroxyphenyl)-5,7-Dihydroxy-3-[(2S,3R,4S,5R,6R)-3,4,5-Trihydroxy-6-(Hydroxymethyl) Oxan-2-Yl]-Oxychromen-4-One, as the Main Phenolic Component of the Extracts of Walnut Leaves by DFT. J. Iran. Chem. Soc. 2021, 18, 1067–1079. [Google Scholar]
- Boshkayeva, A.K.; Omarova, R.A.; Ordabayeva, S.K.; Serikbayeva, A.D.; Umurzakhova, G.G.; Massakbayev, A.J. Modeling of the Structure and Forecasting Properties of DihydroQ Derivatives. Drug Dev. Ind. Pharm. 2022, 48, 52–57. [Google Scholar] [CrossRef]
- Son, N.T.; Thuy, P.T.; Van Trang, N. Antioxidative Capacities of Stilbenoid Suaveolensone A and Flavonoid Suaveolensone B: A Detailed Analysis of Structural-Electronic Properties and Mechanisms. J. Mol. Struct. 2021, 1224, 129025. [Google Scholar] [CrossRef]
- Siddiqa, A.; Tajammal, A.; Irfan, A.; Munawar, M.A.; Azam, M.; Basra, M.A.R. Synthesis, Antioxidant, in Silico and Computational Investigation of 2,5-Dihydroxyacetophenone Derived Chloro-Substituted Hydroxychalcones, Hydroxyflavanones and Hydroxyflavindogenides. J. Biomol. Struct. Dyn. 2022, 40, 10265–10277. [Google Scholar] [CrossRef]
- Martínez-Busi, M.; Arredondo, F.; González, D.; Echeverry, C.; Vega-Teijido, M.A.; Carvalho, D.; Rodríguez-Haralambides, A.; Rivera, F.; Dajas, F.; Abin-Carriquiry, J.A. Purification, Structural Elucidation, Antioxidant Capacity and Neuroprotective Potential of the Main Polyphenolic Compounds Contained in Achyrocline Satureioides (Lam) D.C. (Compositae). Bioorg. Med. Chem. 2019, 27, 2579–2591. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Sun, C.; Wang, L. Understanding Prominent Effects of the Intramolecular Hydrogen Bond on the Photophysical Properties and Antiradical Abilities of Six Flavonoids. J. Mol. Liq. 2023, 386, 122534. [Google Scholar] [CrossRef]
- Abbo, H.S.; Hung Lai, C.; Titinchi, S.J.J. Substituent and Solvent Effects on UV-visible Absorption Spectra of Chalcones Derivatives: Experimental and Computational Studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2023, 303, 123180. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, Y.; Gao, L. Mechanism of Antioxidant Properties of Q and Q-DNA Complex. J. Mol. Model. 2020, 26, 133. [Google Scholar] [CrossRef]
- Nam, G.; Hong, M.; Lee, J.; Jin Lee, H.; Ji, Y.; Kang, J.; Baik, M.-H.; Hee Lim, M. Multiple Reactivities of Flavonoids towards Pathological Elements in Alzheimer’s Disease: Structure–Activity Relationship. Chem. Sci. 2020, 11, 10243–10254. [Google Scholar] [CrossRef] [PubMed]
- Halevas, E.; Matsia, S.; Hatzidimitriou, A.; Geromichalou, E.; Papadopoulos, T.A.; Katsipis, G.; Pantazaki, A.; Litsardakis, G.; Salifoglou, A. A Unique Ternary Ce(III)-Q-Phenanthroline Assembly with Antioxidant and Anti-Inflammatory Properties. J. Inorg. Biochem. 2022, 235, 111947. [Google Scholar] [CrossRef]
- Bulat, F.A.; Murray, J.S.; Politzer, P. Identifying the Most Energetic Electrons in a Molecule: The Highest Occupied Molecular Orbital and the Average Local Ionization Energy. Comput. Theor. Chem. 2021, 1199, 113192. [Google Scholar] [CrossRef]
- Akhtari, K.; Hassanzadeh, K.; Fakhraei, B.; Fakhraei, N.; Hassanzadeh, H.; Zarei, S.A. A Density Functional Theory Study of the Reactivity Descriptors and Antioxidant Behavior of Crocin. Comput. Theor. Chem. 2013, 1013, 123–129. [Google Scholar] [CrossRef]
- Reza Nazifi, S.M.; Asgharshamsi, M.H.; Dehkordi, M.M.; Zborowski, K.K. Antioxidant Properties of Aloe Vera Components: A DFT Theoretical Evaluation. Free Radic. Res. 2019, 53, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.E. Gaussian ~09 Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Volume 32, pp. 5648–5652. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2009; ISBN 978-0-470-74659-2. [Google Scholar]
- Huq, F. Molecular Modelling Analysis of the Metabolism of Fentanyl. J. Pharmacol. Toxicol. 2007, 2, 176–182. [Google Scholar] [CrossRef]
- Zainuri, D.A.; Razak, I.A.; Arshad, S. Crystal Structure, Spectroscopic Characterization and DFT Study of Two New Linear Fused-Ring Chalcones. Acta Crystallogr. Sect. E Crystallogr. Commun. 2018, 74, 1427–1432. [Google Scholar] [CrossRef]
- da Silva, R.R.; Ramalho, T.C.; Santos, J.M.; Figueroa-Villar, J.D. Reply to “Comment on the Paper ‘On the Limits of Highest-Occupied Molecular Orbital Driven Reactions: The Frontier Effective-for-Reaction Molecular Orbital Concept’”. J. Phys. Chem. A 2006, 110, 10653–10654. [Google Scholar] [CrossRef]
- Dewar, M.J.S. A Critique of Frontier Orbital Theory. J. Mol. Struct. Theochem. 1989, 200, 301–323. [Google Scholar] [CrossRef]
- Anderson, J.S.M.; Melin, J.; Ayers, P.W. Conceptual Density-Functional Theory for General Chemical Reactions, Including Those That Are Neither Charge- nor Frontier-Orbital-Controlled. 2. Application to Molecules Where Frontier Molecular Orbital Theory Fails. J. Chem. Theory Comput. 2007, 3, 375–389. [Google Scholar] [CrossRef]
- Svatunek, D.; Denk, C.; Mikula, H. A Computational Model to Predict the Diels–Alder Reactivity of Aryl/Alkyl-Substituted Tetrazines. Monatshefte Chem.-Chem. Mon. 2018, 149, 833–837. [Google Scholar] [CrossRef]
- Sjoberg, P.; Murray, J.S.; Brinck, T.; Politzer, P. Average Local Ionization Energies on the Molecular Surfaces of Aromatic Systems as Guides to Chemical Reactivity. Can. J. Chem. 1990, 68, 1440–1443. [Google Scholar] [CrossRef]
- Brinck, T.; Liljenberg, M. The Use of Quantum Chemistry for Mechanistic Analyses of SEAr Reactions. In Arene Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 83–105. ISBN 978-1-118-75488-7. [Google Scholar]
- Brinck, T.; Stenlid, J.H. The Molecular Surface Property Approach: A Guide to Chemical Interactions in Chemistry, Medicine, and Material Science. Adv. Theory Simul. 2019, 2, 1800149. [Google Scholar] [CrossRef]
- Jakobušić Brala, C.; Fabijanić, I.; Karković Marković, A.; Pilepić, V. The Average Local Ionization Energy and Fukui Function of L-Ascorbate, the Local Reactivity Descriptors of Antioxidant Reactivity. Comput. Theor. Chem. 2014, 1049, 1–6. [Google Scholar] [CrossRef]
- Fabijanić, I.; Jakobušić Brala, C.; Pilepić, V. The DFT Local Reactivity Descriptors of α-Tocopherol. J. Mol. Model. 2015, 21, 99. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, S.; Jeyasingh, V.; Murugesan, K.; Selvapalam, N.; Dass, G. Molecular Electrostatic Potential (MEP) Surface Analysis of Chemo Sensors: An Extra Supporting Hand for Strength, Selectivity & Non-Traditional Interactions. J. Photochem. Photobiol. 2021, 6, 100022. [Google Scholar]
- Liu, S.; Pedersen, L.G. Estimation of Molecular Acidity via Electrostatic Potential at the Nucleus and Valence Natural Atomic Orbitals. J. Phys. Chem. A 2009, 113, 3648–3655. [Google Scholar] [CrossRef] [PubMed]
- Rajan, V.K.; Muraleedharan, K. A Computational Investigation on the Structure, Global Parameters and Antioxidant Capacity of a Polyphenol, Gallic Acid. Food Chem. 2017, 220, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Vikramaditya, T.; Chai, J.-D.; Lin, S.-T. Impact of Non-Empirically Tuning the Range-Separation Parameter of Long-Range Corrected Hybrid Functionals on Ionization Potentials, Electron Affinities, and Fundamental Gaps. J. Comput. Chem. 2018, 39, 2378–2384. [Google Scholar] [CrossRef]
- Fassihi, A.; Hasanzadeh, F.; Attar, A.M.; Saghaie, L.; Mohammadpour, M. Synthesis and Evaluation of Antioxidant Activity of Some Novel Hydroxypyridinone Derivatives: A DFT Approach for Explanation of Their Radical Scavenging Activity. Res. Pharm. Sci. 2020, 15, 515–528. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness: Applications from Molecules to Solids; Wiley: Hoboken, NJ, USA, 1998; ISBN 978-3-527-29482-4. [Google Scholar]
- Yang, W.; Parr, R.G. Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef]
- hossein Asgarshamsi, M.; Fassihi, A.; Hassanzadeh, F.; Saghaei, L.; Attar, A.M.; Mohammad-Beigi, H. Synthesis, Antioxidant Activity, and Density Functional Theory Study of Some Novel 4-[(Benzo[d]Thiazol-2-Ylimino) Methyl] Phenol Derivatives: A Comparative Approach for the Explanation of Their Radical Scavenging Activities. Res. Pharm. Sci. 2020, 16, 35–47. [Google Scholar]
- Foresman, J.B. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems by David Young (Cytoclonal Pharmaceutics Inc.). J. Am. Chem. Soc. 2001, 123, 10142–10143. [Google Scholar] [CrossRef]
- Sheela, N.R.; Muthu, S.; Sampathkrishnan, S. Molecular Orbital Studies (Hardness, Chemical Potential and Electrophilicity), Vibrational Investigation and Theoretical NBO Analysis of 4-4′-(1H-1,2,4-Triazol-1-Yl Methylene)-Dibenzonitrile Based on Abinitio and DFT Methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 120, 237–251. [Google Scholar] [CrossRef]
- Gázquez, J.L.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Chen, D.-F.; Deng, G.; Guo, R. The Substituent Effect on the Radical Scavenging Activity of Apigenin. Molecules 2018, 23, 1989. [Google Scholar] [CrossRef]
- Pal, R.; Chattaraj, P.K. Chemical Reactivity from a Conceptual Density Functional Theory Perspective. J. Indian Chem. Soc. 2021, 98, 100008. [Google Scholar] [CrossRef]
- Weinhold, F. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 978-1-118-22919-4. [Google Scholar]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What Is NBO Analysis and How Is It Useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Resonance Natural Bond Orbitals: Efficient Semilocalized Orbitals for Computing and Visualizing Reactive Chemical Processes. Chem. Theory Comput. 2019, 15, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F. Natural Bond Orbital Analysis: A Critical Overview of Relationships to Alternative Bonding Perspectives. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef] [PubMed]
- Pahari, B.; Chakraborty, S.; Chaudhuri, S.; Sengupta, B.; Sengupta, P.K. Binding and Antioxidant Properties of Therapeutically Important Plant Flavonoids in Biomembranes: Insights from Spectroscopic and Quantum Chemical Studies. Chem. Phys. Lipids 2012, 165, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Cao, B.; Yin, H.; Shi, Y. Relationship between ESIPT Properties and Antioxidant Activities of 5-Hydroxyflavone Derivates. Chin. Phys. B 2020, 29, 058202. [Google Scholar] [CrossRef]
- Daday, C.; Manolescu, A.; Marinescu, D.C.; Gudmundsson, V. Electronic Charge and Spin Density Distribution in a Quantum Ring with Spin-Orbit and Coulomb Interactions. Phys. Rev. B 2011, 84, 115311. [Google Scholar] [CrossRef]
- Brehm, P.C.; Frontera, A.; Streubel, R. On Metal Coordination of Neutral Open-Shell P-Ligands Focusing on Phosphanoxyls, Their Electron Residence and Reactivity. Chem. Commun. 2022, 58, 6270–6279. [Google Scholar] [CrossRef]
- Pluta, T.; Kolaski, M.; Medved’, M.; Budzák, Š. Dipole Moment and Polarizability of the Low-Lying Excited States of Uracil. Chem. Phys. Lett. 2012, 546, 24–29. [Google Scholar] [CrossRef]
- Glevitzky, I.; Dumitrel, G.-A.; Mirel, G.; Pasca, M.B.; Otřísal, P.; Bungau, S.; Cioca, G.; Carmen, P.; Popa, M. Statistical Analysis of the Relationship Between Antioxidant Activity and the Structure of Flavonoid Compounds. Rev. Chim. 2019, 70, 7497. [Google Scholar] [CrossRef]
- Bentz, E.N.; Pomilio, A.B.; Lobayan, R.M. Exploratory Conformational Study of (+)-Catechin. Modeling of the Polarizability and Electric Dipole Moment. J. Mol. Model. 2014, 20, 2522. [Google Scholar] [CrossRef]
- Lobayan, R.M.; Bentz, E.N.; Jubert, A.H.; Pomilio, A.B. Structural and Electronic Properties of Z Isomers of (4α→6´´,2α→O→1´´)-Phenylflavans Substituted with R = H, OH and OCH3 Calculated in Aqueous Solution with PCM Solvation Model. J. Mol. Model. 2012, 18, 1667–1676. [Google Scholar] [CrossRef]
- Chermette, H. Density Functional Theory: A Powerful Tool for Theoretical Studies in Coordination Chemistry. Coord. Chem. Rev. 1998, 178–180, 699–721. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why Is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Wang, C.-S. Atom-Bond Electronegativity Equalization Method and Its Applications Based on Density Functional Theory. J. Theor. Comput. Chem. 2003, 2, 273–299. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Lei, J.; Liu, W.; Tong, M.; Liang, J. The Degradation Pathways of Carbamazepine in Advanced Oxidation Process: A Mini Review Coupled with DFT Calculation. Sci. Total Environ. 2021, 779, 146498. [Google Scholar] [CrossRef]
- Bardak, F. Experimental and DFT Analysis of Structural and Spectroscopic Features of Nitroterephthalic Acid, and Computational Insights into Its Molecular Interactions with hER-α via Molecular Docking. J. Mol. Struct. 2019, 1175, 458–470. [Google Scholar] [CrossRef]
- Abraham, C.S.; Muthu, S.; Prasana, J.C.; Armaković, S.; Armaković, S.J.; Geoffrey, B. Computational Evaluation of the Reactivity and Pharmaceutical Potential of an Organic Amine: A DFT, Molecular Dynamics Simulations and Molecular Docking Approach. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 222, 117188. [Google Scholar] [CrossRef]
- Djeradi, H.; Rahmouni, A.; Cheriti, A. Antioxidant Activity of Flavonoids: A QSAR Modeling Using Fukui Indices Descriptors. J. Mol. Model. 2014, 20, 2476. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R. Atomic Charges. J. Org. Chem. 2018, 83, 15463–15469. [Google Scholar] [CrossRef]
- Bultinck, P.; Langenaeker, W.; Lahorte, P.; De Proft, F.; Geerlings, P.; Waroquier, M.; Tollenaere, J.P. The Electronegativity Equalization Method I: Parametrization and Validation for Atomic Charge Calculations. J. Phys. Chem. A 2002, 106, 7887–7894. [Google Scholar] [CrossRef]
- Amic, D.; Davidovic-Amic, D.; Beslo, D.; Rastija, V.; Lucic, B.; Trinajstic, N. SAR and QSAR of the Antioxidant Activity of Flavonoids. Curr. Med. Chem. 2007, 14, 827–845. [Google Scholar] [CrossRef]
- Rajendran, M.; Ravichandran, R.; Devapiriam, D. Electronic Description of Few Selected Flavonoids by Theoretical Study. Int. J. Comput. Appl. 2013, 77, 18–25. [Google Scholar] [CrossRef]
- Šimunková, M.; Biela, M.; Štekláč, M.; Hlinčík, A.; Klein, E.; Malček, M. Cu(II) Complexes of Flavonoids in Solution: Impact of the Cu(II) Ion on the Antioxidant and DNA-Intercalating Properties. J. Mol. Liq. 2022, 359, 119230. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Comparison of Computational Methods for Atomic Charges. Acta Phys.-Chim. Sin. 2011, 28, 1–18. [Google Scholar]
- Ionescu, C.-M.; Sehnal, D.; Falginella, F.L.; Pant, P.; Pravda, L.; Bouchal, T.; Svobodová Vařeková, R.; Geidl, S.; Koča, J. AtomicChargeCalculator: Interactive Web-Based Calculation of Atomic Charges in Large Biomolecular Complexes and Drug-like Molecules. J. Cheminform. 2015, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Miličević, A.; Miletić, G.I.; Novak Jovanović, I. Electrochemical Oxidation of Flavonoids: PM6 and DFT for Elucidating Electronic Changes and Modelling Oxidation Potential (Part II). J. Mol. Liq. 2019, 295, 111730. [Google Scholar] [CrossRef]
- Park, S.; Kim, M.; Lin, Y.; Hong, M.; Nam, G.; Mieczkowski, A.; Kardos, J.; Lee, Y.-H.; Hee Lim, M. Designing Multi-Target-Directed Flavonoids: A Strategic Approach to Alzheimer’s Disease. Chem. Sci. 2023, 14, 9293–9305. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Agwupuye, J.A.; Louis, H.; Unimuke, T.O.; David, P.; Ubana, E.I.; Moshood, Y.L. Electronic Structure Investigation of the Stability, Reactivity, NBO Analysis, Thermodynamics, and the Nature of the Interactions in Methyl-Substituted Imidazolium-Based Ionic Liquids. J. Mol. Liq. 2021, 337, 116458. [Google Scholar] [CrossRef]
- Gázquez, J.L. Perspectives on the Density Functional Theory of Chemical Reactivity. J. Mex. Chem. Soc. 2008, 52, 3–10. [Google Scholar]
- Reina, M.; Castañeda-Arriaga, R.; Perez-Gonzalez, A.; Guzman-Lopez, E.G.; Tan, D.-X.; Reiter, R.J.; Galano, A. A Computer-Assisted Systematic Search for Melatonin Derivatives with High Potential as Antioxidants. Melatonin Res. 2018, 1, 27–58. [Google Scholar] [CrossRef]
- Castro-González, L.M.; Alvarez-Idaboy, J.R.; Galano, A. Computationally Designed Sesamol Derivatives Proposed as Potent Antioxidants. ACS Omega 2020, 5, 9566–9575. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Vargas, R.; Galano, A. What Is Important to Prevent Oxidative Stress? A Theoretical Study on Electron-Transfer Reactions between Carotenoids and Free Radicals. J. Phys. Chem. B 2009, 113, 12113–12120. [Google Scholar] [CrossRef] [PubMed]
- Burdett, J.K.; McCormick, T.A. Electron Localization in Molecules and Solids: The Meaning of ELF. J. Phys. Chem. A 1998, 102, 6366–6372. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Silvi, B.; Fourré, I.; Alikhani, M.E. The Topological Analysis of the Electron Localization Function. A Key for a Position Space Representation of Chemical Bonds. Monatshefte Chem.-Chem. Mon. 2005, 136, 855–879. [Google Scholar] [CrossRef]
- Matito, E.; Solà, M. The Role of Electronic Delocalization in Transition Metal Complexes from the Electron Localization Function and the Quantum Theory of Atoms in Molecules Viewpoints. Coord. Chem. Rev. 2009, 253, 647–665. [Google Scholar] [CrossRef]
- Sadovskiĭ, M.V. Electron Localization in Disordered Systems: Critical Behavior and Macroscopic Manifestations. Sov. Phys. Uspekhi 1981, 24, 96. [Google Scholar] [CrossRef]
- Schmider, H.L.; Becke, A.D. Chemical Content of the Kinetic Energy Density. J. Mol. Struct. Theochem. 2000, 527, 51–61. [Google Scholar] [CrossRef]
- Jacobsen, H. Localized-Orbital Locator (LOL) Profiles of Chemical Bonding. Can. J. Chem. 2008, 86, 695–702. [Google Scholar] [CrossRef]
- Divya, P.; Muthuraja, P.; Dhandapani, M.; Jothy, V.B. Hydrogen Bonding Interactions on Molecular Properties of Pesticidal Compound 4-Nitrophthalic Acid: Experimental Density Functional Theory Computations, Electron Localized Function, Localized Orbital Locator Analysis and Molecular Docking Scrutiny. Spectrosc. Lett. 2022, 55, 362–372. [Google Scholar] [CrossRef]
- Amić, D.; Stepanić, V.; Lučić, B.; Marković, Z.; Dimitrić Marković, J.M. PM6 Study of Free Radical Scavenging Mechanisms of Flavonoids: Why Does O–H Bond Dissociation Enthalpy Effectively Represent Free Radical Scavenging Activity? J. Mol. Model. 2013, 19, 2593–2603. [Google Scholar] [CrossRef]
- Foti, M.C. Antioxidant Properties of Phenols. J. Pharm. Pharmacol. 2007, 59, 1673–1685. [Google Scholar] [CrossRef]
- Olszowy, M. What Is Responsible for Antioxidant Properties of Polyphenolic Compounds from Plants? Plant Physiol. Biochem. 2019, 144, 135–143. [Google Scholar] [CrossRef]
- Klein, E.; Rimarčík, J.; Senajová, E.; Vagánek, A.; Lengyel, J. Deprotonation of Flavonoids Severely Alters the Thermodynamics of the Hydrogen Atom Transfer. Comput. Theor. Chem. 2016, 1085, 7–17. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Gu, D.; Liu, C.; Wang, Y.; Tang, S.; Yin, Y.; Tian, J. Fermentation of Robinia Pseudoacacia Flower for Improving the Antioxidation: Optimized Conditions, Active Composition, Mechanism, and Biotransformation Process. Prep. Biochem. Biotechnol. 2023, 53, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Lespade, L.; Bercion, S. Theoretical investigation of the effect of sugar substitution on the antioxidant properties of flavonoids. Free Radic. Res. 2012, 46, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Marković, Z.; Milenković, D.; Đorović, J.; Dimitrić Marković, J.M.; Stepanić, V.; Lučić, B.; Amić, D. Free Radical Scavenging Activity of Morin 2′-O− Phenoxide Anion. Food Chem. 2012, 135, 2070–2077. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Deng, G.; Zhang, Y.-C. Multiple Free Radical Scavenging Reactions of Flavonoids. Dyes Pigment. 2022, 198, 109877. [Google Scholar] [CrossRef]
- Amić, A.; Marković, Z.; Dimitrić Marković, J.M.; Stepanić, V.; Lučić, B.; Amić, D. Towards an Improved Prediction of the Free Radical Scavenging Potency of Flavonoids: The Significance of Double PCET Mechanisms. Food Chem. 2014, 152, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Vashistha, V.K.; Das, D.K. Recent Advances in the Antioxidant Activity and Mechanisms of Chalcone Derivatives: A Computational Review. Free Radic. Res. 2022, 56, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.W. Reorganization energies, entropies, and free energy surfaces for electron transfer. J. Phys. Chem. B 2021, 125, 7940–7945. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship—A Quantum Chemical Study. Antioxidants 2020, 9, 461. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Reprint of: Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2014, 66, 45–57. [Google Scholar] [CrossRef]
- Zhang, M.; Vervoort, L.; Moalin, M.; Mommers, A.; Douny, C.; den Hartog, G.J.M.; Haenen, G.R.M.M. The Chemical Reactivity of (-)-Epicatechin Quinone Mainly Resides in Its B-Ring. Free Radic. Biol. Med. 2018, 124, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Sroka, Z.; Żbikowska, B.; Hładyszowski, J. The Antiradical Activity of Some Selected Flavones and Flavonols. Experimental and Quantum Mechanical Study. J. Mol. Model. 2015, 21, 307. [Google Scholar] [CrossRef] [PubMed]
- Parcheta, M.; Świsłocka, R.; Orzechowska, S.; Akimowicz, M.; Choińska, R.; Lewandowski, W. Recent Developments in Effective Antioxidants: The Structure and Antioxidant Properties. Materials 2021, 14, 1984. [Google Scholar] [CrossRef]
- Jabbari, M.; Jabbari, A. Antioxidant Potential and DPPH Radical Scavenging Kinetics of Water-Insoluble Flavonoid Naringenin in Aqueous Solution of Micelles. Colloids Surf. A Physicochem. Eng. Asp. 2016, 489, 392–399. [Google Scholar] [CrossRef]
- Musialik, M.; Kuzmicz, R.; Pawłowski, T.S.; Litwinienko, G. Acidity of Hydroxyl Groups: An Overlooked Influence on Antiradical Properties of Flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Trang, N.V.; Thuy, P.T.; Thanh, D.T.M.; Son, N.T. Benzofuran–Stilbene Hybrid Compounds: An Antioxidant Assessment—A DFT Study. RSC Adv. 2021, 11, 12971–12980. [Google Scholar] [CrossRef]
- Thuy, P.T.; Trang, N.V.; Son, N.T. Antioxidation of 2-Phenylbenzofuran Derivatives: Structural-Electronic Effects and Mechanisms. RSC Adv. 2020, 10, 6315–6332. [Google Scholar] [CrossRef] [PubMed]
- Thuy, P.T.; Son, N.T. Thermodynamic and Kinetic Studies on Antioxidant Capacity of Amentoflavone: A DFT (Density Functional Theory) Computational Approach. Free Radic. Res. 2022, 56, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Erdoğan, Ş.; Özbakır Işın, D. A DFT Study on OH Radical Scavenging Activities of Eriodictyol, Isosakuranetin and Pinocembrin. J. Biomol. Struct. Dyn. 2022, 40, 10802–10811. [Google Scholar] [CrossRef]
- Vo, Q.V.; Nam, P.C.; Thong, N.M.; Trung, N.T.; Phan, C.-T.D.; Mechler, A. Antioxidant Motifs in Flavonoids: O–H versus C–H Bond Dissociation. ACS Omega 2019, 4, 8935–8942. [Google Scholar] [CrossRef]
- Milenković, D.; Dimić, D.; Avdović, E.; Simijonović, D.; Vojinović, R.; Marković, Z. A Thermodynamic and Kinetic HO Radical Scavenging Study and Protein Binding of Baicalein. J. Chem. Thermodyn. 2023, 185, 107110. [Google Scholar] [CrossRef]
- Costentin, C. Electrochemical Approach to the Mechanistic Study of Proton-Coupled Electron Transfer. Chem. Rev. 2008, 108, 2145–2179. [Google Scholar] [CrossRef] [PubMed]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current Status of Transition-State Theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A Program for Real-Space Analysis of Quantum Chemical Interactions in Solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Hovorun, D.M. Energy of the CH⋯O H-Bonds and Others Specific Contacts in the Q Molecule: QM/QTAIM Approximation Formulas. J. Mol. Liq. 2020, 313, 113456. [Google Scholar] [CrossRef]
- Šimunková, M.; Valko, M.; Bučinský, L.; Malček, M. Structure Functionality Relationship of Flavonoids (Myricetin, Morin, Taxifolin and 3′,4′-Dihydroxyflavone). A Computational Study via the Cupric Ion Probe. J. Mol. Struct. 2020, 1222, 128923. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, X. Computational Investigation of Flavonol-Based GLP-1R Agonists Using DFT Calculations and Molecular Docking. Comput. Theor. Chem. 2020, 1190, 113005. [Google Scholar] [CrossRef]
- Zannou, O.; Koca, I. Greener extraction of anthocyanins and antioxidant activity from blackberry (Rubus spp) using natural deep eutectic solvents. LWT 2022, 158, 113184. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Deng, G.; Guo, R.; Fu, Z.-M.; Chen, D.-F. The Influence of the H5⋯OC4 Intramolecular Hydrogen-Bond (IHB) on the Antioxidative Activity of Flavonoid. Phytochemistry 2019, 160, 19–24. [Google Scholar] [CrossRef]
- Zhong, Y.; Chen, Y.; Feng, X.; Sun, Y.; Cui, S.; Li, X.; Jin, X.; Zhao, G. Hydrogen-Bond Facilitated Intramolecular Proton Transfer in Excited State and Fluorescence Quenching Mechanism of Flavonoid Compounds in Aqueous Solution. J. Mol. Liq. 2020, 302, 112562. [Google Scholar] [CrossRef]
- Ji, F.; Guo, Y.; Wang, M.; Wang, C.; Wu, Z.; Wang, S.; Wang, H.; Feng, X.; Zhao, G. New Insights into ESIPT Mechanism of Three Sunscreen Compounds in Solution: A Combined Experimental and Theoretical Study. Colloids Surf. B Biointerfaces 2021, 207, 112039. [Google Scholar] [CrossRef]
- Wu, P.; Chaudret, R.; Hu, X.; Yang, W. Noncovalent Interaction Analysis in Fluctuating Environments. J. Chem. Theory Comput. 2013, 9, 2226–2234. [Google Scholar] [CrossRef]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Aderinwale, T.; Christoffer, C.W.; Sarkar, D.; Alnabati, E.; Kihara, D. Computational Structure Modeling for Diverse Categories of Macromolecular Interactions. Curr. Opin. Struct. Biol. 2020, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.B.; Murphy, R.B.; Sindhikara, D.; Borrelli, K.W.; Grisewood, M.J.; Ranalli, F.; Dixon, S.L.; Jerome, S.; Boyles, N.A.; Day, T.; et al. Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein–Ligand Binding. J. Chem. Theory Comput. 2021, 17, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.E.Z.; Ambarsari, L.; Hariana, K. Children’s Opinion on Vegetables Consumption: A Qualitative Study on School-Agers in City of Semarang. Indones. J. Appl. Res. IJAR 2021, 2, 126–143. [Google Scholar]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Vásquez-Espinal, A.; Yañez, O.; Osorio, E.; Areche, C.; García-Beltrán, O.; Ruiz, L.M.; Cassels, B.K.; Tiznado, W. Theoretical Study of the Antioxidant Activity of Q Oxidation Products. Front. Chem. 2019, 7, 818. [Google Scholar] [CrossRef]
- Kamel, E.M.; Bin-Ammar, A.; El-Bassuony, A.A.; Alanazi, M.M.; Altharawi, A.; Ahmeda, A.F.; Alanazi, A.S.; Lamsabhi, A.M.; Mahmoud, A.M. Molecular Modeling and DFT Studies on the Antioxidant Activity of Centaurea Scoparia Flavonoids and Molecular Dynamics Simulation of Their Interaction with β-Lactoglobulin. RSC Adv. 2023, 13, 12361–12374. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Y.; Zhu, T. The Theoretical Investigation of Monohydroxy Flavone: A Combined DFT and Molecular Docking Study. J. Mol. Struct. 2022, 1250, 131823. [Google Scholar] [CrossRef]
- Erdoğan, Ş.; Özbakır Işın, D. Molecular Docking and A DFT Study on the Antiradical Activity of Naringenin and Hesperetin with Nitric Oxide, Peroxy, and Methoxy Radicals. J. Phys. Org. Chem. 2023, 36, e4479. [Google Scholar] [CrossRef]
- Bamdad, F.; Farrokhpour, H.; Ashrafizaadeh, M.; Najafi, B. Decomposition of the Interaction Energy of Several Flavonoids with Escherichia Coli DNA Gyr Using the SAPT (DFT) Method: The Relation between the Interaction Energy Components, Ligand Structure, and Biological Activity. Biochim. Biophys. Acta BBA-Gen. Subj. 2022, 1866, 130111. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yu, Y.; Geng, S.; Liu, B.; Hao, Y.; Liang, G. Computational Methods for the Interaction between Cyclodextrins and Natural Compounds: Technology, Benefits, Limitations, and Trends. J. Agric. Food Chem. 2022, 70, 2466–2482. [Google Scholar] [CrossRef] [PubMed]
- Raeessi-Babaheydari, E.; Farhadian, S.; Shareghi, B. Evaluation of Interaction between Citrus Flavonoid, Naringenin, and Pepsin Using Spectroscopic Analysis and Docking Simulation. J. Mol. Liq. 2021, 339, 116763. [Google Scholar] [CrossRef]
- Singh, A.; Vanga, S.K.; Orsat, V.; Raghavan, V. Application of Molecular Dynamic Simulation to Study Food Proteins: A Review. Crit. Rev. Food Sci. Nutr. 2018, 58, 2779–2789. [Google Scholar] [CrossRef]
- Chen, Q.; Xiao, Y.; Zhang, W.; Mu, W. Current Methods and Applications in Computational Protein Design for Food Industry. Crit. Rev. Food Sci. Nutr. 2020, 60, 3259–3270. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed]
- Fuguet, E.; Ràfols, C.; Mañé, M.; Ruiz, R.; Bosch, E. Acidity Constants of Hydroxyl Groups Placed in Several Flavonoids: Two Flavanones, Two Flavones and Five Flavonols. Talanta 2023, 253, 124096. [Google Scholar] [CrossRef]
- Toth, A.M.; Liptak, M.D.; Phillips, D.L.; Shields, G.C. Accurate Relative pKa Calculations for Carboxylic Acids Using Complete Basis Set and Gaussian-n Models Combined with Continuum Solvation Methods. J. Chem. Phys. 2001, 114, 4595–4606. [Google Scholar] [CrossRef]
- Ribeiro, A.R.; Schmidt, T.C. Determination of Acid Dissociation Constants (pKa) of Cephalosporin Antibiotics: Computational and Experimental Approaches. Chemosphere 2017, 169, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Lemańska, K.; Szymusiak, H.; Tyrakowska, B.; Zieliński, R.; Soffers, A.E.M.F.; Rietjens, I.M.C.M. The Influence of pH on Antioxidant Properties and the Mechanism of Antioxidant Action of Hydroxyflavones. Free Radic. Biol. Med. 2001, 31, 869–881. [Google Scholar] [CrossRef] [PubMed]
- XLOGP3 (v3.2.2). Available online: http://www.sioc-ccbg.ac.cn/skins/ccbgwebsite/software/xlogp3/ (accessed on 18 March 2024).
- On-Line Lipophilicity/Aqueous Solubility Calculation Software. Available online: https://vcclab.org/lab/alogps/ (accessed on 18 March 2024).
- Marvin. Available online: https://chemaxon.com/marvin (accessed on 18 March 2024).
- Chemaxon. Available online: https://chemaxon.com (accessed on 18 March 2024).
- Hitchcock, S.A.; Pennington, L.D. Structure−Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.A.; Berger, B.K.; Kogelman, K. 8.18 Chromatographic Separations and Analysis: Supercritical Fluid Chromatography for Chiral Analysis and Semi-preparative Purification. Compr. Chirality 2012, 8, 354–392. [Google Scholar]
- Bytheway, I.; Darley, M.G.; Popelier, P.L.A. The Calculation of Polar Surface Area from First Principles: An Application of Quantum Chemical Topology to Drug Design. ChemMedChem 2008, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Markiewicz, A.; Darłak, P.; Sikora, J.; Adla, S.K.; Bagina, S.; Huttunen, K.M. Current chemical, biological, and physiological views in the development of successful brain-targeted pharmaceutics. Neurotherapeutics 2022, 19, 942–976. [Google Scholar] [CrossRef] [PubMed]
- Ronsisvalle, S.; Panarello, F.; Longhitano, G.; Siciliano, E.A.; Montenegro, L.; Panico, A. Natural Flavones and Flavonols: Relationships among Antioxidant Activity, Glycation, and Metalloproteinase Inhibition. Cosmetics 2020, 7, 71. [Google Scholar] [CrossRef]
- Lemmens, K.J.; Vrolijk, M.F.; Bouwman, F.G.; Van der Vijgh, W.J.; Bast, A.; Haenen, G.R. The minor structural difference between the antioxidants quercetin and 4’O-methylquercetin has a major impact on their selective thiol toxicity. Int. J. Mol. Sci. 2014, 15, 7475–7484. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Formula | Description |
|---|---|---|
| Hardness(η) Softness (S) | η = (IP − EA)/2 S = 1/2η | η reflects the reluctance towards deformation or polarization of the electron cloud under slight perturbation [106,107,108]. |
| Chemical potential (μ) | −μ = (IP + EA)/2 | μ indicates the direction of charge flow. Electrons will migrate from high μ to low μ locations [109,110]. |
| Electronegativity (χ) | χ = (IP + EA)/2 | χ gives the tendency of a molecule to attract electrons. |
| Electrophilicity (ω) | ω = μ2/2η | ω reflects the electron-donating ability of a molecule. |
| ω+ | ω+ = (IP + 3EA)2/16(IP − EA) | ω− and ω+ reflect respectively the electron-donating and electron-accepting ability of antioxidants [111,112]. |
| ω− | ω− = (3IP + EA)2/16(IP − EA) |
| Mechanism | Reaction Equation | Enthalpy |
|---|---|---|
| HAT | ArOHoptimized → ArO•optimized + H• | Bond dissociation enthalpy (BDE) |
| SPLET | ArOHoptimized → ArO−optimized + H+ | Proton affinity (PA) |
| ArO−unoptimized → ArO•optimized + e− | Electron transfer enthalpy (ETE) | |
| ETPT | ArOHoptimized → ArOH•+optimized + e− | Ionization potential (IP) |
| ArOH•+unoptimized → ArO•optimized + H+ | Proton dissociation enthalpy (PDE) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, C.; Li, Z.; Moalin, M.; Hartog, G.J.M.d.; Zhang, M. Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids—An Overview. Molecules 2024, 29, 2627. https://doi.org/10.3390/molecules29112627
Wang Y, Li C, Li Z, Moalin M, Hartog GJMd, Zhang M. Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids—An Overview. Molecules. 2024; 29(11):2627. https://doi.org/10.3390/molecules29112627
Chicago/Turabian StyleWang, Yue, Chujie Li, Zhengwen Li, Mohamed Moalin, Gertjan J. M. den Hartog, and Ming Zhang. 2024. "Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids—An Overview" Molecules 29, no. 11: 2627. https://doi.org/10.3390/molecules29112627
APA StyleWang, Y., Li, C., Li, Z., Moalin, M., Hartog, G. J. M. d., & Zhang, M. (2024). Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids—An Overview. Molecules, 29(11), 2627. https://doi.org/10.3390/molecules29112627